Non-Coding RNAs as Key Regulators of Glutaminolysis in Cancer

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
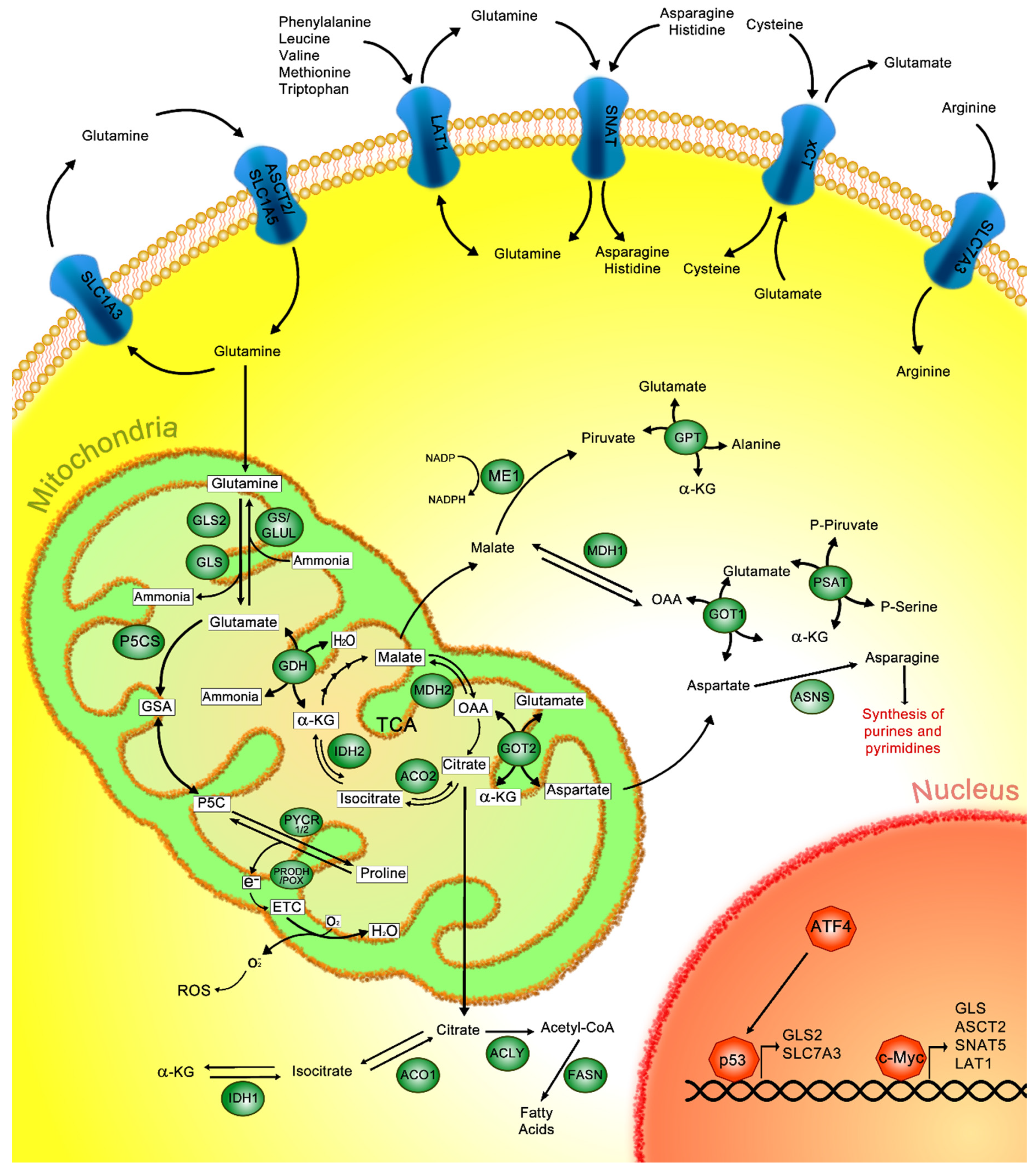
2. Glutamine Metabolism
3. Glutamine Metabolism in Cancer
4. Therapeutic Approaches Targeting the Glutaminolysis Pathway in Cancer
5. Non-Coding RNA (ncRNA)
6. microRNAs
7. Long Non-Coding RNAs
8. Circular RNAs
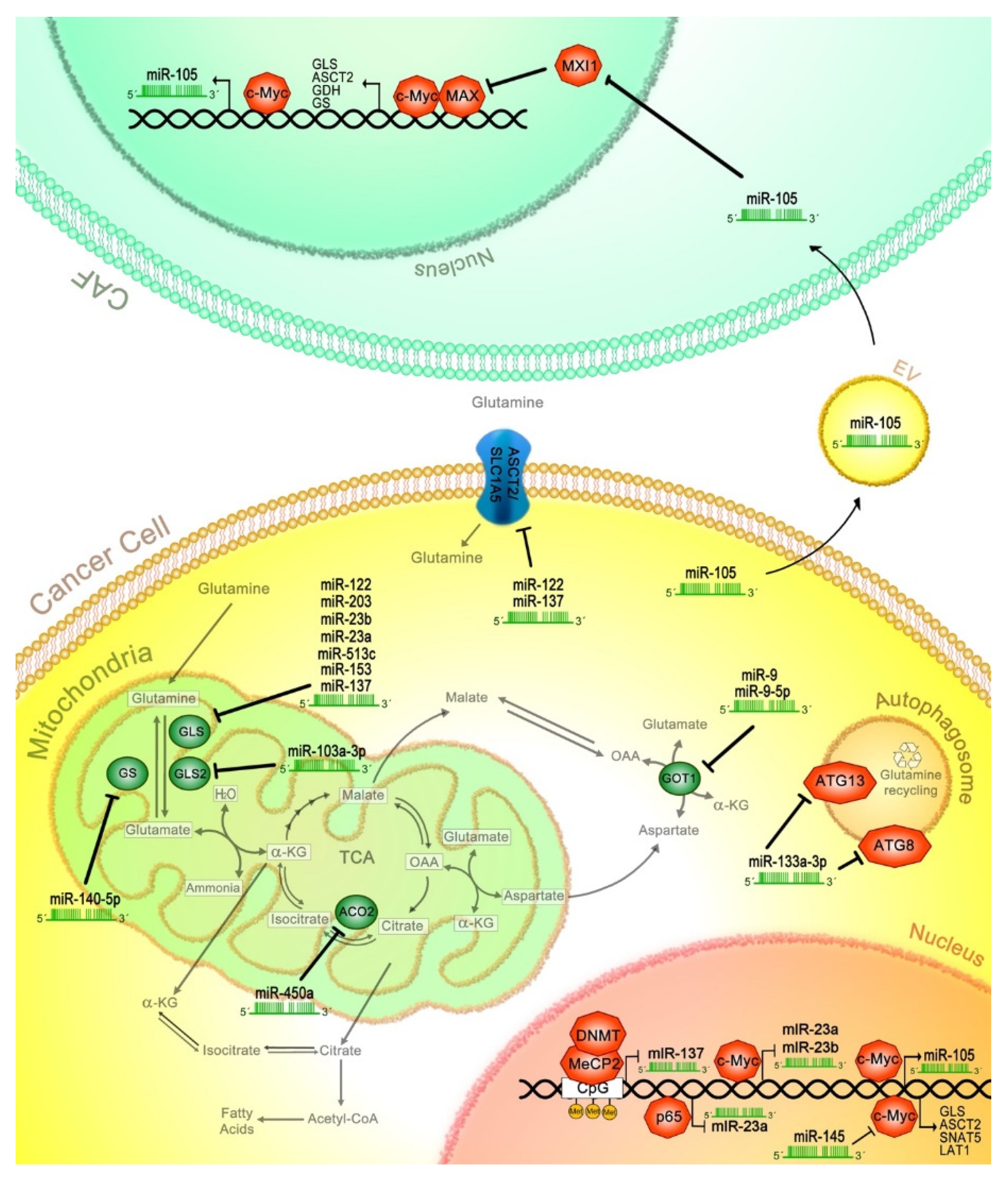
9. The Role of miRNAs in the Regulation of Glutaminolysis in Cancer
9.1. miR-103a-3p
9.2. miR-145
9.3. miR-450a
9.4. miR-137
9.5. miR-9
9.6. miR-105
9.7. miR-153
9.8. miR-513c
9.9. miR-23a/b
9.10. miR-203
9.11. miR-133a-3p
9.12. miR-140-5p
9.13. miR-122
10. The Role of lncRNAs in the Regulation of Glutaminolysis in Cancer
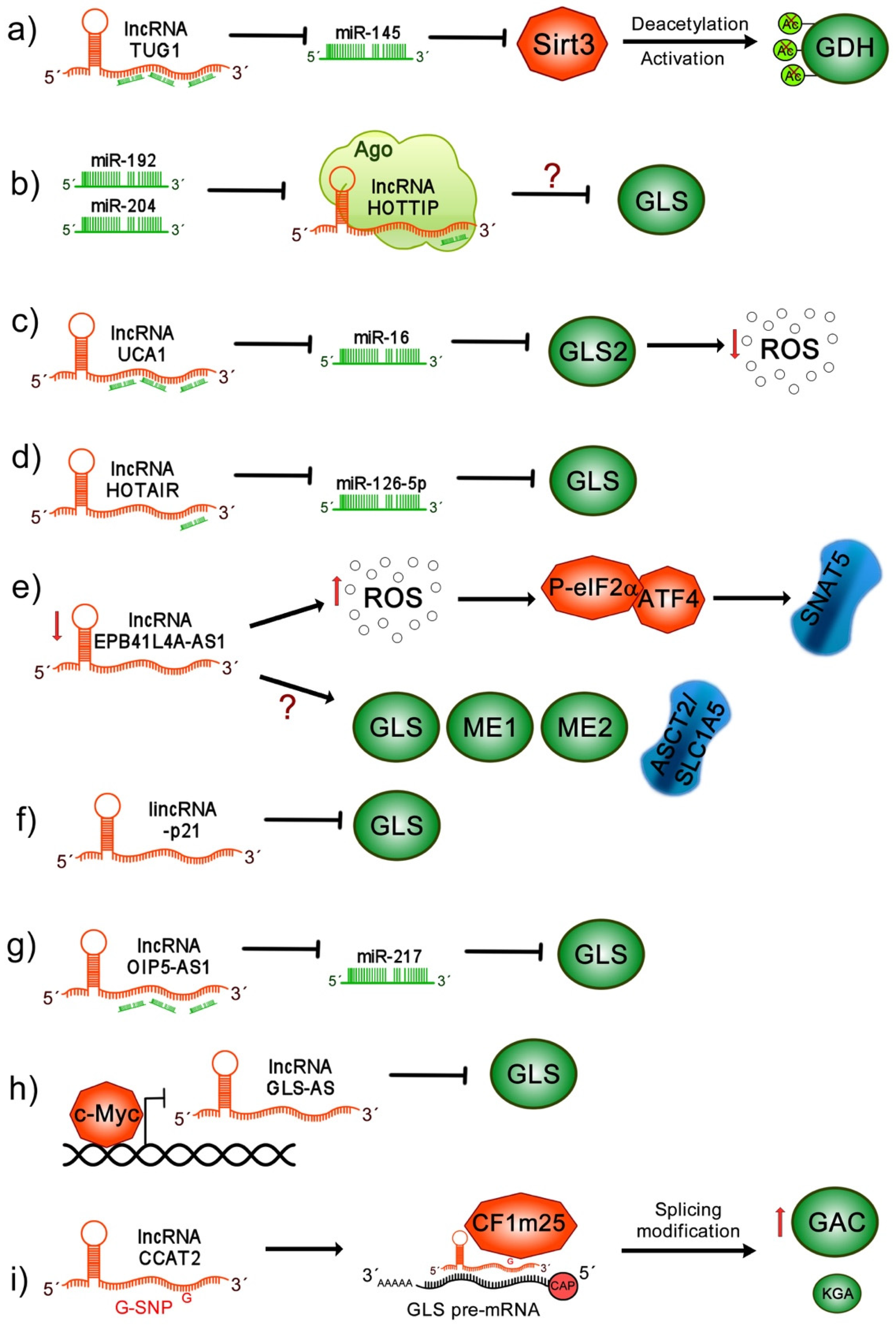
10.1. lncRNA TUG1
10.2. lncRNA HOTTIP
10.3. lncRNA UCA1
10.4. lncRNA HOTAIR
10.5. lncRNA EPB41L4A-AS1
10.6. lincRNA-p21
10.7. lncRNA OIP5-AS1
10.8. lncRNA GLS-AS
10.9. lncRNA CCAT2
11. Implications of CircRNAs in the Glutaminolysis Pathway in Cancer
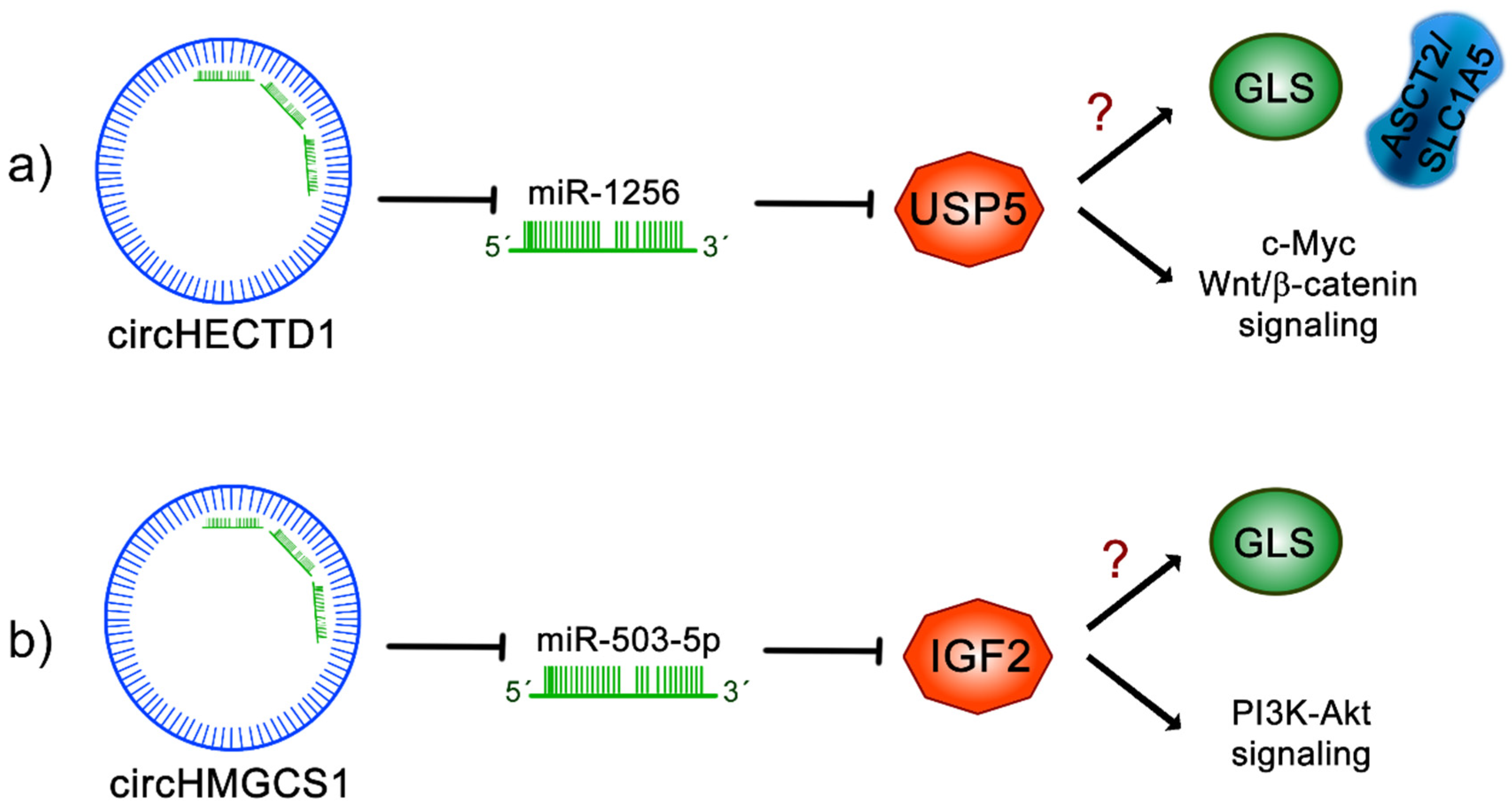
11.1. circHECTD1
11.2. circHMGCS1
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deberardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Park, K.G. Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S.M. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep. 1985, 5, 393–400. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Beltrán-Anaya, F.O.; Cedro-Tanda, A.; Hidalgo-Miranda, A.; Romero-Cordoba, S.L. Insights into the Regulatory Role of Non-coding RNAs in Cancer Metabolism. Front. Physiol. 2016, 7, 342. [Google Scholar] [CrossRef] [Green Version]
- Dumortier, O.; Hinault, C.; Van Obberghen, E. MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab. 2013, 18, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Wang, Y.; Fan, Y.; Fang, N.; Wang, T.; Xu, T.; Shu, Y. CircRNAs in cancer metabolism: A review. J. Hematol. Oncol. 2019, 12, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bott, A.J.; Maimouni, S.; Zong, W.X. The pleiotropic effects of glutamine metabolism in cancer. Cancers (Basel) 2019, 11, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Kim, H. Oncogenes and tumor suppressors regulate glutamine metabolism in cancer cells. J. Cancer Prev. 2013, 18, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2531–2539. [Google Scholar] [CrossRef]
- Gegelashvili, M.; Rodriguez-Kern, A.; Pirozhkova, I.; Zhang, J.; Sung, L.; Gegelashvili, G. High-affinity glutamate transporter GLAST/EAAT1 regulates cell surface expression of glutamine/neutral amino acid transporter ASCT2 in human fetal astrocytes. Neurochem. Int. 2006, 48, 611–615. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine transport and mitochondrial metabolism in cancer cell growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Lu, X. The Role of Large Neutral Amino Acid Transporter (LAT1) in Cancer. Curr. Cancer Drug Targets 2019, 19, 863–876. [Google Scholar] [CrossRef]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Umene, K.; Yamasaki, J.; Suina, K.; Otsuki, Y.; Yoshikawa, M.; Minami, Y.; Masuko, T.; Kawaguchi, S.; Nakayama, H.; et al. Glutaminolysis-related genes determine sensitivity to xCT-targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 2019, 110, 3453–3463. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. (Lond. Engl.) 2018, 38, 12. [Google Scholar] [CrossRef] [Green Version]
- Bröer, S. The SLC38 family of sodium-amino acid co-transporters. Pflug. Arch. Eur. J. Physiol. 2014, 466, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Rahimi, F.; Bröer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolková, K.; Ježek, P. The Role of Mitochondrial NADPH-Dependent Isocitrate Dehydrogenase in Cancer Cells. Int. J. Cell Biol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Michalak, K.P.; Maćkowska-Kędziora, A.; Agnieszka Sobolewski, B.; Woźniak, P. Key Roles of Glutamine Pathways in Reprogramming the Cancer Metabolism. Oxid. Med. Cell. Longev. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Ericksen, R.E.; Escande-Beillard, N.; Lee, Q.Y.; Loh, A.; Denil, S.; Steckel, M.; Haegebarth, A.; Wai Ho, T.S.; Chow, P.; et al. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J. Hepatol. 2020, 72, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwörer, S.; Berisa, M.; Violante, S.; Qin, W.; Zhu, J.; Hendrickson, R.C.; Cross, J.R.; Thompson, C.B. Proline biosynthesis is a vent for TGFβ-induced mitochondrial redox stress. EMBO J. 2020, 39, e103334. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korla, K.; Vadlakonda, L.; Mitra, C.K. Kinetic simulation of malate-aspartate and citrate-pyruvate shuttles in association with Krebs cycle. J. Biomol. Struct. Dyn. 2015, 33, 2390–2403. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhou, L.; Stanley, W.C.; Cabrera, M.E.; Saidel, G.M.; Yu, X. Role of the malate-aspartate shuttle on the metabolic response to myocardial ischemia. J. Theor. Biol. 2008, 254, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Castegna, A.; Menga, A. Glutamine Synthetase: Localization Dictates Outcome. Genes (Basel) 2018, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.W.; Cerione, R.A. Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget 2010, 1, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, M.; Kaira, K.; Ohshima, Y.; Ishioka, N.S.; Shino, M.; Sakakura, K.; Takayasu, Y.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; et al. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer 2014, 110, 2506–2513. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanein, M.; Hoeksema, M.D.; Shiota, M.; Qian, J.; Harris, B.K.; Chen, H.; Clark, J.E.; Alborn, W.E.; Eisenberg, R.; Massion, P.P. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 2013, 19, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; Van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, H.; Kaira, K.; Oriuchi, N.; Shimizu, K.; Tominaga, H.; Yanagitani, N.; Sunaga, N.; Ishizuka, T.; Nagamori, S.; Promchan, K.; et al. Inhibition of L-type amino acid transporter 1 has antitumor activity in non-small cell lung cancer. Anticancer Res. 2010, 30, 4819–4828. [Google Scholar]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef] [Green Version]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Kamarajan, P.; Rajendiran, T.M.; Kinchen, J.; Bermúdez, M.; Danciu, T.; Kapila, Y.L. Head and Neck Squamous Cell Carcinoma Metabolism Draws on Glutaminolysis, and Stemness Is Specifically Regulated by Glutaminolysis via Aldehyde Dehydrogenase. J. Proteome Res. 2017, 16, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K.H.; Hu, W.; Feng, Z. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 2014, 5, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.K.; Islam, S.M.R.; Abdullah-Al-Wadud, M.; Islam, S.; Ali, F.; Park, K.S. Multiomics Analysis Reveals that GLS and GLS2 Differentially Modulate the Clinical Outcomes of Cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Jutabha, P.; Endou, H.; Anzai, N. c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol. Rep. 2012, 28, 862–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.Q.; Lowman, X.H.; Reid, M.A.; Mendez-Dorantes, C.; Pan, M.; Yang, Y.; Kong, M. Tumor-associated mutant p53 promotes cancer cell survival upon glutamine deprivation through p21 induction. Oncogene 2017, 36, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736.e6. [Google Scholar] [CrossRef] [Green Version]
- Lowman, X.H.; Hanse, E.A.; Yang, Y.; Ishak Gabra, M.B.; Tran, T.Q.; Li, H.; Kong, M. p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep. 2019, 26, 3051–3060.e4. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and nutrient homeostasis: The central role of the lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [Green Version]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis Activates Rag-mTORC1 Signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Guo, Y.; Seo, W.; Zhang, R.; Lu, C.; Wang, Y.; Luo, L.; Paul, B.; Yan, W.; Saxena, D.; et al. Targeting cellular metabolism to reduce head and neck cancer growth. Sci. Rep. 2019, 9, 4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Tran Hoang, C.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grkovski, M.; Goel, R.; Krebs, S.; Staton, K.D.; Harding, J.J.; Mellinghoff, I.K.; Humm, J.L.; Dunphy, M.P.S. Pharmacokinetic Assessment of 18F-(2S,4R)-4-Fluoroglutamine in Patients with Cancer. J. Nucl. Med. 2020, 61, 357–366. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Luo, Z.; Xu, J.; Sun, J.; Huang, H.; Zhang, Z.; Ma, W.; Wan, Z.; Liu, Y.; Pardeshi, A.; Li, S. Co-delivery of 2-Deoxyglucose and a glutamine metabolism inhibitor V9302 via a prodrug micellar formulation for synergistic targeting of metabolism in cancer. Acta Biomater. 2020, 105, 239–252. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding rnas in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Benmoussa, A.; Provost, P. Small Non-Coding RNAs Derived From Eukaryotic Ribosomal RNA. Non-Coding RNA 2019, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Wang, F.; Nazarali, A.J.; Ji, S. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am. J. Cancer Res. 2016, 6, 1167–1176. [Google Scholar]
- Hsiao, K.-Y.; Lin, Y.-C.; Gupta, S.K.; Chang, N.; Yen, L.; Sun, H.S.; Tsai, S.-J. Noncoding Effects of Circular RNA CCDC66 Promote Colon Cancer Growth and Metastasis. Cancer Res. 2017, 77, 2339–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Li, J.; Wang, H.; Su, X.; Hou, J.; Gu, Y.; Qian, C.; Lin, Y.; Liu, X.; Huang, M.; et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology 2017, 66, 1151–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Zheng, Q.; Bao, C.; He, J.; Chen, B.; Lyu, D.; Zheng, B.; Xu, Y.; Long, Z.; et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017, 388, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Ardekani, A.M.; Naeini, M.M. The role of microRNAs in human diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar] [PubMed]
- Misiewicz-Krzeminska, I.; Krzeminski, P.; Corchete, L.A.; Quwaider, D.; Rojas, E.A.; Herrero, A.B.; Gutiérrez, N.C. Factors regulating microRNA expression and function in multiple myeloma. Non-Coding RNA 2019, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 287–314. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [Green Version]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res 2018, 46, 2169–2184. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA–LncRNA interactions. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 1402, pp. 271–286. [Google Scholar]
- Cajal, S.R.; Segura, M.F.; Hümmer, S. Interplay between ncRNAs and cellular communication: A proposal for understanding cell-specific signaling pathways. Front. Genet. 2019, 10, 281. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular Intronic Long Noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sharp, P.A. A circuitous route to noncoding RNA. Science 2013, 340, 440–441. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- He, R.; Liu, P.; Xie, X.; Zhou, Y.; Liao, Q.; Xiong, W.; Li, X.; Li, G.; Zeng, Z.; Tang, H. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J. Exp. Clin. Cancer Res. 2017, 36, 145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, S.; Wang, H.; Cao, J.; Huang, X.; Chen, Z.; Xu, P.; Sun, G.; Xu, J.; Lv, J.; et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol. Cancer 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Li, F.Q.; Tian, L.L.; Shang, D.S.; Guo, Y.; Zhang, J.R.; Liu, M. Comprehensive analysis of the whole coding and non-coding RNA transcriptome expression profiles and construction of the circRNA–lncRNA co-regulated ceRNA network in laryngeal squamous cell carcinoma. Funct. Integr. Genom. 2019, 19, 109–121. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, R.; Zhang, X.; Wu, Y.; Li, X.; Zhang, S.; Hou, W.; Ding, Y.; Tian, J.; Sun, L.; et al. Comprehensive analysis of circRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of atherosclerosis in rabbits. Aging (Albany. N.Y.) 2018, 10, 2266–2283. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhang, J.; Tong, Y.; Li, J.; Liu, B. Physcion 8-O-β-glucopyranoside induced ferroptosis via regulating miR-103a-3p/GLS2 axis in gastric cancer. Life Sci. 2019, 237, 116893. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Miao, J.; Zhang, M.; Wang, X.; Wang, Z.; Han, J.; Tong, D.; Huang, C. miRNA-103a-3p promotes human gastric cancer cell proliferation by targeting and suppressing ATF7 in vitro. Mol. Cells 2018, 41, 390–400. [Google Scholar] [PubMed]
- Li, J.; Li, X.; Wu, L.; Pei, M.; Li, H.; Jiang, Y. miR-145 inhibits glutamine metabolism through c-myc/GLS1 pathways in ovarian cancer cells. Cell Biol. Int. 2019, 43, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Muys, B.R.; Sousa, J.F.; Plaça, J.R.; de Araújo, L.F.; Sarshad, A.A.; Anastasakis, D.G.; Wang, X.; Li, X.L.; de Molfetta, G.A.; Ramão, A.; et al. miR-450a Acts as a Tumor Suppressor in Ovarian Cancer by Regulating Energy Metabolism. Cancer Res. 2019, 79, 3294–3305. [Google Scholar] [CrossRef]
- Luan, W.; Zhou, Z.; Zhu, Y.; Xia, Y.; Wang, J.; Xu, B. miR-137 inhibits glutamine catabolism and growth of malignant melanoma by targeting glutaminase. Biochem. Biophys. Res. Commun. 2018, 495, 46–52. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Xiao, D.; Zhao, Z.; Ren, P.; Li, C.; Hu, Y.; Shi, J.; Su, H.; Wang, L.; Liu, H.; et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis 2017, 6, e356. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, B.; Ren, H.; Chen, W. miR-9-5p inhibits pancreatic cancer cell proliferation, invasion and glutamine metabolism by targeting GOT1. Biochem. Biophys. Res. Commun. 2019, 509, 241–248. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, L.; Zhang, P.; Luo, M.; Du, J.; Gao, T.; O’Connell, D.; Wang, G.; Wang, H.; Yang, Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018, 57, 1566–1576. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wang, J.; Li, Y.; Fan, J.; Chen, L.; Xu, R. MicroRNA-153 regulates glutamine metabolism in glioblastoma through targeting glutaminase. Tumour Biol. 2017, 39, 1010428317691429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.-L.; Lv, Y.; Xu, C.-W.; Fu, M.-C.; Zhang, T.; Yan, X.-M.; Dai, S.; Xiong, Q.-W.; Zhou, Y.; Wang, J.; et al. MiR-513c suppresses neuroblastoma cell migration, invasion, and proliferation through direct targeting glutaminase (GLS). Cancer Biomark. 2017, 20, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, M.G.; Saumet, A.; Rossi, J.-F.; de Bettignies, C.; Tempé, D.; Lecellier, C.-H.; Villalba, M. The NF-κB member p65 controls glutamine metabolism through miR-23a. Int. J. Biochem. Cell Biol. 2012, 44, 1448–1456. [Google Scholar] [CrossRef]
- Chang, X.; Zhu, W.; Zhang, H.; Lian, S. Sensitization of melanoma cells to temozolomide by overexpression of microRNA 203 through direct targeting of glutaminase-mediated glutamine metabolism. Clin. Exp. Dermatol. 2017, 42, 614–621. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Z.; Xuan, Z.; Xu, P.; Wang, W.; Chen, Z.; Wang, S.; Sun, G.; Xu, J.; Xu, Z. Novel role of miR-133a-3p in repressing gastric cancer growth and metastasis via blocking autophagy-mediated glutaminolysis. J. Exp. Clin. Cancer Res. 2018, 37, 320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhu, J.C.; Hu, H.; Lin, Q.Y.; Shao, W.; Ji, T.H. MicroRNA-140-5p suppresses invasion and proliferation of glioma cells by targeting glutamate-ammonia ligase (GLUL). Neoplasma 2020. [Google Scholar] [CrossRef]
- Sengupta, D.; Cassel, T.; Teng, K.-Y.; Aljuhani, M.; Chowdhary, V.K.; Hu, P.; Zhang, X.; Fan, T.W.-M.; Ghoshal, K. Regulation of hepatic glutamine metabolism by miR-122. Mol. Metab. 2020, 34, 174–186. [Google Scholar] [CrossRef]
- Liu, H.; Luo, J.; Luan, S.; He, C.; Li, Z. Long non-coding RNAs involved in cancer metabolic reprogramming. Cell. Mol. Life Sci. 2019, 76, 495–504. [Google Scholar] [CrossRef]
- Zeng, B.; Ye, H.; Chen, J.; Cheng, D.; Cai, C.; Chen, G.; Chen, X.; Xin, H.; Tang, C.; Zeng, J. LncRNA TUG1 sponges miR-145 to promote cancer progression and regulate glutamine metabolism via Sirt3/GDH axis. Oncotarget 2017, 8, 113650–113661. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.-L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Yan, X.; Jin, Y.; Yang, X.; Yu, X.; Zhou, L.; Han, S.; Yuan, Q.; Yang, M. MiRNA-192 [corrected] and miRNA-204 Directly Suppress lncRNA HOTTIP and Interrupt GLS1-Mediated Glutaminolysis in Hepatocellular Carcinoma. PLoS Genet. 2015, 11, e1005726. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-J.; Li, X.; Pang, H.; Pan, J.-J.; Xie, X.-J.; Chen, W. Long non-coding RNA UCA1 promotes glutamine metabolism by targeting miR-16 in human bladder cancer. Jpn. J. Clin. Oncol. 2015, 45, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cui, S.; Wan, T.; Li, X.; Tian, W.; Zhang, R.; Luo, L.; Shi, Y. Long non-coding RNA HOTAIR acts as a competing endogenous RNA to promote glioma progression by sponging miR-126-5p. J. Cell. Physiol. 2018, 233, 6822–6831. [Google Scholar] [CrossRef]
- Liao, M.; Liao, W.; Xu, N.; Li, B.; Liu, F.; Zhang, S.; Wang, Y.; Wang, S.; Zhu, Y.; Chen, D.; et al. LncRNA EPB41L4A-AS1 regulates glycolysis and glutaminolysis by mediating nucleolar translocation of HDAC2. EBioMedicine 2019, 41, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Zhan, H.; Lin, F.; Liu, Y.; Yang, K.; Gao, Q.; Ding, M.; Liu, Y.; Huang, W.; Cai, Z. LincRNA-p21 suppresses glutamine catabolism and bladder cancer cell growth through inhibiting glutaminase expression. Biosci. Rep. 2019, 39, BSR20182372. [Google Scholar] [CrossRef] [Green Version]
- Luan, W.; Zhang, X.; Ruan, H.; Wang, J.; Bu, X. Long noncoding RNA OIP5-AS1 acts as a competing endogenous RNA to promote glutamine catabolism and malignant melanoma growth by sponging miR-217. J. Cell. Physiol. 2019, 234, 16609–16618. [Google Scholar] [CrossRef]
- Deng, S.-J.; Chen, H.-Y.; Zeng, Z.; Deng, S.; Zhu, S.; Ye, Z.; He, C.; Liu, M.-L.; Huang, K.; Zhong, J.-X.; et al. Nutrient Stress-Dysregulated Antisense lncRNA GLS-AS Impairs GLS-Mediated Metabolism and Represses Pancreatic Cancer Progression. Cancer Res. 2019, 79, 1398–1412. [Google Scholar] [CrossRef]
- Redis, R.S.; Vela, L.E.; Lu, W.; Ferreira de Oliveira, J.; Ivan, C.; Rodriguez-Aguayo, C.; Adamoski, D.; Pasculli, B.; Taguchi, A.; Chen, Y.; et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol. Cell 2016, 61, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Chen, Z.; Wang, J.; Wang, J.; Chen, X.; Liang, L.; Huang, M.; Zhang, Z.; Zuo, X. circHECTD1 facilitates glutaminolysis to promote gastric cancer progression by targeting miR-1256 and activating β-catenin/c-Myc signaling. Cell Death Dis. 2019, 10, 576. [Google Scholar] [CrossRef]
- Zhen, N.; Gu, S.; Ma, J.; Zhu, J.; Yin, M.; Xu, M.; Wang, J.; Huang, N.; Cui, Z.; Bian, Z.; et al. CircHMGCS1 Promotes Hepatoblastoma Cell Proliferation by Regulating the IGF Signaling Pathway and Glutaminolysis. Theranostics 2019, 9, 900–919. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Pedraza, Y.; Muñoz-Bello, J.O.; Olmedo-Nieva, L.; Contreras-Paredes, A.; Martínez-Ramírez, I.; Langley, E.; Lizano, M. Non-Coding RNAs as Key Regulators of Glutaminolysis in Cancer. Int. J. Mol. Sci. 2020, 21, 2872. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082872
Ortiz-Pedraza Y, Muñoz-Bello JO, Olmedo-Nieva L, Contreras-Paredes A, Martínez-Ramírez I, Langley E, Lizano M. Non-Coding RNAs as Key Regulators of Glutaminolysis in Cancer. International Journal of Molecular Sciences. 2020; 21(8):2872. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082872
Chicago/Turabian StyleOrtiz-Pedraza, Yunuen, J. Omar Muñoz-Bello, Leslie Olmedo-Nieva, Adriana Contreras-Paredes, Imelda Martínez-Ramírez, Elizabeth Langley, and Marcela Lizano. 2020. "Non-Coding RNAs as Key Regulators of Glutaminolysis in Cancer" International Journal of Molecular Sciences 21, no. 8: 2872. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082872