Ethanol Intoxication Alleviates the Inflammatory Response of Remote Organs to Experimental Traumatic Brain Injury


 , and
, and 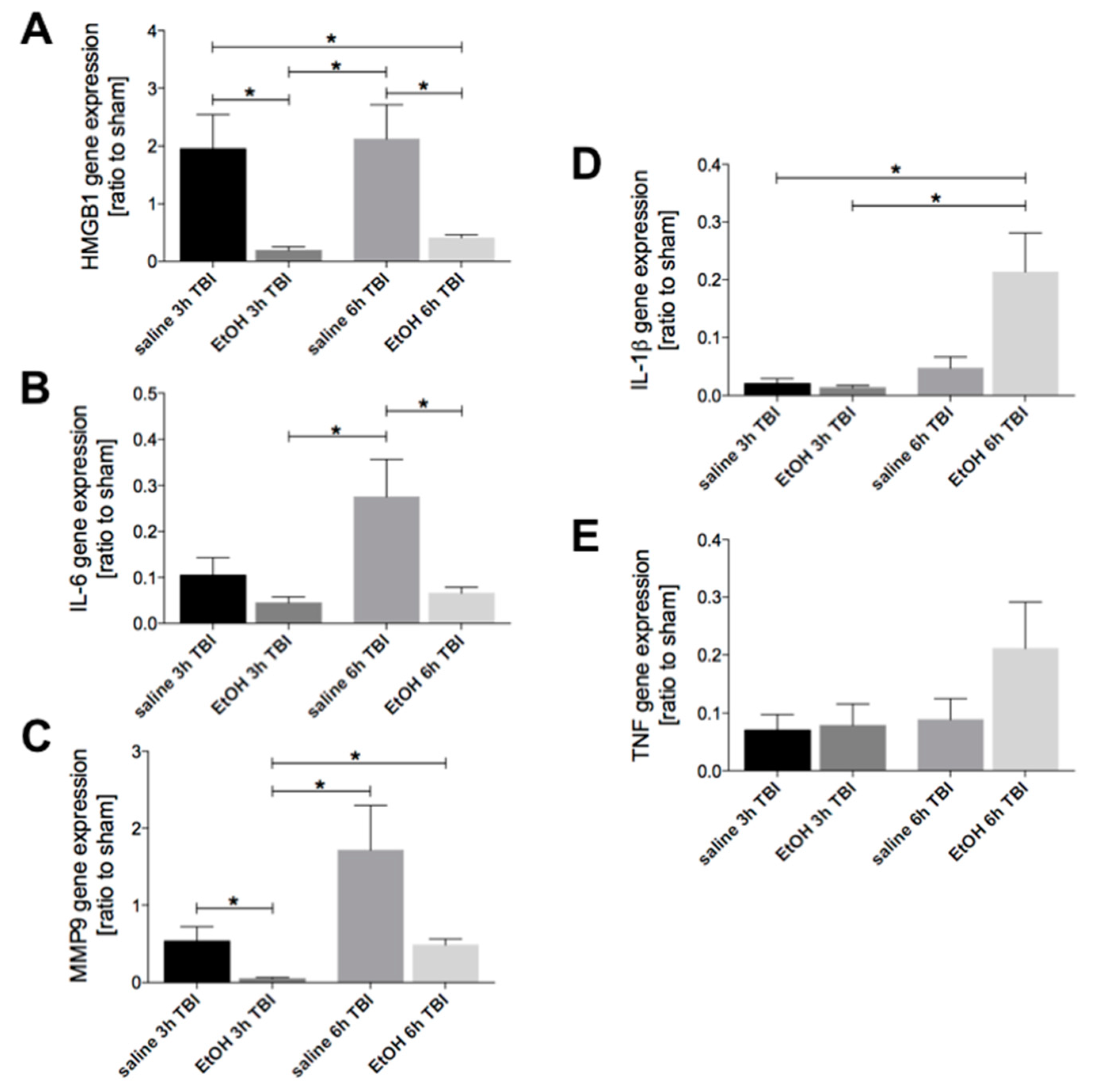{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Ethanol Intoxication Downregulates TBI-Induced HMGB1, IL-6, and MMP9 but Enhances IL-1β Gene Expression in Liver Tissue
2.2. Ethanol Intoxication Modulates the Early Inflammatory Response in Liver Tissue upon TBI
2.3. Ethanol Intoxication Reduces the TBI-Induced Protein HMGB1 in Liver Tissue
2.4. Ethanol Intoxication Downregulates the TBI-Induced HMGB1, IL-6, IL-1β and TNF Gene Expression in Lungs
2.5. Elevation of IL-10 In Lung Tissue Early after Experimental TBI
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Model
4.2. Group Allocation
4.3. Quantification of Homogenate Protein Expression Levels by ELISA
4.4. Immunohistological Analysis of HMGB1
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rubiano, A.M.; Carney, N.A.; Chesnut, R.M.; Puyana, J.C. Global neurotrauma research challenges and opportunities. Nature 2015, 527, S193–S197. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, I.; Wood, R.L.; Phillips, C.J.; Macey, S. The costs of traumatic brain injury: A literature review. Clin. Outcomes Res. 2013, 5, 281–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Peeters, W.; Feigin, V.L.; Theadom, A.; Holkovic, L.; et al. Epidemiology of Traumatic Brain Injury in Europe: A Living Systematic Review. J. Neurotrauma 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savola, O.; Niemelä, O.; Hillbom, M. Alcohol intake and the pattern of trauma in young adults and working aged people admitted after trauma. Alcohol Alcohol. 2005, 40, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Wang, Z.; Shen, M.; Su, Z.; Shen, L. Acute Alcohol Exposure and Risk of Mortality of Patients with Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Alcohol. Clin. Exp. Res. 2017, 41, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.J.; Sharkey, J.M.; Sun, M.; Kaukas, L.M.; Shultz, S.R.; Turner, R.J.; Leonard, A.V.; Brady, R.D.; Corrigan, F. Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 770–781. [Google Scholar] [CrossRef]
- Ott, L.; McClain, C.J.; Gillespie, M.; Young, B. Cytokines and Metabolic Dysfunction After Severe Head Injury. J. Neurotrauma 1994, 11, 447–472. [Google Scholar] [CrossRef]
- Nicolls, M.R.; Laubach, V.E. Traumatic brain injury: Lungs in a RAGE. Sci. Transl. Med. 2014, 6, 252fs34. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, T.; Morganti-Kossmann, M.C. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Gyoneva, S.; Ransohoff, R.M. Inflammatory reaction after traumatic brain injury: Therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharm. Sci. 2015, 36, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, T.M.; Nguyen, A.H.; Rabang, J.R.; Patil, A.-A.; Agrawal, D.K. The danger zone: Systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj. 2016, 31, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, X.F. Ghrelin alleviates traumatic brain injury-induced acute lung injury through pyroptosis/NF-kappaB pathway. Int. Immunopharmacol 2020, 79, 106175. [Google Scholar] [CrossRef]
- Kerr, N.A.; Vaccari, J.P.D.R.; Abbassi, S.; Kaur, H.; Zambrano, R.; Wu, S.; Dietrich, W.D.; Keane, R.W. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J. Neurotrauma 2018, 35, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ma, Y.; Liu, Y.; Que, H.; Zhu, C.; Liu, S. Elevated serum haptoglobin after traumatic brain injury is synthesized mainly in liver. J. Neurosci. Res. 2012, 91, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Arredouani, M.; Matthijs, P.; Van Hoeyveld, E.; Kasran, A.; Baumann, H.; Ceuppens, J.L.; Stevens, E. Haptoglobin directly affects T cells and suppresses T helper cell type 2 cytokine release. Immunology 2003, 108, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Gottesfeld, Z.; Moore, A.N.; Dash, P.K. Acute Ethanol Intake Attenuates Inflammatory Cytokines after Brain Injury in Rats: A Possible Role for Corticosterone. J. Neurotrauma 2002, 19, 317–326. [Google Scholar] [CrossRef]
- Zink, B.J.; Feustel, P.J. Effects of ethanol on respiratory function in traumatic brain injury. J. Neurosurg. 1995, 82, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Zink, B.J.; Walsh, R.F.; Feustel, P.J. Effects of Ethanol in Traumatic Brain Injury. J. Neurotrauma 1993, 10, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Kany, S.; Janicova, A.; Relja, B. Kany Innate Immunity and Alcohol. J. Clin. Med. 2019, 8, 1981. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Akbarpour, A.; Mörs, K.; Voth, M.; Störmann, P.; Auner, B.; Lehnert, M.; Marzi, I.; Relja, B. Alcohol Intoxication Reduces Systemic Interleukin-6 Levels and Leukocyte Counts After Severe TBI Compared With Not Intoxicated TBI Patients. Shock 2016, 46, 261–269. [Google Scholar] [CrossRef]
- Relja, B.; Menke, J.; Wagner, N.; Auner, B.; Voth, M.; Nau, C.; Marzi, I. Effects of positive blood alcohol concentration on outcome and systemic interleukin-6 in major trauma patients. Injury 2016, 47, 640–645. [Google Scholar] [CrossRef]
- Relja, B.; Höhn, C.; Bormann, F.; Seyboth, K.; Henrich, D.; Marzi, I.; Lehnert, M. Acute alcohol intoxication reduces mortality, inflammatory responses and hepatic injury after haemorrhage and resuscitationin vivo. Br. J. Pharmacol. 2012, 165, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, A.; Heuvel, F.O.; Palmer, A.; Linkus, B.; Ludolph, A.C.; Boeckers, T.M.; Relja, B.; Huber-Lang, M.; Roselli, F. Acute ethanol administration results in a protective cytokine and neuroinflammatory profile in traumatic brain injury. Int. Immunopharmacol. 2017, 51, 66–75. [Google Scholar] [CrossRef]
- Heuvel, F.O.; Holl, S.; Chandrasekar, A.; Li, Z.; Wang, Y.; Rehman, R.; Förstner, P.; Sinske, D.; Palmer, A.; Wiesner, D.; et al. STAT6 mediates the effect of ethanol on neuroinflammatory response in TBI. Brain, Behav. Immun. 2019, 81, 228–246. [Google Scholar] [CrossRef]
- Weber, D.J.; Allette, Y.M.; Wilkes, D.S.; White, F.A. The HMGB1-RAGE Inflammatory Pathway: Implications for Brain Injury-Induced Pulmonary Dysfunction. Antioxidants Redox Signal. 2015, 23, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, J.; Ribeiro, C.T.; Bortolin, R.C.; Somensi, N.; Fernandes, H.S.; Teixeira, A.A.; Guasselli, M.O.R.; Agani, C.A.J.O.; Souza, N.C.; Grings, M.; et al. Anti-RAGE antibody selectively blocks acute systemic inflammatory responses to LPS in serum, liver, CSF and striatum. Brain Behav. Immun. 2017, 62, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, Á.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef] [Green Version]
- Au, A.K.; Aneja, R.K.; Bell, M.J.; Bayir, H.; Feldman, K.; Adelson, P.D.; Fink, E.L.; Kochanek, P.M.; Clark, R.S. Cerebrospinal Fluid Levels of High-Mobility Group Box 1 and Cytochrome C Predict Outcome after Pediatric Traumatic Brain Injury. J. Neurotrauma 2012, 29, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Harris, H.E.; Andersson, U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 2004, 34, 1503–1512. [Google Scholar] [CrossRef]
- Fang, W.-H.; Yao, Y.-M.; Shi, Z.-G.; Yu, Y.; Wu, Y.; Lu, L.-R.; Sheng, Z.-Y. The Significance of Changes in High Mobility Group-1 Protein mRNA Expression in Rats After Thermal Injury. Shock 2002, 17, 329–333. [Google Scholar] [CrossRef]
- Teng, S.X.; Katz, P.S.; Maxi, J.K.; Mayeux, J.P.; Gilpin, N.W.; Molina, P.E. Alcohol exposure after mild focal traumatic brain injury impairs neurological recovery and exacerbates localized neuroinflammation. Brain Behav. Immun. 2015, 45, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Ge, X. High. mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J. Biol. Chem. 2014, 289, 22672–22691. [Google Scholar] [CrossRef] [Green Version]
- Lowe, P.P.; Gyongyosi, B.; Satishchandran, A.; Iracheta-Vellve, A.; Cho, Y.; Ambade, A.; Szabo, G. Reduced gut microbiome protects from alcohol-induced neuroinflammation and alters intestinal and brain inflammasome expression. J. Neuroinflammation 2018, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.; McAlister, A.; Willoughby, T.; Sivaraman, V. Alcohol-dependent pulmonary inflammation: A role for HMGB-1. Alcohol 2019, 80, 45–52. [Google Scholar] [CrossRef]
- Teng, S.X.; Molina, P.E. Acute Alcohol Intoxication Prolongs Neuroinflammation without Exacerbating Neurobehavioral Dysfunction following Mild Traumatic Brain Injury. J. Neurotrauma 2014, 31, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.D.; Makley, A.T.; Campion, E.M.; Friend, L.A.W.; Lentsch, A.B.; Pritts, T.A. Preinjury alcohol exposure attenuates the neuroinflammatory response to traumatic brain injury. J. Surg. Res. 2013, 184, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- El-Guindy, N.B.D.; De Villiers, W.J.; Doherty, D.E. Acute alcohol intake impairs lung inflammation by changing pro- and anti-inflammatory mediator balance. Alcohol 2007, 41, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, B.W.; Volkmer, D.L.; Yong, S.L.; Himes, R.D.; Lauing, K.L.; Morgan, M.; Stover, M.D.; Callaci, J.J. Binge Alcohol Exposure Modulates Rodent Expression of Biomarkers of the Immunoinflammatory Response to Orthopaedic Trauma. J. Bone Jt. Surg.-Am. Vol. 2011, 93, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Khoruts, A.; Stahnke, L.; McClain, C.J.; Logan, G.; Allen, J.I. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology 1991, 13, 267–276. [Google Scholar] [CrossRef]
- Pang, M.; Bala, S.; Kodys, K.; Catalano, D.; Szabo, G. Inhibition of TLR8- and TLR4-induced Type I IFN induction by alcohol is different from its effects on inflammatory cytokine production in monocytes. BMC Immunol. 2011, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Liu, T. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Huang, M.; Xie, L.; Shen, J.; Xiao, T.; Wang, R. IVIG inhibits TNF-α-induced MMP9 expression and activity in monocytes by suppressing NF-κB and P38 MAPK activation. Int. J. Clin. Exp. Pathol. 2015, 8, 15879–15886. [Google Scholar]
- Hoyt, L.; Ather, J.L.; Randall, M.J.; Depuccio, D.P.; Landry, C.C.; Wewers, M.D.; Gavrilin, M.A.; Poynter, M.E. Ethanol and Other Short-Chain Alcohols Inhibit NLRP3 Inflammasome Activation through Protein Tyrosine Phosphatase Stimulation. J. Immunol. 2016, 197, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Nurmi, K.; Virkanen, J.; Rajamäki, K.; Niemi, K.; Kovanen, P.T.; Eklund, K.K. Ethanol Inhibits Activation of NLRP3 and AIM2 Inflammasomes in Human Macrophages–A Novel Anti-Inflammatory Action of Alcohol. PLoS ONE 2013, 8, e78537. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; Depuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; Van Der Vliet, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. New Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Cerami, A. Cachectin/tumor necrosis factor: An endogenous mediator of shock and inflammation. Immunol. Res. 1986, 5, 281–293. [Google Scholar] [CrossRef]
- Szabo, G.; Mandrekar, P.; Girouard, L.; Catalano, D. Regulation of Human Monocyte Functions by Acute Ethanol Treatment: Decreased Tumor Necrosis Factor? Interleukin-l? and Elevated Interleukin-10, and Transforming Growth Factor-? Production. Alcohol. Clin. Exp. Res. 1996, 20, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Shanley, T.P.; Schmal, H.; Friedl, H.P.; Jones, M.L.; Ward, P.A. Regulatory effects of intrinsic IL-10 in IgG immune complex-induced lung injury. J. Immunol. 1995, 154, 3454–3460. [Google Scholar]
- Kobbe, P.; Stoffels, B.; Schmidt, J.; Tsukamoto, T.; Gutkin, D.W.; Bauer, A.J.; Pape, H.-C. IL-10 deficiency augments acute lung but not liver injury in hemorrhagic shock. Cytokine 2009, 45, 26–31. [Google Scholar] [CrossRef]
- Flierl, M.A.; Stahel, P.F.; Beauchamp, K.M.; Morgan, S.J.; Smith, W.R.; Shohami, E. Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 2009, 4, 1328–1337. [Google Scholar] [CrossRef]
- Bouchard, J.C.; Kim, J.; Beal, D.R.; Vaickus, L.J.; Craciun, F.L.; Remick, D. Acute oral ethanol exposure triggers asthma in cockroach allergen-sensitized mice. Am. J. Pathol. 2012, 181, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Namjoshi, D.R.; Good, C.; Cheng, W.H.; Panenka, W.J.; Richards, D.; Cripton, P.A.; Wellington, C. Towards clinical management of traumatic brain injury: A review of models and mechanisms from a biomechanical perspective. Dis. Model. Mech. 2013, 6, 1325–1338. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Dixon, C.E.; Clifton, G.L.; Lighthall, J.W.; Yaghmai, A.A.; Hayes, R.L. A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods 1991, 39, 253–262. [Google Scholar] [CrossRef]
- Dixon, C.E.; Lyeth, B.G.; Povlishock, J.T.; Findling, R.L.; Hamm, R.J.; Marmarou, A.; Young, H.F.; Hayes, R.L. A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 1987, 67, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Feeney, D.M.; Boyeson, M.G.; Linn, R.T.; Murray, H.M.; Dail, W.G. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981, 211, 67–77. [Google Scholar] [CrossRef]
- Albert-Weißenberger, C.; Várrallyay, C.; Raslan, F.; Kleinschnitz, C.; Sirén, A.-L. An experimental protocol for mimicking pathomechanisms of traumatic brain injury in mice. Exp. Transl. Stroke Med. 2012, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Constantini, S.; Trembovler, V.; Weinstock, M.; Shohami, E. An Experimental Model of Closed Head Injury in Mice: Pathophysiology, Histopathology, and Cognitive Deficits. J. Neurotrauma 1996, 13, 557–568. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, B.; Chandrasekar, A.; olde Heuvel, F.; Powerski, M.; Nowak, A.; Noack, L.; Omari, J.; Huber-Lang, M.; Roselli, F.; Relja, B. Ethanol Intoxication Alleviates the Inflammatory Response of Remote Organs to Experimental Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 8181. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218181
Xu B, Chandrasekar A, olde Heuvel F, Powerski M, Nowak A, Noack L, Omari J, Huber-Lang M, Roselli F, Relja B. Ethanol Intoxication Alleviates the Inflammatory Response of Remote Organs to Experimental Traumatic Brain Injury. International Journal of Molecular Sciences. 2020; 21(21):8181. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218181
Chicago/Turabian StyleXu, Baolin, Akila Chandrasekar, Florian olde Heuvel, Maciej Powerski, Aleksander Nowak, Laurens Noack, Jazan Omari, Markus Huber-Lang, Francesco Roselli, and Borna Relja. 2020. "Ethanol Intoxication Alleviates the Inflammatory Response of Remote Organs to Experimental Traumatic Brain Injury" International Journal of Molecular Sciences 21, no. 21: 8181. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218181