Interferon Response in Hepatitis C Virus-Infected Hepatocytes: Issues to Consider in the Era of Direct-Acting Antivirals


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Canonical and Non-Canonical Types I and III IFN Signaling Pathways in Hepatocytes
2.1. Canonical Types I and III IFN Signaling
2.2. Non-Canonical Types I and III IFN Signaling
2.3. Regulation of Types I and III IFN Signaling
3. Types I and III IFN Responses in HCV Infections
3.1. Host Factors Involved in HCV Sensing and IFN Production
3.2. Viral Evasion from Endogenous IFN Responses in HCV-Infected Cells
3.3. Type III IFNs in HCV-Infected Hepatocytes
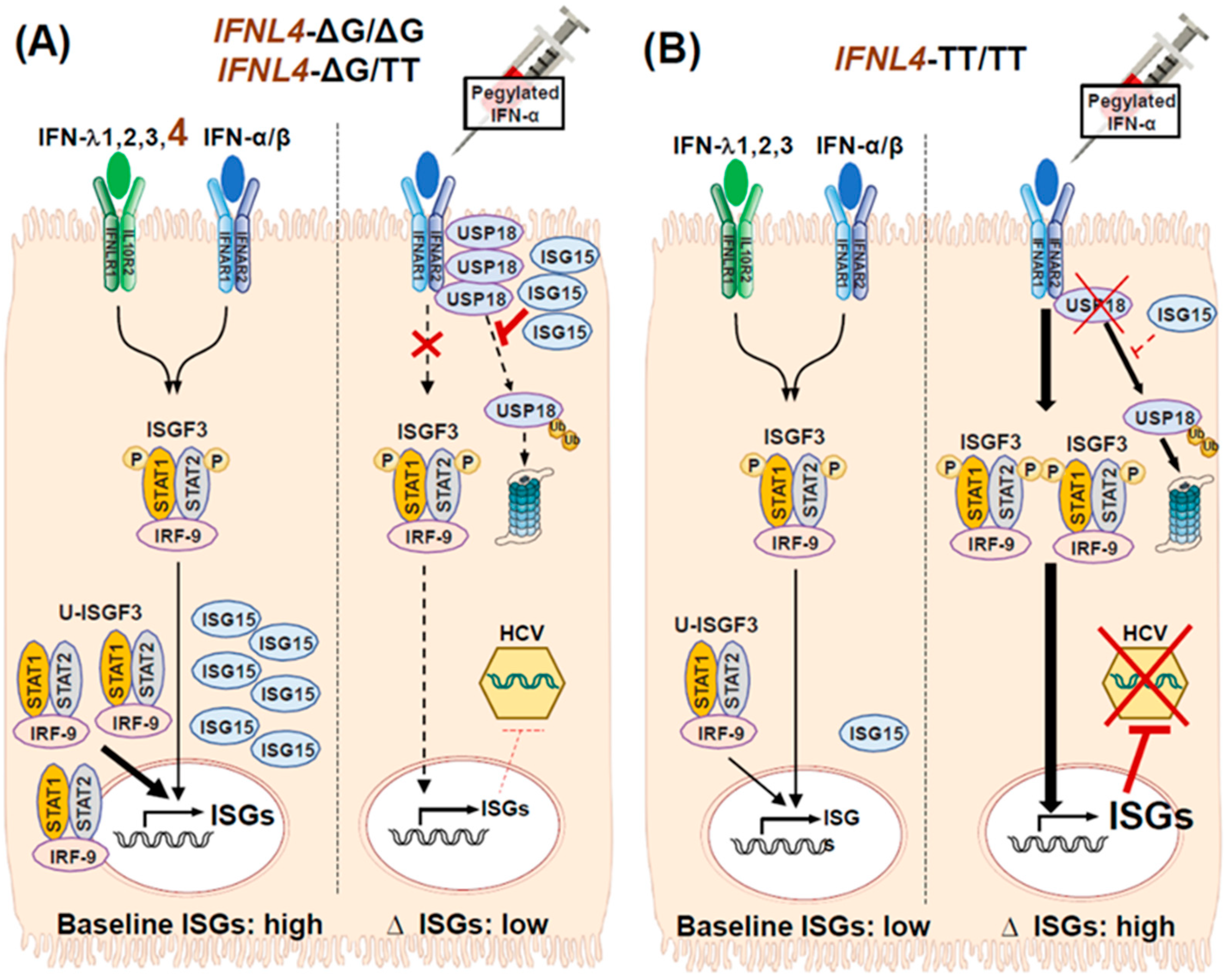
3.4. Impact of IFNL4 Genotype on the Outcome of HCV Infection
4. Impact of DAA Treatment on Types I and III IFN Responses to HCV Infections
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.R.; Prokunina-Olsson, L.; Donnelly, R.P. IFN-lambda4: The paradoxical new member of the interferon lambda family. J. Interferon Cytokine Res. 2014, 34, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamming, O.J.; Terczynska-Dyla, E.; Vieyres, G.; Dijkman, R.; Jorgensen, S.E.; Akhtar, H.; Siupka, P.; Pietschmann, T.; Thiel, V.; Hartmann, R. Interferon lambda 4 signals via the IFNlambda receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. 2013, 32, 3055–3065. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laidlaw, S.M.; Dustin, L.B. Interferon lambda: Opportunities, risks, and uncertainties in the fight against HCV. Front. Immunol. 2014, 5, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-lambda orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.S.; Hong, S.H.; Chung, J.H.; Kim, S.; Park, S.H.; Kim, H.M.; Yoon, S.K.; Shin, E.C. IFN-lambda4 potently blocks IFN-alpha signalling by ISG15 and USP18 in hepatitis C virus infection. Sci. Rep. 2017, 7, 3821. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.C.; Sung, P.S.; Park, S.H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef]
- Sung, P.S.; Cheon, H.; Cho, C.H.; Hong, S.H.; Park, D.Y.; Seo, H.I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443–10448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeon, J.E. Recent update of the 2017 Korean Association for the Study of the Liver (KASL) treatment guidelines of chronic hepatitis C: Comparison of guidelines from other continents, 2017 AASLD/IDSA and 2016 EASL. Clin. Mol. Hepatol. 2018, 24, 278–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marascio, N.; Quirino, A.; Barreca, G.S.; Galati, L.; Costa, C.; Pisani, V.; Mazzitelli, M.; Matera, G.; Liberto, M.C.; Foca, A.; et al. Discussion on critical points for a tailored therapy to cure hepatitis C virus infection. Clin. Mol. Hepatol. 2019, 25, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Parigi, T.L.; Torres, M.C.P.; Aghemo, A. Upcoming direct acting antivirals for hepatitis C patients with a prior treatment failure. Clin. Mol. Hepatol. 2019, 25, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Amaddeo, G.; Nguyen, C.T.; Maille, P.; Mule, S.; Luciani, A.; Machou, C.; Rodrigues, A.; Regnault, H.; Mallat, A.; Laurent, A.; et al. Intrahepatic immune changes after hepatitis c virus eradication by direct-acting antiviral therapy. Liver Int. 2020, 40, 74–82. [Google Scholar] [CrossRef]
- Ramamurthy, N.; Marchi, E.; Ansari, M.A.; Pedergnana, V.; McLean, A.; Hudson, E.; STOP HCV consortium; Bowden, R.; Spencer, C.C.A.; Barnes, E.; et al. Impact of IFNL4 genotype on Interferon-stimulated Gene Expression during DAA therapy for Hepatitis C. Hepatology 2018. [Google Scholar] [CrossRef]
- Hayes, C.N.; Chayama, K. Interferon stimulated genes and innate immune activation following infection with hepatitis B and C viruses. J. Med. Virol. 2017, 89, 388–396. [Google Scholar] [CrossRef]
- Noureddin, M.; Rotman, Y.; Zhang, F.; Park, H.; Rehermann, B.; Thomas, E.; Liang, T.J. Hepatic expression levels of interferons and interferon-stimulated genes in patients with chronic hepatitis C: A phenotype-genotype correlation study. Genes Immun. 2015. [Google Scholar] [CrossRef]
- Dill, M.T.; Duong, F.H.; Vogt, J.E.; Bibert, S.; Bochud, P.Y.; Terracciano, L.; Papassotiropoulos, A.; Roth, V.; Heim, M.H. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology 2011, 140, 1021–1031. [Google Scholar] [CrossRef]
- Honda, M.; Sakai, A.; Yamashita, T.; Nakamoto, Y.; Mizukoshi, E.; Sakai, Y.; Yamashita, T.; Nakamura, M.; Shirasaki, T.; Horimoto, K.; et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology 2010, 139, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Lanford, R.E.; Feng, Z.; Chavez, D.; Guerra, B.; Brasky, K.M.; Zhou, Y.; Yamane, D.; Perelson, A.S.; Walker, C.M.; Lemon, S.M. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 11223–11228. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.S.; Shin, E.C.; Yoon, S.K. Interferon Response in Hepatitis C Virus (HCV) Infection: Lessons from Cell Culture Systems of HCV Infection. Int. J. Mol. Sci. 2015, 16, 23683–23694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terczynska-Dyla, E.; Bibert, S.; Duong, F.H.; Krol, I.; Jorgensen, S.; Collinet, E.; Kutalik, Z.; Aubert, V.; Cerny, A.; Kaiser, L.; et al. Reduced IFNlambda4 activity is associated with improved HCV clearance and reduced expression of interferon-stimulated genes. Nat. Commun. 2014, 5, 5699. [Google Scholar] [CrossRef]
- Aka, P.V.; Kuniholm, M.H.; Pfeiffer, R.M.; Wang, A.S.; Tang, W.; Chen, S.; Astemborski, J.; Plankey, M.; Villacres, M.C.; Peters, M.G.; et al. Association of the IFNL4-DeltaG Allele With Impaired Spontaneous Clearance of Hepatitis C Virus. J. Infect. Dis. 2014, 209, 350–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, T.R.; Pfeiffer, R.M.; Paquin, A.; Lang Kuhs, K.A.; Chen, S.; Bonkovsky, H.L.; Edlin, B.R.; Howell, C.D.; Kirk, G.D.; Kuniholm, M.H.; et al. Comparison of functional variants in IFNL4 and IFNL3 for association with HCV clearance. J. Hepatol. 2015, 63, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, T.R.; Kottilil, S.; Pfeiffer, R.M. IFNL4 Genotype Is Associated with Virologic Relapse After 8-Week Treatment With Sofosbuvir, Velpatasvir, and Voxilaprevir. Gastroenterology 2017, 153, 1694–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, T.R.; Lang Kuhs, K.A.; Pfeiffer, R.M. Subgroup differences in response to 8 weeks of ledipasvir/sofosbuvir for chronic hepatitis C. Open Forum Infect. Dis. 2014, 1, ofu110. [Google Scholar] [CrossRef] [Green Version]
- Meissner, E.G.; Bon, D.; Prokunina-Olsson, L.; Tang, W.; Masur, H.; O’Brien, T.R.; Herrmann, E.; Kottilil, S.; Osinusi, A. IFNL4-DeltaG genotype is associated with slower viral clearance in hepatitis C, genotype-1 patients treated with sofosbuvir and ribavirin. J. Infect. Dis. 2014, 209, 1700–1704. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, G.R.; Cheon, H.; Wang, Y. Responses to Cytokines and Interferons that Depend upon JAKs and STATs. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Jilg, N.; Lin, W.; Hong, J.; Schaefer, E.A.; Wolski, D.; Meixong, J.; Goto, K.; Brisac, C.; Chusri, P.; Fusco, D.N.; et al. Kinetic differences in the induction of interferon stimulated genes by interferon-alpha and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 2014, 59, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Casazza, R.L.; Lazear, H.M. Why Is IFN-lambda Less Inflammatory? One IRF Decides. Immunity 2019, 51, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Forero, A.; Ozarkar, S.; Li, H.; Lee, C.H.; Hemann, E.A.; Nadjsombati, M.S.; Hendricks, M.R.; So, L.; Green, R.; Roy, C.N.; et al. Differential Activation of the Transcription Factor IRF1 Underlies the Distinct Immune Responses Elicited by Type I and III Interferons. Immunity 2019. [Google Scholar] [CrossRef] [PubMed]
- Broggi, A.; Granucci, F.; Zanoni, I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; Feng, H.; Rivera-Serrano, E.E.; Selitsky, S.R.; Hirai-Yuki, A.; Das, A.; McKnight, K.L.; Misumi, I.; Hensley, L.; Lovell, W.; et al. Basal expression of interferon regulatory factor 1 drives intrinsic hepatocyte resistance to multiple RNA viruses. Nat. Microbiol. 2019, 4, 1096–1104. [Google Scholar] [CrossRef]
- Nan, Y.; Wu, C.; Zhang, Y.J. Interferon Independent Non-Canonical STAT Activation and Virus Induced Inflammation. Viruses 2018, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef] [Green Version]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef]
- Wang, W.; Yin, Y.; Xu, L.; Su, J.; Huang, F.; Wang, Y.; Boor, P.P.C.; Chen, K.; Wang, W.; Cao, W.; et al. Unphosphorylated ISGF3 drives constitutive expression of interferon-stimulated genes to protect against viral infections. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Szappanos, D.; Cheon, H.; Vogl, C.; Shukla, P.; Stark, G.R.; Sexl, V.; Schreiber, R.; Schindler, C.; et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO Rep. 2016, 17, 367–382. [Google Scholar] [CrossRef]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 2015, 466, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testoni, B.; Vollenkle, C.; Guerrieri, F.; Gerbal-Chaloin, S.; Blandino, G.; Levrero, M. Chromatin dynamics of gene activation and repression in response to interferon alpha (IFN(alpha)) reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. J. Biol. Chem. 2011, 286, 20217–20227. [Google Scholar] [CrossRef] [Green Version]
- Honke, N.; Shaabani, N.; Zhang, D.E.; Hardt, C.; Lang, K.S. Multiple functions of USP18. Cell Death Dis. 2016, 7, e2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dustin, L.B.; Rice, C.M. Flying under the radar: The immunobiology of hepatitis C. Annu. Rev. Immunol. 2007, 25, 71–99. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef]
- Zhang, X.; Bogunovic, D.; Payelle-Brogard, B.; Francois-Newton, V.; Speer, S.D.; Yuan, C.; Volpi, S.; Li, Z.; Sanal, O.; Mansouri, D.; et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 2015, 517, 89–93. [Google Scholar] [CrossRef]
- Speer, S.D.; Li, Z.; Buta, S.; Payelle-Brogard, B.; Qian, L.; Vigant, F.; Rubino, E.; Gardner, T.J.; Wedeking, T.; Hermann, M.; et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 2016, 7, 11496. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAKSTAT 2013, 2, e24053. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and Autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Sharrocks, A.D. PIAS proteins and transcriptional regulation—More than just SUMO E3 ligases? Genes Dev. 2006, 20, 754–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungureanu, D.; Vanhatupa, S.; Kotaja, N.; Yang, J.; Aittomaki, S.; Janne, O.A.; Palvimo, J.J.; Silvennoinen, O. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood 2003, 102, 3311–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droescher, M.; Begitt, A.; Marg, A.; Zacharias, M.; Vinkemeier, U. Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO). J. Biol. Chem. 2011, 286, 18731–18746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef]
- Hoffmann, F.S.; Schmidt, A.; Dittmann Chevillotte, M.; Wisskirchen, C.; Hellmuth, J.; Willms, S.; Gilmore, R.H.; Glas, J.; Folwaczny, M.; Muller, T.; et al. Polymorphisms in melanoma differentiation-associated gene 5 link protein function to clearance of hepatitis C virus. Hepatology 2015, 61, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Hiet, M.S.; Bauhofer, O.; Zayas, M.; Roth, H.; Tanaka, Y.; Schirmacher, P.; Willemsen, J.; Grunvogel, O.; Bender, S.; Binder, M.; et al. Control of temporal activation of hepatitis C virus-induced interferon response by domain 2 of nonstructural protein 5A. J. Hepatol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Hilgenfeld, R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [Google Scholar] [CrossRef] [Green Version]
- Hei, L.; Zhong, J. Laboratory of genetics and physiology 2 (LGP2) plays an essential role in hepatitis C virus infection-induced interferon responses. Hepatology 2017, 65, 1478–1491. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, N.; Dabo, S.; Akazawa, D.; Fukasawa, M.; Shinkai-Ouchi, F.; Hugon, J.; Wakita, T.; Meurs, E.F. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog. 2011, 7, e1002289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Li, N.L.; Wei, D.; Pfeffer, S.R.; Fan, M.; Pfeffer, L.M. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 2012, 55, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Cao, X.; Ding, Q.; Zhao, Y.; He, Z.; Zhong, J. Hepatitis C virus NS4B induces the degradation of TRIF to inhibit TLR3-mediated interferon signaling pathway. PLoS Pathog. 2018, 14, e1007075. [Google Scholar] [CrossRef]
- Namineni, S.; O’Connor, T.; Faure-Dupuy, S.; Johansen, P.; Riedl, T.; Liu, K.; Xu, H.; Singh, I.; Shinde, P.; Li, F.; et al. A dual role for hepatocyte-intrinsic canonical NF-kappaB signaling in virus control. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadkhan, S.; Fortes, P. Regulation of the Interferon Response by lncRNAs in HCV Infection. Front. Microbiol. 2018, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Chen, S.; Tian, R.; Huang, X.; Deng, R.; Xue, B.; Qin, Y.; Xu, Y.; Wang, J.; Guo, M.; et al. Long Noncoding RNA ITPRIP-1 Positively Regulates the Innate Immune Response through Promotion of Oligomerization and Activation of MDA5. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Duan, X.; Holmes, J.A.; Li, W.; Lee, S.H.; Tu, Z.; Zhu, C.; Salloum, S.; Lidofsky, A.; Schaefer, E.A.; et al. A Long Noncoding RNA Regulates Hepatitis C Virus Infection Through Interferon Alpha-Inducible Protein 6. Hepatology 2019, 69, 1004–1019. [Google Scholar] [CrossRef]
- Horner, S.M.; Gale, M., Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 2013, 19, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef] [PubMed]
- Bellecave, P.; Sarasin-Filipowicz, M.; Donze, O.; Kennel, A.; Gouttenoire, J.; Meylan, E.; Terracciano, L.; Tschopp, J.; Sarrazin, C.; Berg, T.; et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 2010, 51, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Loo, Y.M.; Horner, S.M.; Gale, M., Jr.; Malik, H.S. Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol. 2012, 10, e1001282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.; Wen, Y.; Shu, C.; Han, Q.; Konan, K.V.; Li, P.; Kao, C.C. Hepatitis C Virus NS4B Can Suppress STING Accumulation to Evade Innate Immune Responses. J. Virol. 2016, 90, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitta, S.; Sakamoto, N.; Nakagawa, M.; Kakinuma, S.; Mishima, K.; Kusano-Kitazume, A.; Kiyohashi, K.; Murakawa, M.; Nishimura-Sakurai, Y.; Azuma, S.; et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 2013, 57, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M., Jr.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [Green Version]
- Gagne, B.; Tremblay, N.; Park, A.Y.; Baril, M.; Lamarre, D. Importin beta1 targeting by hepatitis C virus NS3/4A protein restricts IRF3 and NF-kappaB signaling of IFNB1 antiviral response. Traffic 2017, 18, 362–377. [Google Scholar] [CrossRef]
- Park, H.; Serti, E.; Eke, O.; Muchmore, B.; Prokunina-Olsson, L.; Capone, S.; Folgori, A.; Rehermann, B. IL-29 is the dominant type III interferon produced by hepatocytes during acute hepatitis C virus infection. Hepatology 2012, 56, 2060–2070. [Google Scholar] [CrossRef]
- Thomas, E.; Gonzalez, V.D.; Li, Q.; Modi, A.A.; Chen, W.; Noureddin, M.; Rotman, Y.; Liang, T.J. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 2012, 142, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Israelow, B.; Narbus, C.M.; Sourisseau, M.; Evans, M.J. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 2014, 60, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, T.; Imanaka, N.; Marukian, S.; Dorner, M.; Liu, P.; Ploss, A.; Rice, C.M. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 2014, 15, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Prieto, A.M.; Dorner, M. Immune Evasion Strategies during Chronic Hepatitis B and C Virus Infection. Vaccines 2017, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.J.; Arora, S.; Everson, G.; Flisiak, R.; George, J.; Ghalib, R.; Gordon, S.C.; Gray, T.; Greenbloom, S.; Hassanein, T.; et al. A randomized phase 2b study of peginterferon lambda-1a for the treatment of chronic HCV infection. J. Hepatol. 2014, 61, 1238–1246. [Google Scholar] [CrossRef]
- Wieland, S.; Makowska, Z.; Campana, B.; Calabrese, D.; Dill, M.T.; Chung, J.; Chisari, F.V.; Heim, M.H. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2014, 59, 2121–2130. [Google Scholar] [CrossRef]
- Chen, L.; Borozan, I.; Feld, J.; Sun, J.; Tannis, L.L.; Coltescu, C.; Heathcote, J.; Edwards, A.M.; McGilvray, I.D. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology 2005, 128, 1437–1444. [Google Scholar] [CrossRef]
- Feld, J.J.; Nanda, S.; Huang, Y.; Chen, W.; Cam, M.; Pusek, S.N.; Schweigler, L.M.; Theodore, D.; Zacks, S.L.; Liang, T.J.; et al. Hepatic gene expression during treatment with peginterferon and ribavirin: Identifying molecular pathways for treatment response. Hepatology 2007, 46, 1548–1563. [Google Scholar] [CrossRef] [Green Version]
- Katsounas, A.; Hubbard, J.J.; Wang, C.H.; Zhang, X.; Dou, D.; Shivakumar, B.; Winter, S.; Schlaak, J.F.; Lempicki, R.A.; Masur, H.; et al. High interferon-stimulated gene ISG-15 expression affects HCV treatment outcome in patients co-infected with HIV and HCV. J. Med. Virol. 2013, 85, 959–963. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L. Genetics of the Human Interferon Lambda Region. J. Interferon Cytokine Res. 2019, 39, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Hong, S.H.; Seo, N.; Kim, T.S.; An, H.J.; Lee, P.; Shin, E.C.; Kim, H.M. Structure-based glycoengineering of interferon lambda 4 enhances its productivity and anti-viral potency. Cytokine 2020, 125, 154833. [Google Scholar] [CrossRef] [PubMed]
- Onabajo, O.O.; Porter-Gill, P.; Paquin, A.; Rao, N.; Liu, L.; Tang, W.; Brand, N.; Prokunina-Olsson, L. Expression of Interferon Lambda 4 Is Associated with Reduced Proliferation and Increased Cell Death in Human Hepatic Cells. J. Interferon Cytokine Res. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obajemu, A.A.; Rao, N.; Dilley, K.A.; Vargas, J.M.; Sheikh, F.; Donnelly, R.P.; Shabman, R.S.; Meissner, E.G.; Prokunina-Olsson, L.; Onabajo, O.O. IFN-lambda4 Attenuates Antiviral Responses by Enhancing Negative Regulation of IFN Signaling. J. Immunol. 2017, 199, 3808–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.F.; Goldstein, D.B.; Urban, T.J.; Bradrick, S.S. Interferon-lambda4 is a cell-autonomous type III interferon associated with pre-treatment hepatitis C virus burden. Virology 2015, 476, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, C.; Vieyres, G.; Terczyńska-Dyla, E.; Dijkman, R.; Gad, H.H.; Akhtar, H.; Pietschmann, T. Transcriptome analysis reveals a classical interferon signature induced by IFNlambda4 in human primary cells. Genes Immun. 2015, 16, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, M.A.; Aranday-Cortes, E.; Ip, C.L.; da Silva Filipe, A.; Lau, S.H.; Bamford, C.; Bonsall, D.; Trebes, A.; Piazza, P.; Sreenu, V.; et al. Interferon lambda 4 impacts the genetic diversity of hepatitis C virus. Elife 2019, 8. [Google Scholar] [CrossRef]
- Alao, H.; Cam, M.; Keembiyehetty, C.; Zhang, F.; Serti, E.; Suarez, D.; Park, H.; Fourie, N.H.; Wright, E.C.; Henderson, W.A.; et al. Baseline Intrahepatic and Peripheral Innate Immunity are Associated with Hepatitis C Virus Clearance During Direct-Acting Antiviral Therapy. Hepatology 2018, 68, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, B.R.; Freije, C.A.; Imanaka, N.; Chen, S.T.; Eitson, J.L.; Caron, R.; Uhl, S.A.; Zeremski, M.; Talal, A.; Jacobson, I.M.; et al. Genetic Variation at IFNL4 Influences Extrahepatic Interferon-Stimulated Gene Expression in Chronic HCV Patients. J. Infect. Dis. 2018, 217, 650–655. [Google Scholar] [CrossRef]
- Emmanuel, B.; El-Kamary, S.S.; Magder, L.S.; Stafford, K.A.; Charurat, M.E.; Chairez, C.; McLaughlin, M.; Hadigan, C.; Prokunina-Olsson, L.; O’Brien, T.R.; et al. Metabolic Changes in Chronic Hepatitis C Patients Who Carry IFNL4-DeltaG and Achieve Sustained Virologic Response With Direct-Acting Antiviral Therapy. J. Infect. Dis. 2020, 221, 102–109. [Google Scholar] [CrossRef]
- Petta, S.; Rosso, C.; Leung, R.; Abate, M.L.; Booth, D.; Salomone, F.; Gambino, R.; Rizzetto, M.; Caviglia, P.; Smedile, A.; et al. Effects of IL28B rs12979860 CC genotype on metabolic profile and sustained virologic response in patients with genotype 1 chronic hepatitis C. Clin. Gastroenterol. Hepatol. 2013, 11, 311–317.e1. [Google Scholar] [CrossRef]
- Meissner, E.G.; Wu, D.; Osinusi, A.; Bon, D.; Virtaneva, K.; Sturdevant, D.; Porcella, S.; Wang, H.; Herrmann, E.; McHutchison, J.; et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J. Clin. Invest. 2014, 124, 3352–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, J.A.; Carlton-Smith, C.; Kim, A.Y.; Dumas, E.O.; Brown, J.; Gustafson, J.L.; Lauer, G.M.; Silva, S.T.; Robidoux, M.; Kvistad, D.; et al. Dynamic changes in innate immune responses during direct-acting antiviral therapy for HCV infection. J. Viral Hepat. 2019, 26, 362–372. [Google Scholar] [CrossRef]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, T.R.; Kottilil, S.; Feld, J.J.; Morgan, T.R.; Pfeiffer, R.M. Race or genetic makeup for hepatitis C virus treatment decisions? Hepatology 2017, 65, 2124–2125. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, I.M.; Lawitz, E.; Gane, E.J.; Willems, B.E.; Ruane, P.J.; Nahass, R.G.; Borgia, S.M.; Shafran, S.D.; Workowski, K.A.; Pearlman, B.; et al. Efficacy of 8 Weeks of Sofosbuvir, Velpatasvir, and Voxilaprevir in Patients With Chronic HCV Infection: 2 Phase 3 Randomized Trials. Gastroenterology 2017, 153, 113–122. [Google Scholar] [CrossRef]
- Peiffer, K.H.; Sommer, L.; Susser, S.; Vermehren, J.; Herrmann, E.; Doring, M.; Dietz, J.; Perner, D.; Berkowski, C.; Zeuzem, S.; et al. Interferon lambda 4 genotypes and resistance-associated variants in patients infected with hepatitis C virus genotypes 1 and 3. Hepatology 2016, 63, 63–73. [Google Scholar] [CrossRef]
- Naveed, M.; Ali, A.; Sheikh, N.; Rafique, S.; Idrees, M. Expression of TRIM22 mRNA in Chronic Hepatitis C Patients Treated with Direct-Acting Antiviral Drugs. APMIS 2019. [Google Scholar] [CrossRef]
- Sung, P.S.; Lee, E.B.; Park, D.J.; Lozada, A.; Jang, J.W.; Bae, S.H.; Choi, J.Y.; Yoon, S.K. Interferon-free treatment for hepatitis C virus infection induces normalization of extrahepatic type I interferon signaling. Clin. Mol. Hepatol. 2018, 24, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Carlton-Smith, C.; Holmes, J.A.; Naggie, S.; Lidofsky, A.; Lauer, G.M.; Kim, A.Y.; Chung, R.T.; ACTG A5327 study group. IFN-free therapy is associated with restoration of type I IFN response in HIV-1 patients with acute HCV infection who achieve SVR. J. Viral Hepat. 2018, 25, 465–472. [Google Scholar] [CrossRef]
- Sansonno, D.; Russi, S.; Serviddio, G.; Conteduca, V.; D’Andrea, G.; Sansonno, L.; Pavone, F.; Lauletta, G.; Mariggio, M.A.; Dammacco, F. Interleukin 28B gene polymorphisms in hepatitis C virus-related cryoglobulinemic vasculitis. J. Rheumatol. 2014, 41, 91–98. [Google Scholar] [CrossRef]
- Stattermayer, A.F.; Rutter, K.; Beinhardt, S.; Scherzer, T.M.; Stadlmayr, A.; Hofer, H.; Wrba, F.; Steindl-Munda, P.; Krebs, M.; Datz, C.; et al. Association of the IL28B genotype with insulin resistance in patients with chronic hepatitis C. J. Hepatol. 2012, 57, 492–498. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sung, P.S.; Shin, E.-C. Interferon Response in Hepatitis C Virus-Infected Hepatocytes: Issues to Consider in the Era of Direct-Acting Antivirals. Int. J. Mol. Sci. 2020, 21, 2583. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072583
Sung PS, Shin E-C. Interferon Response in Hepatitis C Virus-Infected Hepatocytes: Issues to Consider in the Era of Direct-Acting Antivirals. International Journal of Molecular Sciences. 2020; 21(7):2583. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072583
Chicago/Turabian StyleSung, Pil Soo, and Eui-Cheol Shin. 2020. "Interferon Response in Hepatitis C Virus-Infected Hepatocytes: Issues to Consider in the Era of Direct-Acting Antivirals" International Journal of Molecular Sciences 21, no. 7: 2583. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072583