In-Depth Understanding of Camellia oleifera Self-Incompatibility by Comparative Transcriptome, Proteome and Metabolome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
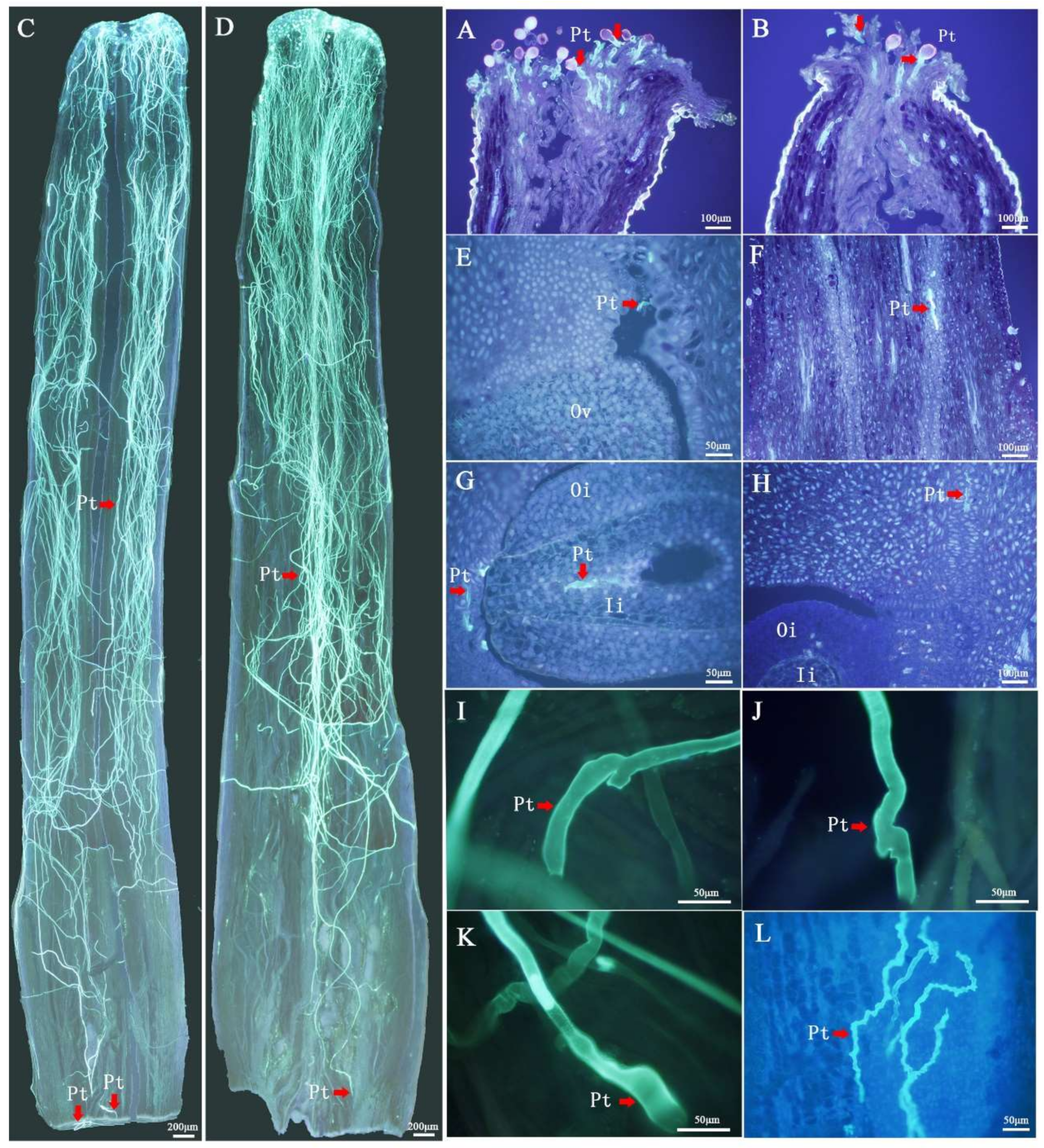
2.1. Cytological Observation of SI Type in C. Oleifera
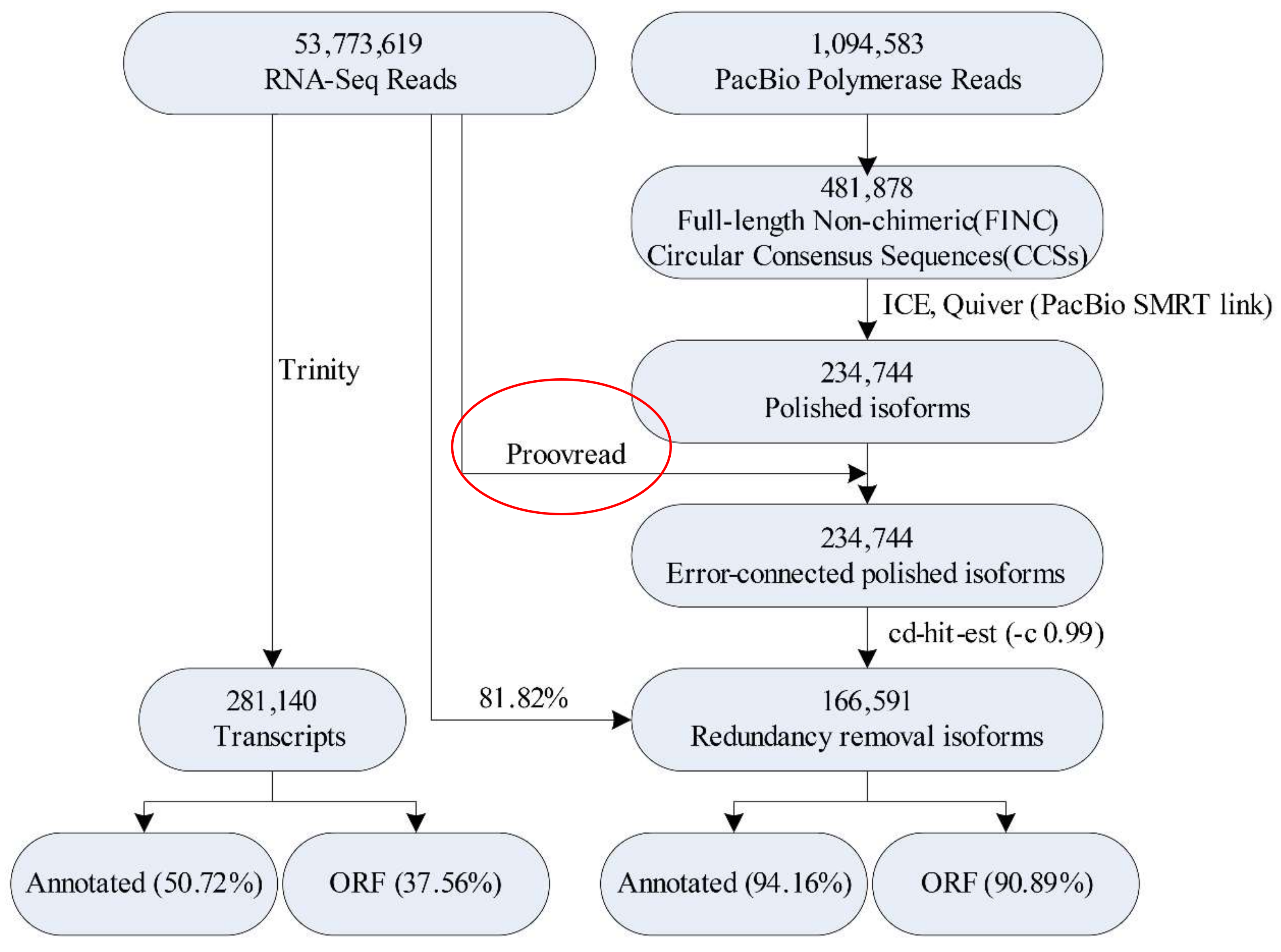
2.2. Illumina- and SMRT-Based RNA Sequencing and Error Correction
2.3. Functional Annotation and Categorization of Transcripts
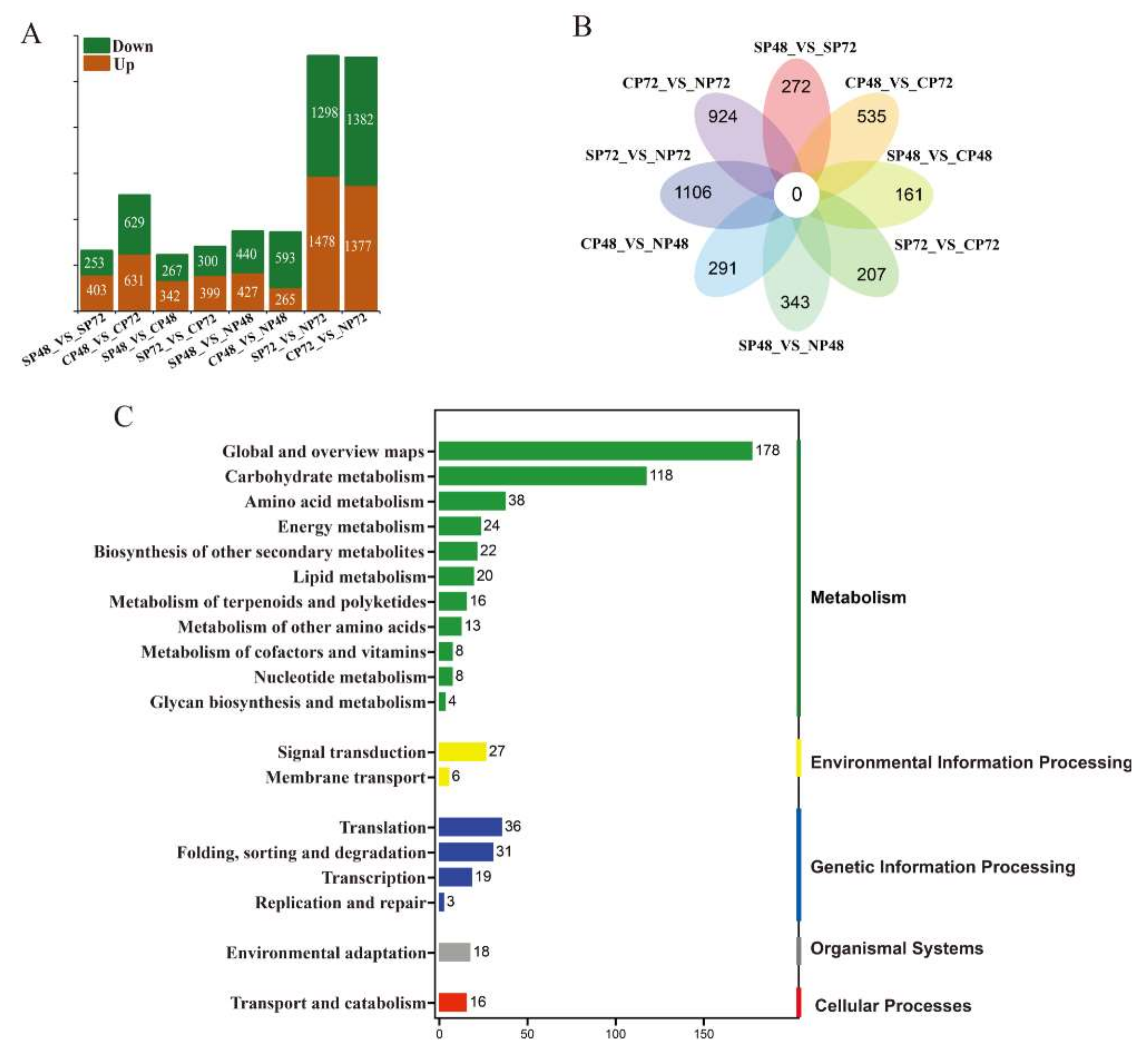
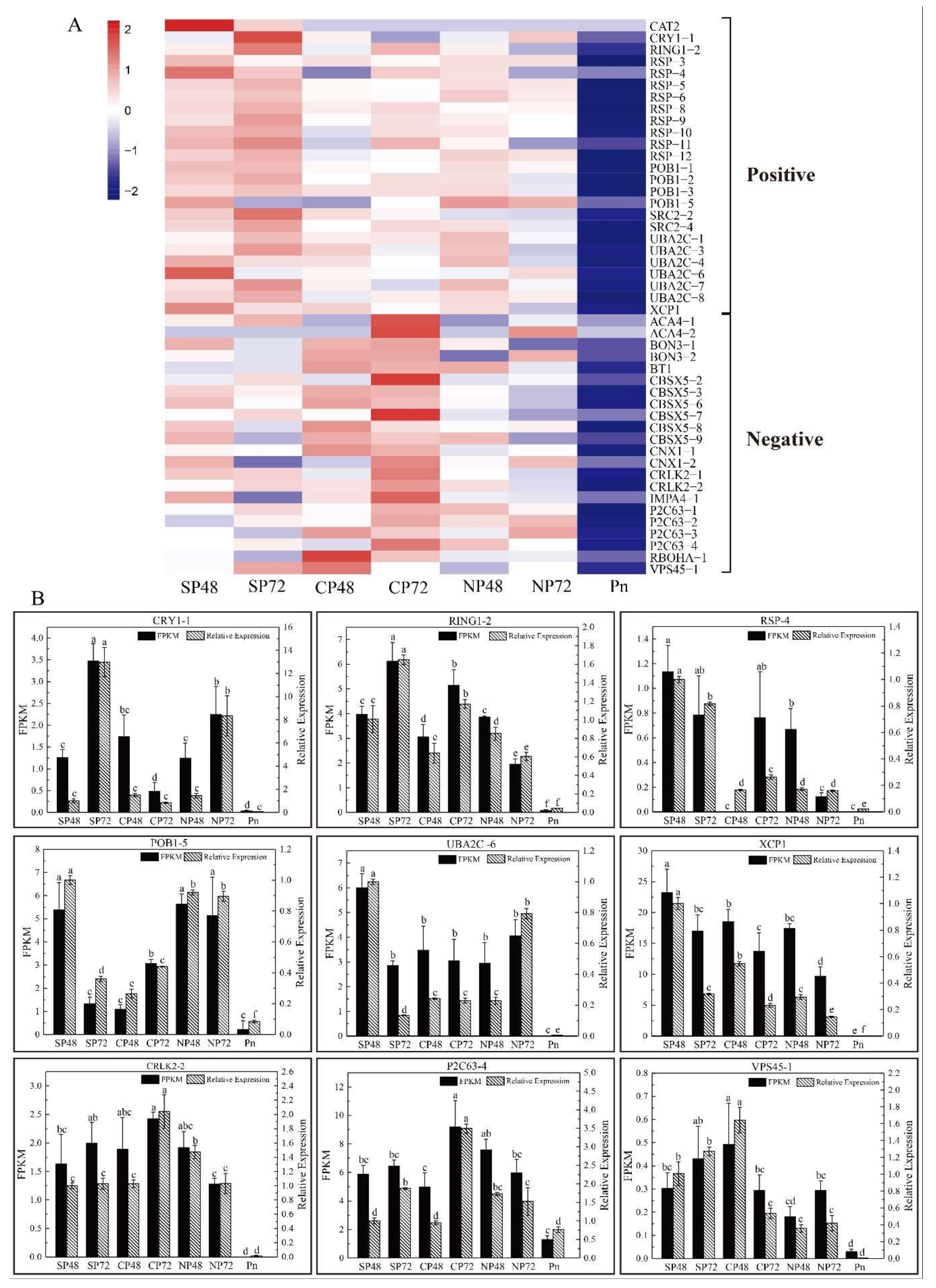
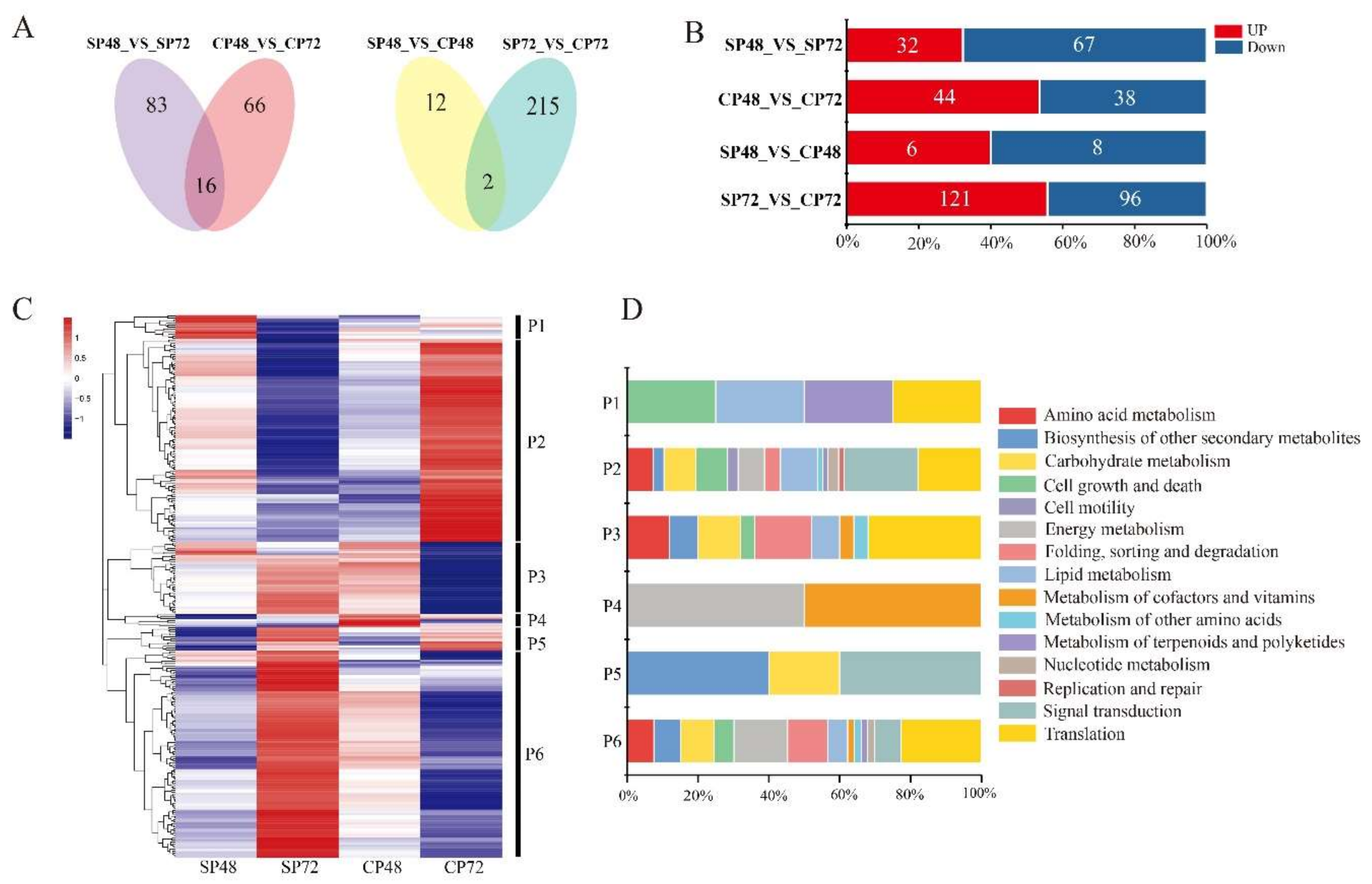
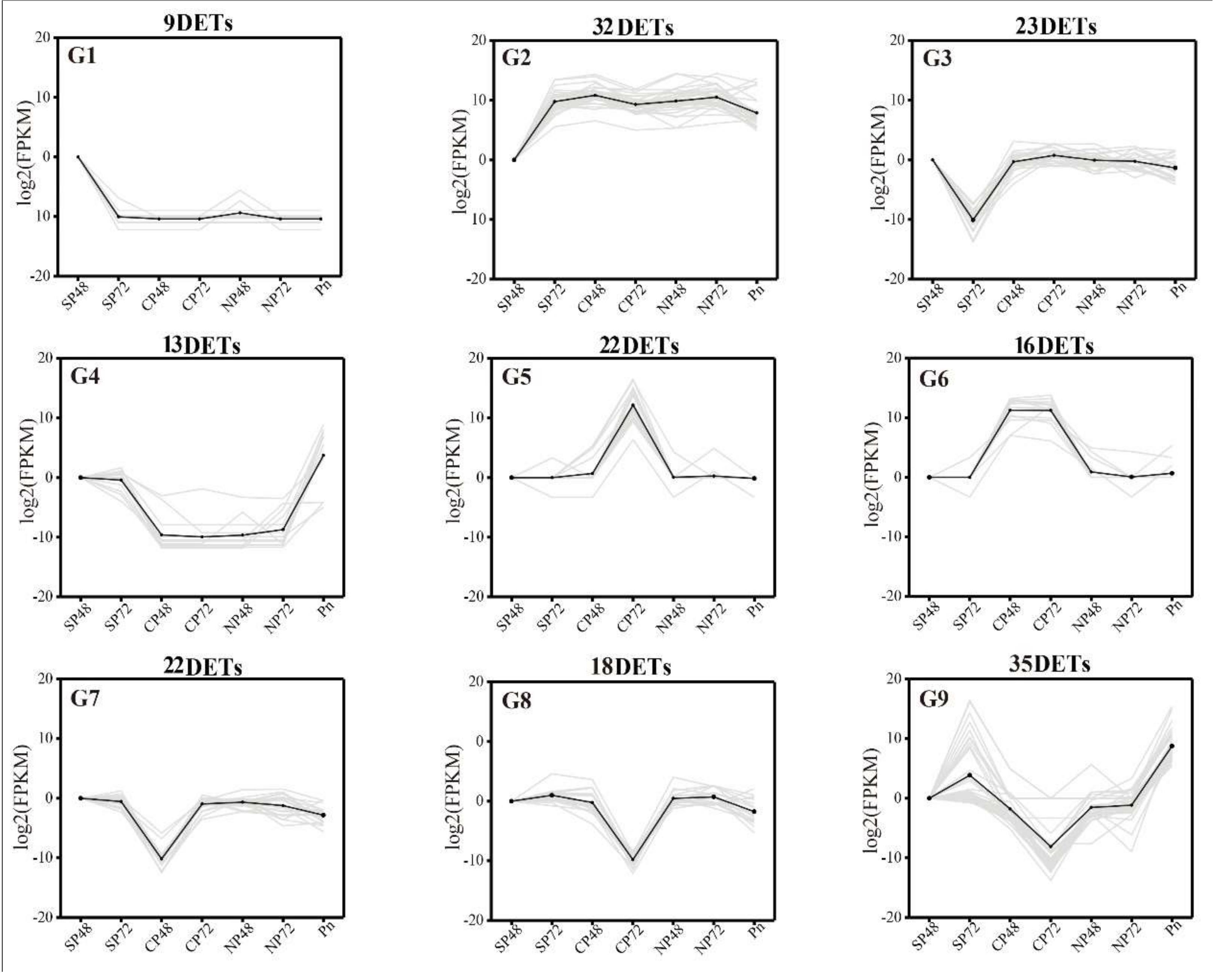
2.4. Transcript Expression Profiles and Enrichment in Self- and Cross-Pollinated Plants
2.5. Identification of PCD Candidate Transcriptsin Self- and Cross-Pollination
2.6. Proteomic Changes of C. oleifera in Response to SI
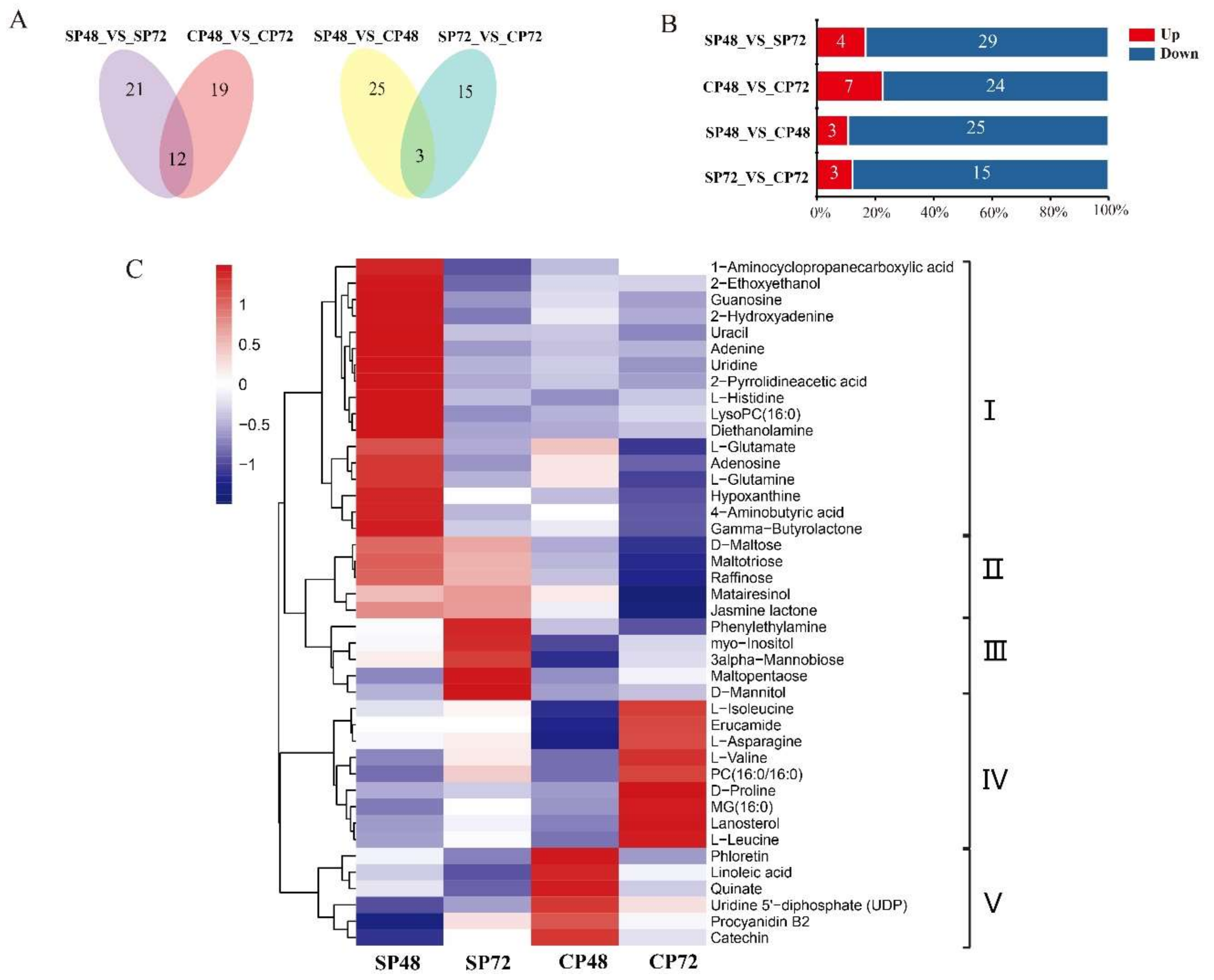
2.7. Overall Metabolite in Different Groups of C. Oleifera
2.8. Integrated Transcriptomic, Proteomic and Metabolic Datasets
3. Discussion
3.1. PCD-Regulated Genes Regulate the Inhibition of Pollen Tube in Self-Pollination
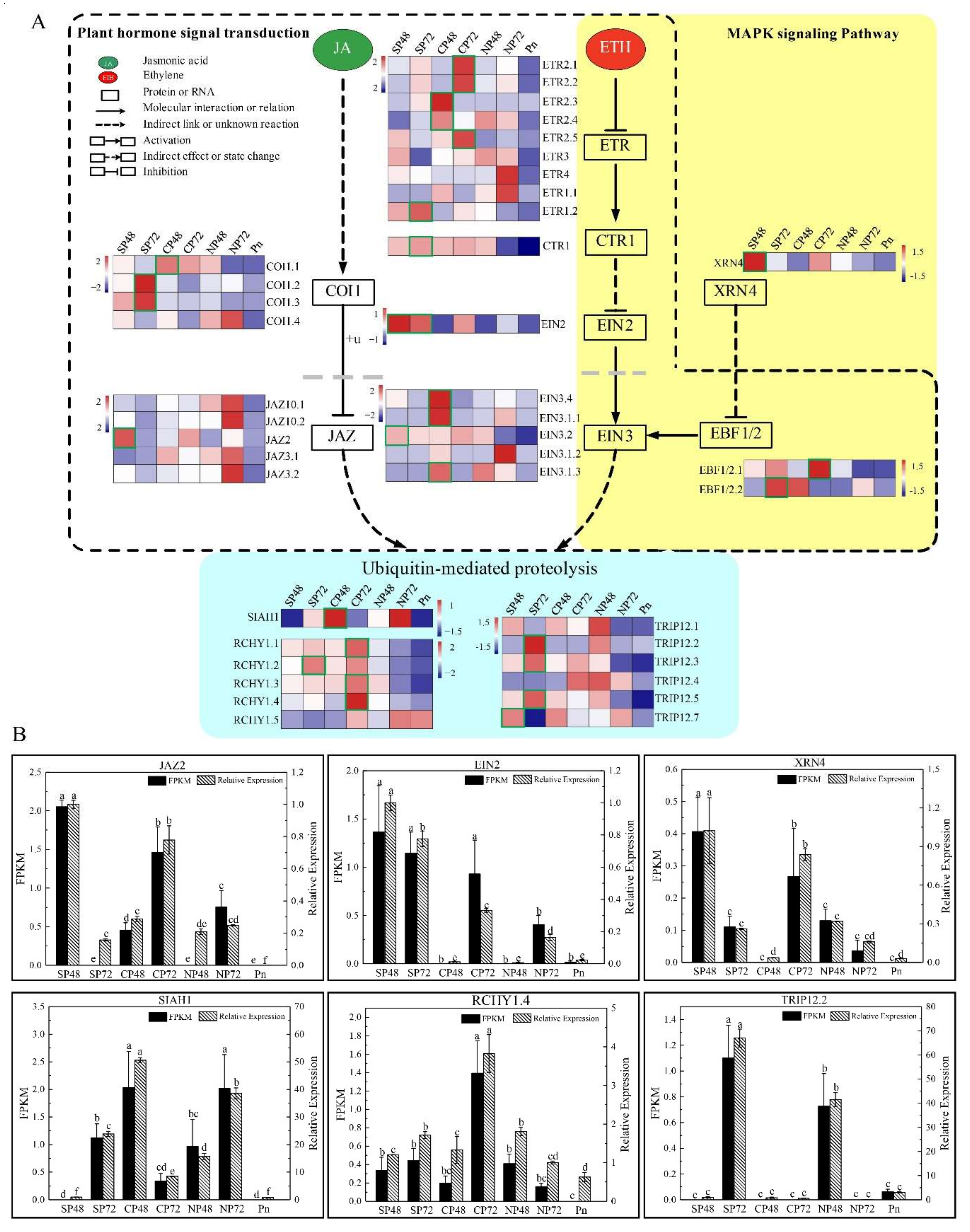
3.2. MAPK Signaling Pathway Involvement in SI of C. Oleifera
3.3. Plant Hormone Signal Transduction Involvement in SI of C. Oleifera
3.4. ABC Transporters Involvement in SI of C. Oleifera
3.5. Ubiquitin-Mediated Proteolysis Involvement in SI of C. Oleifera
4. Materials and Methods
4.1. Plant Materials
4.2. Pollination Treatment, Sample Collection and Cytological Observation
4.3. RNA Preparation
4.4. PacBio cDNA Library Construction and Third-Generation Sequencing
4.5. Illumina cDNA Library Construction and Second-Generation Sequencing
4.6. Proteomic Analysis
4.7. Metabolomic Analysis
4.8. Transcript, Protein and Metabolite Correlation Analysis
4.9. Quantitative RT-PCR
4.10. Statistical Analysis
4.11. Availability of Sequence Data
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COI1 | coronatine-insensitive protein 1 (K13463) |
| JAZ | jasmonate-zim-domain protein (K13464) |
| ETR | ethylene receptor (K14509) |
| CTR1 | serine/threonine-protein kinase 1 (K08856) |
| EIN2 | ethylene-insensitive protein 2 (K14513) |
| EIN3 | EIN3-like protein (K14514) |
| EBF1/2 | EIN3-binding F-box protein (K14515) |
| CALM | calmodulin (K02183) |
| XRN4 | 5′-3′ exoribonuclease 4 (K20553) |
| SIAH1 | E3 ubiquitin-protein ligase SIAH1 (K04506) |
| RCHY1 | RING finger and CHY zinc finger domain-containing protein 1 (K10144) |
| TRIP12 | E3 ubiquitin-protein ligase UPL4 (K10590) |
| WRKY33 | WRKY transcription factor 33 (K13424) |
| ARR-B | two-component response regulator ARR2 isoform (K14491) |
| ABA2 | xanthoxin dehydrogenase (EC:1.1.1.288) |
| TRA2 | transformer-2 protein |
| TUBA | tubulin alpha |
| TGA | transcription factor TGA |
| PLCD | phosphatidylinositol phospholipase C, delta (EC:3.1.4.11) |
| PFN | profilin |
| MAPK1_3 | mitogen-activated protein kinase 1/3 (EC:2.7.11.24) |
| SF3B4 | splicing factor 3B subunit 4 |
| EIF4A | translation initiation factor 4A |
| UBE1 | ubiquitin-activating enzyme E1 (EC:6.2.1.45) |
| WDR61 | WD repeat-containing protein 61 |
References
- Zhuang, R.L. Comprehensive Utilization of Tea-Oil Fruits, Tea-Oil Tree (Camellia oleifera Abel.) of China; Chinese Forestry Publish House: Beijing, China, 2008; pp. 1–3. [Google Scholar]
- Xiao, Y. Investigation and prospect of bio-active components in vegetable oil. Cereal Food Ind. 2006, 13, 1–5. [Google Scholar]
- Li, H.; Fang, X.Z.; Zhong, H.Y.; Fei, X.Q.; Luo, F. Variation of physicochemical properties and nutritional components of oil-tea Camellia seeds during riping. For. Res. 2014, 27, 86–91. [Google Scholar] [CrossRef]
- Li, H.; Zhou, G.; Zhang, H.; He, Y. Chemical constituents and biological activities of saponin from the seed of Camellia oleifera. Sci. Res. Essays 2010, 5, 4088–4092. [Google Scholar]
- Goldberg, E.E.; Kohn, J.R.; Lande, R.; Robertson, K.A.; Smith, S.A.; Igić, B. Species selection maintains self-incompatibility. Science 2010, 330, 493–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin-Tong, N.; Franklin, F.C.H. Gametophytic self-incompatibility inhibits pollen tube growth using different mechanisms. Trends Plant Sci. 2003, 8, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Waligórski, P.; Szaleniec, M. Prediction of white cabbage (Brassica oleracea var. capitata) self-incompatibility based on neural network and discriminant analysis of complex electrophoretic patterns. Comput. Biol. Chem. 2010, 34, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E. Late-acting self-incompatibility—The pariah breeding system in flowering plants. New Phytol. 2014, 203, 717–734. [Google Scholar] [CrossRef]
- Shi, G.J.; Hou, X.L. Measurement of Self-incompatible by Fluoroscope Observation in Non-heading Chinese Cabbage. J. Wuhan Bot. Res. 2004, 22, 197–200. [Google Scholar]
- Xu, Y.; Zhang, S. Characterization and molecular mechanism of gametophytic self-incompatibility in pears. J. Fruit Sci. 2003, 20, 59–63. [Google Scholar]
- Li, X.; Zhang, S.; Wu, J.; Wu, H. Studies on S genotypes and molecular mechanism of self-incompatibility in cherry. Biotechnol. Bull. 2006, 2006, 28–33. [Google Scholar] [CrossRef]
- Stein, J.C.; Howlett, B.; Boyes, D.C.; Nasrallah, M.E.; Nasrallah, J.B. Molecular cloning of a putative receptor protein kinase gene encoded at the self-incompatibility locus of Brassica oleracea. Proc. Natl. Acad. Sci. USA 1991, 88, 8816–8820. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Suzuki, G.; Takayama, S. Milestones Identifying Self-Incompatibility Genes in Brassica Species: From Old Stories to New Findings. Self-Incompat. Flower. Plants 2008, 4527, 157–182. [Google Scholar] [CrossRef]
- Schopfer, C.R.; Nasrallah, M.E.; Nasrallah, J.B. The male determinant of self-incompatibility in Brassica. Science 1999, 286, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, T.; Hatakeyama, K.; Suzuki, G.; Watanabe, M.; Isogai, A.; Hinata, K. The S receptor kinase determines self-incompatibility in Brassica stigma. Nature 2000, 403, 913. [Google Scholar] [CrossRef] [PubMed]
- Kohji, M.; Hiroshi, S.; Megumi, I.; Fang-Sik, C.; Masao, W.; Akira, I.; Seiji, T. A membrane-anchored protein kinase involved in Brassica self-incompatibility signaling. Science 2004, 303, 1516–1519. [Google Scholar]
- Kakita, M.; Murase, K.; Iwano, M.; Matsumoto, T.; Watanabe, M.; Shiba, H.; Isogai, A.; Takayama, S. Two distinct forms of M-locus protein kinase localize to the plasma membrane and interact directly with S-locus receptor kinase to transduce self-incompatibility signaling in Brassica rapa. Plant Cell 2007, 19, 3961–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murfett, J.; Atherton, T.L.; Mou, B.; Gassert, C.S.; McClure, B.A. S-RNase expressed in transgenic Nicotiana causes S-allele-specific pollen rejection. Nature 1994, 367, 563–566. [Google Scholar] [CrossRef]
- Sijacic, P.; Wang, X.; Skirpan, A.L.; Wang, Y.; Dowd, P.E.; McCubbin, A.G.; Huang, S.; Kao, T.H. Identification of the pollen determinant of S-RNase-mediated self-incompatibility. Nature 2004, 429, 302–305. [Google Scholar] [CrossRef]
- McClure, B. New views of S-RNase-based self-incompatibility. Curr. Opin. Plant Biol. 2006, 9, 639–646. [Google Scholar] [CrossRef]
- Chen, J.; Wang, P.; Graaf, B.H.J.D.; Zhang, H.; Wu, J. Phosphatidic Acid Counteracts S-RNase Signaling in Pollen by Stabilizing the Actin Cytoskeleton. Plant Cell 2018, 30, tpc.00021.2018. [Google Scholar] [CrossRef]
- Shi, D.Q.; Tang, C.; Wang, R.Z.; Gu, C.; Wu, X.; Hu, S.; Jiao, J.; Zhang, S.L. Transcriptome and phytohormone analysis reveals a comprehensive phytohormone and pathogen defence response in pear self-/cross-pollination. Plant Cell Rep. 2017, 36, 1785–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.Y.; Li, W.; Doughty, J.; Meng, D.; Yang, Q.; Yuan, H.; Li, Y.; Chen, Q.J.; Yu, J.; Liu, C.S.; et al. A gamma-thionin protein from apple, MdD1, is required for defense against S-RNase-induced inhibition of pollen tube prior to self/non-self recognition. Plant Biotechnol. J. 2019, 17, 2184–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, F.W. The mechanism of pollen incompatibility in Theobroma cacao L. Heredity (Edinb.) 1962, 17, 157–182. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.S.; Wilkinson, M.J. Confocal observations of late-acting self-incompatibility in Theobroma Cacao L. Sex. Plant Reprod. 2012, 25, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Kenrick, J.; Kaul, V.; Williams, E.G. Self-incompatibility in Acacia retinodes: Site of pollen-tube arrest is the nucellus. Planta 1986, 169, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hao, S.; Wang, L.; Fang, W.; Wang, Y.; Li, X. Late-acting self-incompatibility in tea plant (Camellia sinensis). Biologia 2012, 67, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Chao, G.; Yuan, D.; Yang, Y.; Wang, B.; Liu, D.; Feng, Z.; Tan, X. Anatomical Characteristics of Self-Incompatibility in Camellia oleifera. Sci. Silvae Sin. 2015, 51, 60–68. [Google Scholar]
- Zhou, Q.; Zheng, Y. ComparativeDe NovoTranscriptome Analysis of Fertilized Ovules in Xanthoceras sorbifolium Uncovered a Pool of Genes Expressed Specifically or Preferentially in the Selfed Ovule That Are Potentially Involved in Late-Acting Self-Incompatibility. PLoS ONE 2015, 10, e0140507. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.C.; Wang, L.Y.; Wei, K.; Wu, L.Y.; Li, H.L.; Zhang, F.; Cheng, H.; Ni, D.J. Transcriptome analysis reveals self-incompatibility in the tea plant (Camellia sinensis) might be under gametophytic control. Bmc Genom. 2016, 17, 359. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.C.; Tan, L.Q.; Wang, L.Y.; Wei, K.; Wu, L.Y.; Zhang, F.; Cheng, H.; Ni, D.J. Cloning and characterization of an S-RNase gene in Camellia sinensis. Sci. Hortic. 2016, 207, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Lanaud, C.; Fouet, O.; Legavre, T.; Lopes, U.; Sounigo, O.; Eyango, M.C.; Mermaz, B.; Da Silva, M.R.; Loor Solorzano, R.G.; Argout, X.; et al. Deciphering the Theobroma cacao self-incompatibility system: From genomics to diagnostic markers for self-compatibility. J. Exp. Bot. 2017, 68, 4775–4790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C. The Cytological Study on Late-acting Self-incompatibility in Camellia oleifera. PhD Thesis, Central South University of Forestry and Technology, China, 2017. [Google Scholar]
- Wang, C.L.; Xu, G.H.; Jiang, X.T.; Gong, C.; Zhang, S.L. S-RNase triggers mitochondrial alteration and DNA degradation in the incompatible pollen tube of Pyrus pyrifolia in vitro. Plant J. 2009, 57, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Tilgner, H.; Grubert, F.; Snyder, M. A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol. 2013, 31, 1009–1014. [Google Scholar] [CrossRef]
- Minoche, A.E.; Dohm, J.C.; Schneider, J.; Holtgräwe, D.; Viehöver, P.; Montfort, M.; Rosleff Sörensen, T.; Weisshaar, B.; Himmelbauer, H. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 2015, 16, 184. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, Y.; Song, J.; Xu, H.; Xu, J.; Zhu, Y.; Li, X.; Gao, H.; Dong, L.; Qian, J.; et al. High-accuracyde novoassembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 2014, 204, 1041–1049. [Google Scholar] [CrossRef]
- Šamaj, J.; Ovecka, M.; Hlavacka, A.; Lecourieux, F.; Meskiene, I.; Lichtscheidl, I.; Lenart, P.; Salaj, J.; Volkmann, D.; Bögre, L.; et al. Involvement of the mitogen-activated protein kinase SIMK in regulation of root hair tip growth. Embo J. 2002, 21, 3296–3306. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Frankling-Tong, V. Modulating and Monitoring MAPK Activity During Programmed Cell Death in Pollen. Methods Mol. Biol. 2011, 779, 165–183. [Google Scholar]
- Ligterink, W.; Kroj, T.; Nieden, U.; Hirt, H.; Scheel, D. Receptor-Mediated Activation of a MAP Kinase in Pathogen Defense of Plants. Science 1997, 276, 2054–2057. [Google Scholar] [CrossRef] [PubMed]
- Windels, D.; Bucher, E. The 5′-3′ Exoribonuclease XRN4 Regulates Auxin Response via the Degradation of Auxin Receptor Transcripts. Genes 2018, 9, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidhyasekaran, P. Ubiquitin-Proteasome System-Mediated Protein Degradation in Defense Signaling. In Signaling and Communication in Plants; Springer: Dordrecht, The Netherlands.
- Liu, M.; Li, W.; Zhao, G.; Fan, X.; Long, H.; Fan, Y.; Shi, M.; Tan, X.; Zhang, L. New Insights of Salicylic Acid Into Stamen Abortion of Female Flowers in Tung Tree (Vernicia fordii). Front. Genet. 2019, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.G.A.; Gao, M.G. Yariv reagent treatment induces programmed cell death in Arabidopsis cell cultures and implicates arabinogalactan protein involvement. Plant J. 1999, 19, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Williams, B.; Dickman, M. Arabidopsis B-cell lymphoma2 (Bcl-2)-associated athanogene 7 (BAG7)-mediated heat tolerance requires translocation, sumoylation and binding to WRKY29. New Phytol. 2017, 214, 695–705. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, J.; Li, W.; Zhang, L.; Cui, J.; He, Q.; Wang, L.; Jin, B. Transcriptomic Analysis Reveals Mechanisms of Sterile and Fertile Flower Differentiation and Development in Viburnum macrocephalum f. keteleeri. Front. Plant Sci. 2017, 8, 261. [Google Scholar] [CrossRef] [Green Version]
- Sala, K.; Malarz, K.; Barlow, P.W.; Kurczyńska, E.U. Distribution of some pectic and arabinogalactan protein epitopes during Solanum lycopersicum (L.) adventitious root development. Bmc Plant Biol. 2017, 17, 25. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.A.; Dangl, J.L.; Jones, J.D.G. Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc. Natl. Acad. Sci. USA 2002, 99, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Meng, D.; Gu, Z.; Li, W.; Wang, A.; Yuan, H.; Yang, Q.; Li, T. Apple MdABCF assists in the transportation of S-RNase into pollen tubes. Plant J. 2014, 78, 990–1002. [Google Scholar] [CrossRef]
- Liao, T.; Yuan, D.Y.; Zou, F.; Gao, C.; Yang, Y.; Zhang, L.; Tan, X.F. Self-sterility in Camellia oleifera may be due to the prezygotic late-acting self-incompatibility. PLoS ONE 2014, 9, e99639. [Google Scholar] [CrossRef]
- Gao, C.; Yuan, D.; Yang, Y.; Wang, B.; Liu, D.; Zou, F. Pollen tube growth and double fertilization in Camellia oleifera. J. Am. Soc. Hortic. Sci. 2015, 140, 12–18. [Google Scholar] [CrossRef] [Green Version]
- X, C. Identification of Self-incompatibility Model, cloning and expression of Correlative Gene in Camellia sinensis. Ph.D. Thesis, Nanjing Agricultural University, Nanjing, China, 2010; pp. 57–58. [Google Scholar]
- Liu, Z.Q.; Xu, G.H.; Zhang, S.L. Pyrus pyrifolia stylar S-RNase induces alterations in the actin cytoskeleton in self-pollen and tubes in vitro. Protoplasma 2007, 232, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Eaves, D.J.; Flores-Ortiz, C.; Haque, T.; Lin, Z.; Teng, N.; Franklin-Tong, V.E. Self-incompatibility in Papaver: Advances in integrating the signalling network. Biochem. Soc. Trans. 2014, 42, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, K.A.; Poulter, N.S.; Franklin-Tong, V.E. Taking one for the team: Self-recognition and cell suicide in pollen. J. Exp. Bot. 2014, 65, 1331–1342. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.Y.; Liu, Y.; Zhang, S. Activation of a mitogen-activated protein kinase pathway is involved in disease resistance in tobacco. Proc. Natl. Acad. Sci. USA 2001, 98, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Kroj, T.; Rudd, J.J.; Nürnberger, T.; Gäbler, Y.; Lee, J.; Scheel, D. Mitogen-activated protein kinases play an essential role in oxidative burst-independent expression of pathogenesis-related genes in parsley. J. Biol. Chem. 2003, 278, 2256–2264. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, J.B. Recognition and rejection of self in plant self-incompatibility: Comparisons to animal histocompatibility. Trends Immunol. 2005, 26, 412–418. [Google Scholar] [CrossRef]
- Rudd, J.J.; Franklin-Tong, V.E. Signals and targets of the self-incompatibility response in pollen of Papaver rhoeas. J. Exp. Bot. 2003, 54, 141–148. [Google Scholar] [CrossRef]
- Franklin-Tong, V.E.; Holdaway-Clarke, T.L.; Straatman, K.R.; Kunkel, J.G.; Hepler, P.K. Involvement of extracellular calcium influx in the self-incompatibility response of Papaver rhoeas. Plant J. 2002, 29, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Uluhan, E.; Keleş, E.N.; Tufan, F. Analysis of WRKY Transcription Factors in Barley Cultivars Infected with Fusarium culmorum. Int. J. Life Sci. 2019, 2, 165–174. [Google Scholar]
- Young, T.E.; Gallie, D.R. Regulation of programmed cell death in maize endosperm by abscisic acid. Plant Mol. Biol. 2000, 42, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Gunawardena, A.H.; Pearce, D.M.; Jackson, M.B.; Hawes, C.R.; Evans, D.E. Characterisation of programmed cell death during aerenchyma formation induced by ethylene or hypoxia in roots of maize (Zea mays L.). Planta 2001, 212, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Jong, A.J.D.; Yakimova, E.T.; Kapchina, V.M.; Woltering, E.J. A critical role for ethylene in hydrogen peroxide release during programmed cell death in tomato suspension cells. Planta 2002, 214, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, L.F.; Manners, J.M.; Kazan, K. Fusarium oxysporumhijacks COI1-mediated jasmonate signaling to promote disease development in Arabidopsis. Plant J. 2009, 58, 927–939. [Google Scholar] [CrossRef]
- Daiki, M.; Hisayo, Y.; Kazuyuki, A.; Ryutaro, T. Identification of a Skp1-like protein interacting with SFB, the pollen S determinant of the gametophytic self-incompatibility in Prunus. Plant Physiol. 2012, 159, 1252–1262. [Google Scholar]
- Chi, X.; Maofu, L.; Junkai, W.; Han, G.; Qun, L.; Yu’E, Z.; Jijie, C.; Tianzhong, L.; Yongbiao, X. Identification of a canonical SCF(SLF) complex involved in S-RNase-based self-incompatibility of Pyrus (Rosaceae). Plant Mol. Biol. 2013, 81, 245–257. [Google Scholar]
- Martinoia, E.; Klein, M.; Geisler, M.; Bovet, L.; Forestier, C.; Kolukisaoglu, Ü.; Müller-Röber, B.; Schulz, B. Multifunctionality of plant ABC transporters—More than just detoxifiers. Planta 2002, 214, 345–355. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, L.; Chen, P.; Cai, H.; Hou, Z.; Jin, X.; Aslam, M.; Chai, M.; Lai, L.; He, Q.; et al. ATP binding cassette transporters ABCG1 and ABCG16 affect reproductive development via auxin signaling in Arabidopsis. Plant J. 2020. [Google Scholar] [CrossRef]
- Takayama, S.; Shimosato, H.; Shiba, H.; Funato, M.; Che, F.-S.; Watanabe, M.; Iwano, M.; Isogai, A. Direct ligand–receptor complex interaction controls Brassica self-incompatibility. Nature 2001, 413, 534–538. [Google Scholar] [CrossRef]
- Haasen, K.E.; Goring, D.R. The recognition and rejection of self-incompatible pollen in the Brassicaceae. Bot. Stud. 2010, 51, 1–6. [Google Scholar]
- Yuan, H.; Meng, D.; Gu, Z.; Li, W.; Wang, A.; Yang, Q.; Zhu, Y.; Li, T. A novel gene, MdSSK1, as a component of the SCF complex rather than MdSBP1 can mediate the ubiquitination of S-RNase in apple. J. Exp. Bot. 2014, 65, 3121–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; Gu, Z.; Wang, A.; Hui, Y.; Wei, L.; Yang, Q.; Duan, X.; Li, T. Screening and characterization of apple Rho-like GTPase (MdROPs) genes related to S-RNase mediated self-incompatibility. Plant Cell Tiss. Org. 2014, 117, 465–476. [Google Scholar] [CrossRef]
- Jong-Jin, P.; Jakyung, Y.; Jinmi, Y.; Lae-Hyeon, C.; Jin, P.; Hee Joong, J.; Seok Keun, C.; Woo Taek, K.; Gynheung, A. OsPUB15, an E3 ubiquitin ligase, functions to reduce cellular oxidative stress during seedling establishment. Plant J. 2011, 65, 194–205. [Google Scholar]
- Potocký, M.; Jones, M.; Bezvoda, R.; Smirnoff, N.; Žárský, V. Reactive oxygen species produced by NADPH oxidase are involved in pollen tube growth. New Phytol. 2007, 146, S269–S270. [Google Scholar]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. Bmc Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting biomolecular knowledge via a protein database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar] [PubMed]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinform. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Doelling, J.H.; Arendt, C.S.; Hochstrasser, M.; Vierstra, R.D. Molecular organization of the 20S proteasome gene family from Arabidopsis thaliana. Genetics 1998, 149, 677. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Stefan, G.; Miguel, G.G.J.; Javier, T.; Williams, T.D.; Nagaraj, S.H.; José, N.M.; Montserrat, R.; Manuel, T.; Joaquín, D.; Ana, C. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 16. [Google Scholar]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef] [Green Version]
- Decourcelle, M.; Perez-Fons, L.; Baulande, S.; Steiger, S.; Couvelard, L.; Hem, S.; Zhu, C.; Capell, T.; Christou, P.; Fraser, P.; et al. Combined transcript, proteome, and metabolite analysis of transgenic maize seeds engineered for enhanced carotenoid synthesis reveals pleotropic effects in core metabolism. J. Exp. Bot. 2015, 66, 3141–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smart, K.F.; Aggio, R.B.M.; Van Houtte, J.R.; Villas-Bôas, S.G. Analytical platform for metabolome analysis of microbial cells using methyl chloroformate derivatization followed by gas chromatography–mass spectrometry. Nat. Protoc. 2010, 5, 1709–1729. [Google Scholar] [CrossRef] [PubMed]
- Bölling, C.; Fiehn, O. Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 2005, 139, 1995–2005. [Google Scholar] [CrossRef] [Green Version]
- Core, R.; Null, R.D.C.T.; Team, R.; Team, T.S.U.D.I.S.; Coreteam, R. R: A Language and Environment for Statistical Computating. Computing 2011, 14, 12–21. [Google Scholar] [CrossRef]
- Zeng, Y.; Tan, X.; Lin, Z.; Long, H.; Wang, B.; Li, Z.; Zhen, Y. A fructose-1,6-biphosphate aldolase gene from Camellia oleifera: Molecular characterization and impact on salt stress tolerance. Mol. Breed. 2015, 35, 17. [Google Scholar] [CrossRef]
- Livak, K.; Schmittgen, T. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-△△Ct Method. Methods 2000, 25, 402–408. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Lu, M.; Yu, S.; Liu, Y.; Yang, J.; Tan, X. In-Depth Understanding of Camellia oleifera Self-Incompatibility by Comparative Transcriptome, Proteome and Metabolome. Int. J. Mol. Sci. 2020, 21, 1600. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051600
Zhou J, Lu M, Yu S, Liu Y, Yang J, Tan X. In-Depth Understanding of Camellia oleifera Self-Incompatibility by Comparative Transcriptome, Proteome and Metabolome. International Journal of Molecular Sciences. 2020; 21(5):1600. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051600
Chicago/Turabian StyleZhou, Junqin, Mengqi Lu, Shushu Yu, Yiyao Liu, Jin Yang, and Xiaofeng Tan. 2020. "In-Depth Understanding of Camellia oleifera Self-Incompatibility by Comparative Transcriptome, Proteome and Metabolome" International Journal of Molecular Sciences 21, no. 5: 1600. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051600