Spontaneous Mutations in HIV-1 Gag, Protease, RT p66 in the First Replication Cycle and How They Appear: Insights from an In Vitro Assay on Mutation Rates and Types

 ,
,
Abstract
:1. Introduction
2. Results
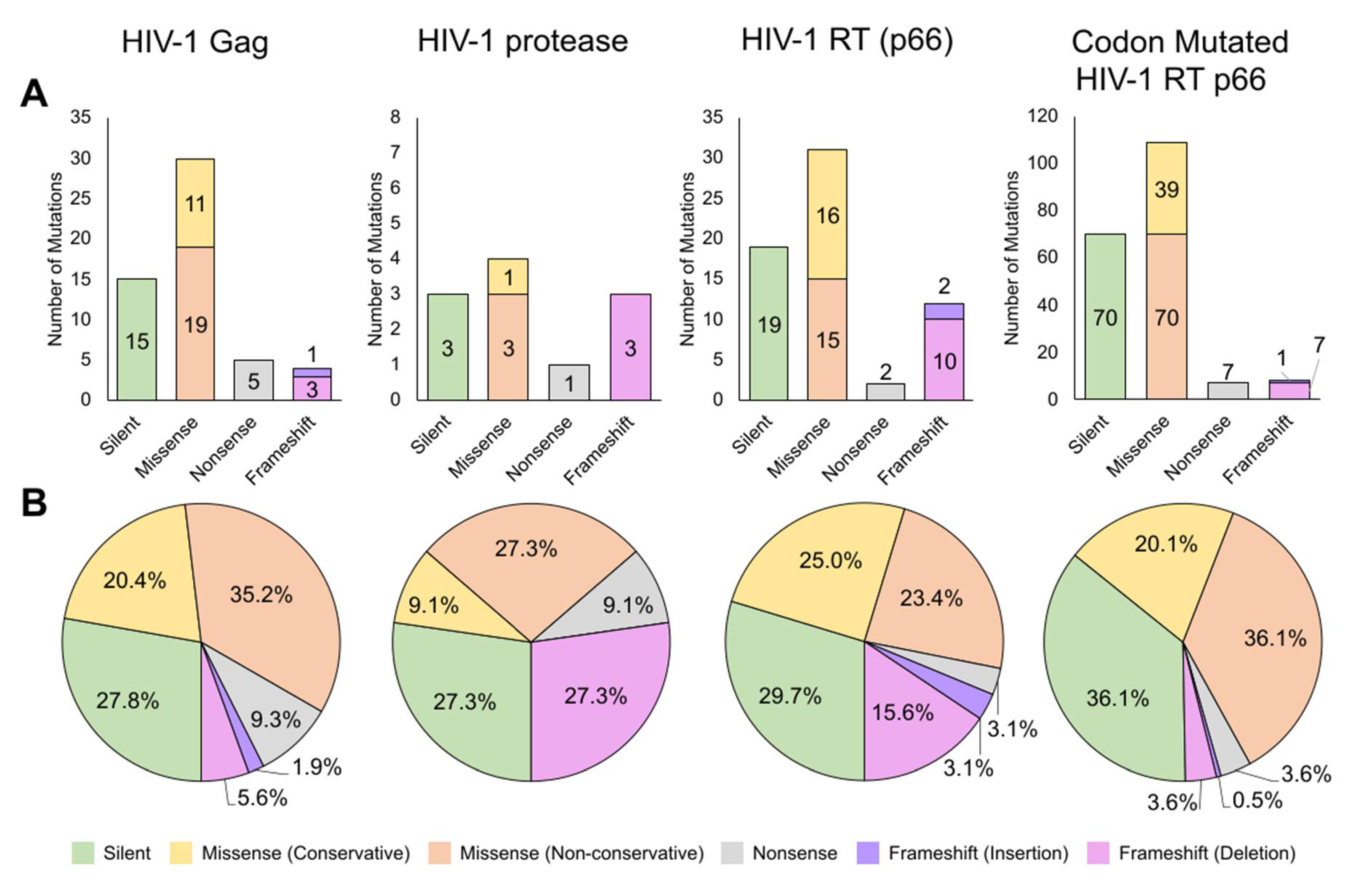
2.1. Characterization of HIV-1 Gag, Protease, RT p66, and Codon Mutated RT p66 Mutant Variants
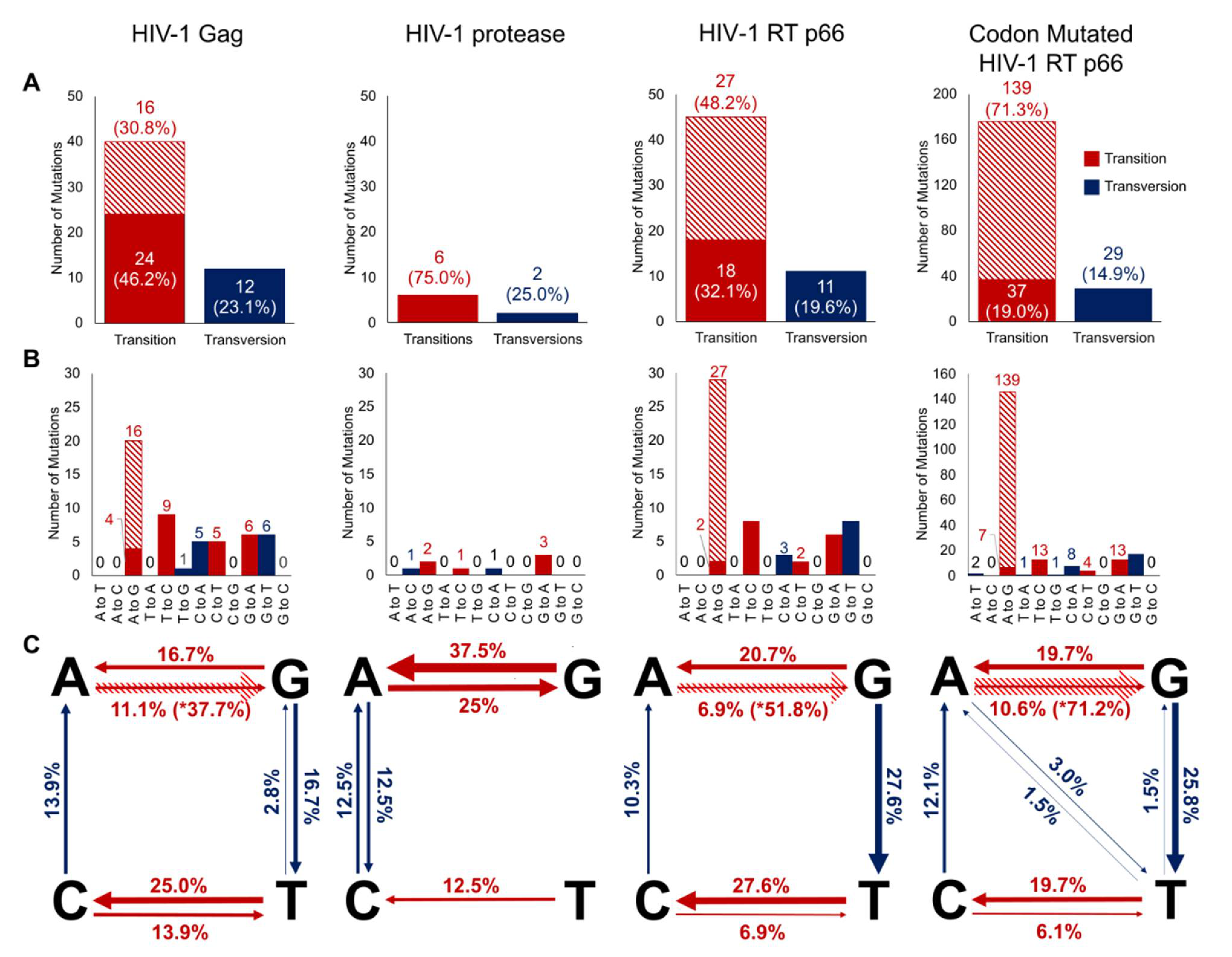
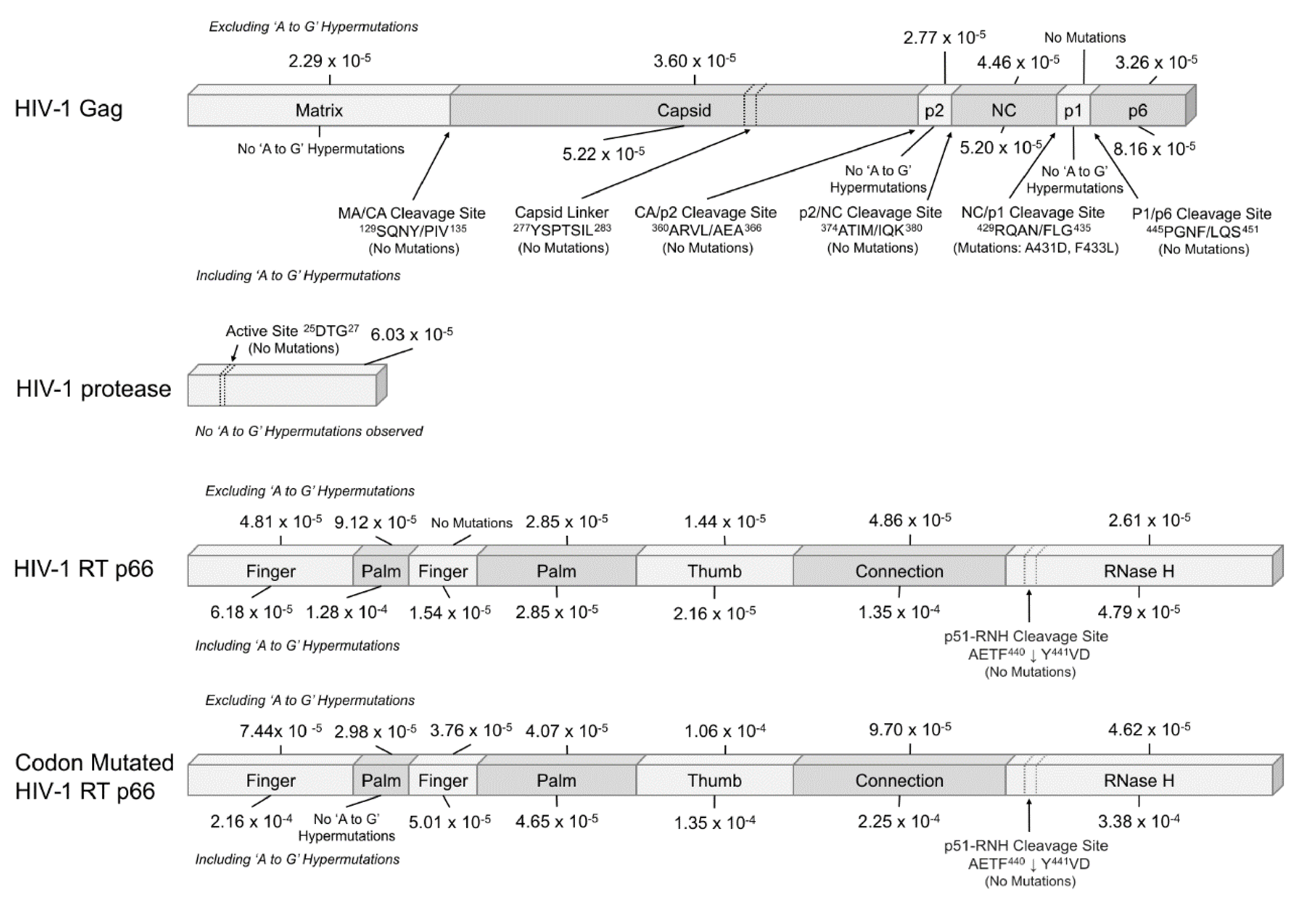
2.2. Mutation Rates of the Domains of HIV-1 Gag, Protease and RT p66
2.3. Flanking Sequences of ‘A to G’ Mutation Sites
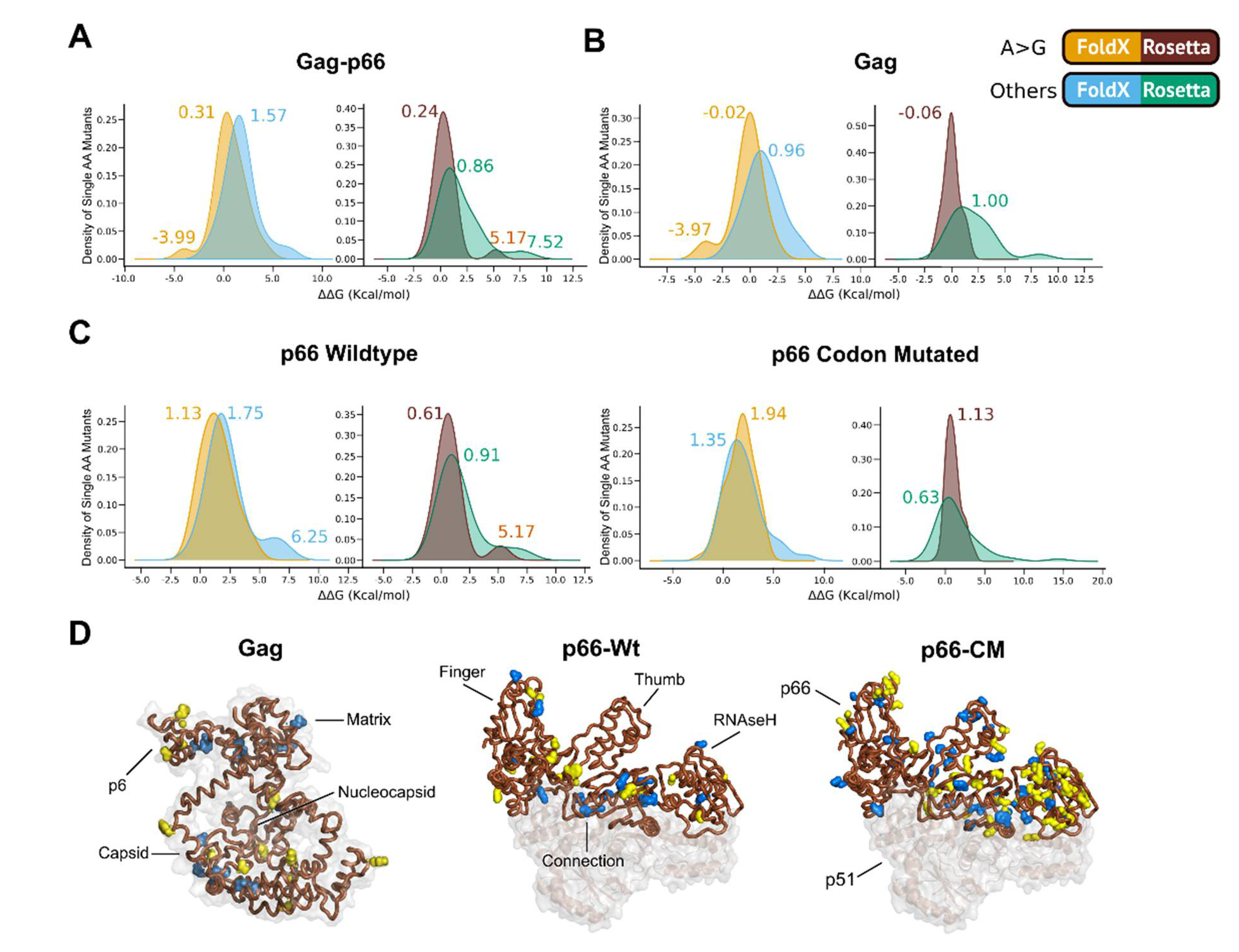
2.4. Effects of ‘A to G’ Mutations on Protein Thermostability
3. Discussion
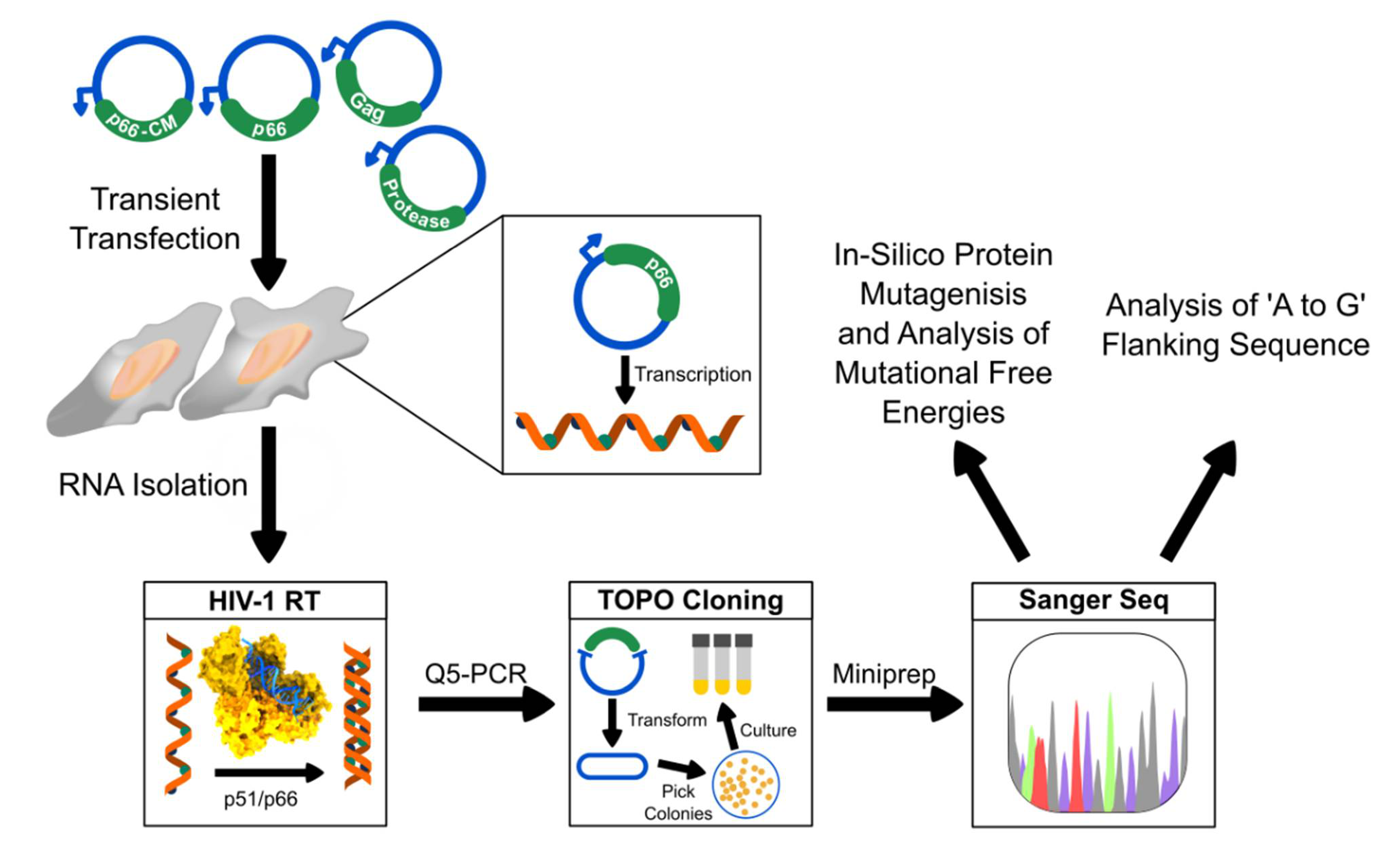
4. Materials and Methods
4.1. Transfection of HIV-1 Gag, Pr and RT Plasmids
4.2. RNA Extraction and cDNA Synthesis
4.3. Amplification of cDNA and TOPO Cloning
4.4. Sequence Analysis
4.5. In-Silico Assessment on Protein Thermostability Using FoldX and Rosetta Cartesian_ddg
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woolhouse, M.E.J.; Haydon, D.T.; Antia, R. Emerging pathogens: The epidemiology and evolution of species jumps. Trends Ecol. Evol. 2005, 20, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.L.; Parrish, C.R.; Cobey, S.; Glass, G.E.; Bush, R.M.; Leighton, T.J. Anticipating the Species Jump: Surveillance for Emerging Viral Threats. Zoonoses Public Health 2012, 59, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailes, E.; Gao, F.; Bibollet-Ruche, F.; Courgnaud, V.; Peeters, M.; Marx, P.A.; Hahn, B.H.; Sharp, P.M. Hybrid Origin of SIV in Chimpanzees. Science 2003, 300, 1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Luckay, A.; Sodora, D.L.; Telfer, P.; Reed, P.; Gettie, A.; Kanu, J.M.; Sadek, R.F.; Yee, J.; Ho, D.D.; et al. Human immunodeficiency virus type 2 (HIV-2) seroprevalence and characterization of a distinct HIV-2 genetic subtype from the natural range of simian immunodeficiency virus-infected sooty mangabeys. J. Virol. 1997, 71, 3953–3960. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Bailes, E.; Robertson, D.L.; Chen, Y.; Rodenburg, C.M.; Michael, S.F.; Cummins, L.B.; Arthur, L.O.; Peeters, M.; Shaw, G.M.; et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 1999, 397, 436–441. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Pettit, S.C.; Everitt, L.E.; Choudhury, S.; Dunn, B.M.; Kaplan, A.H. Initial Cleavage of the Human Immunodeficiency Virus Type 1 GagPol Precursor by Its Activated Protease Occurs by an Intramolecular Mechanism. J. Virol. 2004, 78, 8477–8485. [Google Scholar] [CrossRef] [Green Version]
- Fun, A.; van Maarseveen, N.M.; Pokorná, J.; Maas, R.E.M.; Schipper, P.J.; Konvalinka, J.; Nijhuis, M. HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology 2011, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Fun, A.; Wensing, A.M.J.; Verheyen, J.; Nijhuis, M. Human Immunodeficiency Virus gag and protease: Partners in resistance. Retrovirology 2012, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Clavel, F.; Mammano, F. Role of Gag in HIV Resistance to Protease Inhibitors. Viruses 2010, 2, 1411–1426. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Shao, Q.; Shi, J.; Zhu, W. Effects of drug-resistant mutations on the dynamic properties of HIV-1 protease and inhibition by Amprenavir and Darunavir. Sci. Rep. 2015, 5, 10517. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar] [PubMed]
- Park, J.; Sayer, J.; Aniana, A.; Yu, X.; Weber, I.; Harrison, R.; Louis, J. Binding of clinical inhibitors to a model precursor of a rationally selected multidrug resistant HIV-1 protease is significantly weaker than that to the released mature enzyme. Biochemistry 2016, 55, 2390–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.T.-T.; Kwoh, C.-K.; Verma, C.S.; Gan, S.K.-E. Modeling the full length HIV-1 Gag polyprotein reveals the role of its p6 subunit in viral maturation and the effect of non-cleavage site mutations in protease drug resistance. J. Biomol. Struct. Dyn. 2017, 1–12. [Google Scholar] [CrossRef]
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 drug resistance and resistance testing. Infect. Genet. Evol. 2016, 46, 292–307. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Kohli, A.; McCormick, A.L.; Towers, G.J.; Pillay, D.; Parry, C.M. Full-length HIV-1 Gag determines protease inhibitor susceptibility within in-vitro assays. AIDS 2010, 24, 1651–1655. [Google Scholar] [CrossRef] [Green Version]
- Dam, E.; Quercia, R.; Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Kräusslich, H.-G.; Hance, A.J.; Clavel, F.; Group, A.S. Gag Mutations Strongly Contribute to HIV-1 Resistance to Protease Inhibitors in Highly Drug-Experienced Patients besides Compensating for Fitness Loss. PLoS Pathog. 2009, 5, e1000345. [Google Scholar] [CrossRef] [Green Version]
- Gatanaga, H.; Suzuki, Y.; Tsang, H.; Yoshimura, K.; Kavlick, M.K.; Nagashima, K.; Gorelick, R.J.; Mardy, S.; Tang, C.; Summers, M.F.; et al. Amino Acid Substitutions in Gag Protein at Non-cleavage sites Are Indispensable for the Development of a high Multitude of HIV-1 Resistance against Protease Inhibitors. J. Biol. Chem. 2002, 277, 5952–5961. [Google Scholar] [CrossRef] [Green Version]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.; Wrin, T.; et al. Changes in Human Immunodeficiency Virus Type 1 Gag at Positions L449 and P453 Are Linked to I50V Protease Mutants in vivo and Cause Reduction of Sensitivity to Amprenavir and Improved Viral Fitness in vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [Green Version]
- Tamiya, S.; Mardy, S.; Kavlick, M.F.; Yoshimura, K.; Mitsuya, H. Amino acid insertions near Gag cleavage sites restore the otherwise compromised replication of human immunodeficiency virus type 1 variants resistant to Protease Inhibitors. J. Virol. 2004, 78, 12030–12040. [Google Scholar] [CrossRef] [Green Version]
- Parry, C.M.; Kohli, A.; Boinett, C.J.; Towers, G.J.; McCormick, A.L.; Pillay, D. Gag Determinants of Fitness and Drug Susceptibility in Protease Inhibitor-Resistant Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 9094–9101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightfoote, M.M.; Coligan, J.E.; Folks, T.M.; Fauci, A.S.; Martin, M.A.; Venkatesan, S. Structural characterization of reverse transcriptase and endonuclease polypeptides of the acquired immunodeficiency syndrome retrovirus. J. Virol. 1986, 60, 771–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostomsky, Z.; Hostomska, Z.; Fu, T.B.; Taylor, J. Reverse transcriptase of human immunodeficiency virus type 1: Functionality of subunits of the heterodimer in DNA synthesis. J. Virol. 1992, 66, 3179–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [Green Version]
- Le Grice, S.F.; Naas, T.; Wohlgensinger, B.; Schatz, O. Subunit-selective mutagenesis indicates minimal polymerase activity in heterodimer-associated p51 HIV-1 reverse transcriptase. Embo J. 1991, 10, 3905–3911. [Google Scholar] [CrossRef]
- Yeo, J.Y.; Goh, G.R.; Su, C.T.-T.; Gan, S.K.-E. The Determination of HIV-1 RT Mutation Rate, Its Possible Allosteric Effects, and Its Implications on Drug Resistance. Viruses 2020, 12, 297. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Arias, L. Mutation Rates and Intrinsic Fidelity of Retroviral Reverse Transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef] [Green Version]
- Eggington, J.M.; Greene, T.; Bass, B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011, 2, 319. [Google Scholar] [CrossRef] [Green Version]
- Tokuriki, N.; Oldfield, C.J.; Uversky, V.N.; Berezovsky, I.N.; Tawfik, D.S. Do viral proteins possess unique biophysical features? Trends Biochem. Sci. 2009, 34, 53–59. [Google Scholar] [CrossRef]
- Strokach, A.; Corbi-Verge, C.; Kim, P.M. Predicting changes in protein stability caused by mutation using sequence-and structure-based methods in a CAGI5 blind challenge. Hum. Mutat. 2019, 40, 1414–1423. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [PubMed]
- Potapov, V.; Ong, J.L. Examining sources of error in PCR by single-molecule sequencing. PLoS ONE 2017, 12, e0169774. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Domingo-Calap, P.; Cuevas, J.M.; Rossolillo, P.; Negroni, M.; Sanjuán, R. The external domains of the HIV-1 envelope are a mutational cold spot. Nat. Commun. 2015, 6, 8571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Loeb, L.A. Fidelity of HIV-1 Reverse Transcriptase Copying a Hypervariable Region of the HIV-1 env Gene. Virology 1994, 199, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Evidence for the Selective Basis of Transition-to-Transversion Substitution Bias in Two RNA Viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoltzfus, A.; Norris, R.W. On the Causes of Evolutionary Transition:Transversion Bias. Mol. Biol. Evol. 2016, 33, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, M.S. Patterns of Transitional Mutation Biases Within and Among Mammalian Genomes. Mol. Biol. Evol. 2003, 20, 988–993. [Google Scholar] [CrossRef]
- Chan, K.-F.; Koukouravas, S.; Yeo, J.Y.; Koh, D.W.-S.; Gan, S.K.-E. Probability of change in life: Amino acid changes in single nucleotide substitutions. Biosystems 2020, 193–194, 104135. [Google Scholar] [CrossRef]
- Dawson, T.R.; Sansam, C.L.; Emeson, R.B. Structure and Sequence Determinants Required for the RNA Editing of ADAR2 Substrates. J. Biol. Chem. 2004, 279, 4941–4951. [Google Scholar] [CrossRef] [Green Version]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, Position, and Frequency of Mutations Made in a Single Cycle of HIV-1 Replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, A.M.; Linial, M.L. Modification of retroviral RNA by double-stranded RNA adenosine deaminase. J. Virol. 1995, 69, 5878–5882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gélinas, J.-F.; Clerzius, G.; Shaw, E.; Gatignol, A. Enhancement of Replication of RNA Viruses by ADAR1 via RNA Editing and Inhibition of RNA-Activated Protein Kinase. J. Virol. 2011, 85, 8460–8466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, N.L.; Birlouez, E.; Wain-Hobson, S.; Mahieux, R.; Vartanian, J.-P. Hyperediting of human T-cell leukemia virus type 2 and simian T-cell leukemia virus type 3 by the dsRNA adenosine deaminase ADAR-1. J. Gen. Virol. 2012, 93, 2646–2651. [Google Scholar] [CrossRef] [PubMed]
- Samsudin, F.; Gan, S.K.-E.; Bond, P.J. The Impact of Gag Non-Cleavage Site Mutations on HIV-1 Viral Fitness from Integrative Modelling and Simulations. Comput. Struct. Biotechnol. J. 2020. [Google Scholar] [CrossRef]
- Sanchez-Merino, V.; Farrow, M.A.; Brewster, F.; Somasundaran, M.; Luzuriaga, K. Identification and Characterization of HIV-1 CD8+ T Cell Escape Variants with Impaired Fitness. J. Infect. Dis. 2008, 197, 300–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Merino, V.; Nie, S.; Luzuriaga, K. HIV-1-Specific CD8+ T Cell Responses and Viral Evolution in Women and Infants. J. Immunol. 2005, 175, 6976–6986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.; Penaranda, G.; Gonzalez, D.; Camus, C.; Khiri, H.; Boulmé, R.; Sayada, C.; Philibert, P.; Olive, D.; Halfon, P. Comparison of ultra-deep versus Sanger sequencing detection of minority mutations on the HIV-1 drug resistance interpretations after virological failure. AIDS 2014, 28, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Parkin, N.T.; Gupta, S.; Chappey, C.; Petropoulos, C.J. The K101P and K103R/V179D Mutations in Human Immunodeficiency Virus Type 1 Reverse Transcriptase Confer Resistance to Nonnucleoside Reverse Transcriptase Inhibitors. Antimicrob. Agents Chemother. 2006, 50, 351–354. [Google Scholar] [CrossRef] [Green Version]
- De Iaco, A.; Luban, J. Cyclophilin A promotes HIV-1 reverse transcription but its effect on transduction correlates best with its effect on nuclear entry of viral cDNA. Retrovirology 2014, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Banke, S.; Lillemark, M.R.; Gerstoft, J.; Obel, N.; Jørgensen, L.B. Positive Selection Pressure Introduces Secondary Mutations at Gag Cleavage Sites in Human Immunodeficiency Virus Type 1 Harboring Major Protease Resistance Mutations. J. Virol. 2009, 83, 8916–8924. [Google Scholar] [CrossRef] [Green Version]
- Fisher, T.S.; Darden, T.; Prasad, V.R. Substitutions at Phe61 in the β3-β4 Hairpin of HIV-1 Reverse Transcriptase Reveal a Role for the Fingers Subdomain in Strand Displacement DNA Synthesis. J. Mol. Biol. 2003, 325, 443–459. [Google Scholar] [CrossRef]
- Ceccherini-Silberstein, F.; Gago, F.; Santoro, M.; Gori, C.; Svicher, V.; Rodríguez-Barrios, F.; d’Arrigo, R.; Ciccozzi, M.; Bertoli, A.; d’Arminio Monforte, M.; et al. High Sequence Conservation of Human Immunodeficiency Virus Type 1 Reverse Transcriptase under Drug Pressure despite the Continuous Appearance of Mutations. J. Virol. 2005, 79, 10718–10729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auwerx, J.; Van Nieuwenhove, J.; Rodríguez-Barrios, F.; de Castro, S.; Velázquez, S.; Ceccherini-Silberstein, F.; De Clercq, E.; Camarasa, M.-J.; Perno, C.-F.; Gago, F.; et al. The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes of anti-HIV-1 drugs. FEBS Lett. 2005, 579, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Perno, C.F. HIV-1 Genetic Variability and Clinical Implications. ISRN Microbiol. 2013, 2013, 481314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessilly, G.; Goeminne, L.; Vandenbroucke, A.-T.; Dufrasne, F.E.; Martin, A.; Kabamba-Mukabi, B. First evaluation of the Next-Generation Sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLoS ONE 2019, 13, e0209561. [Google Scholar] [CrossRef]
- Li, M.; Stoneking, M. A new approach for detecting low-level mutations in next-generation sequence data. Genome Biol. 2012, 13, R34. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Miller, J.T.; Lapkouski, M.; Tian, L.; Yang, W.; Le Grice, S.F. Examining the role of the HIV-1 reverse transcriptase p51 subunit in positioning and hydrolysis of RNA/DNA hybrids. J. Biol. Chem. 2013, 288, 16177–16184. [Google Scholar] [CrossRef] [Green Version]
- Betancor, G.; Alvarez, M.; Marcelli, B.; Andrés, C.; Martínez, M.A.; Menéndez-Arias, L. Effects of HIV-1 reverse transcriptase connection subdomain mutations on polypurine tract removal and initiation of (+)-strand DNA synthesis. Nucleic Acids Res. 2015, 43, 2259–2270. [Google Scholar] [CrossRef] [Green Version]
- Capon, D.J.; Ward, R.H.R. The CD4-gpl20 Interaction and Aids Pathogenesis. Annu. Rev. Immunol. 1991, 9, 649–678. [Google Scholar] [CrossRef]
- Hu, W.-S.; Temin, H.M. Genetic consequences of packaging two RNA genomes in one retroviral particle: Pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 1990, 87, 1556–1560. [Google Scholar] [CrossRef] [Green Version]
- Yuste, E.; Sánchez-Palomino, S.; Casado, C.; Domingo, E.; López-Galíndez, C. Drastic fitness loss in human immunodeficiency virus type 1 upon serial bottleneck events. J. Virol. 1999, 73, 2745–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormond, L.; Liu, P.; Matuszewski, S.; Renzette, N.; Bank, C.; Zeldovich, K.; Bolon, D.N.; Kowalik, T.F.; Finberg, R.W.; Jensen, J.D. The combined effect of oseltamivir and favipiravir on influenza A virus evolution. Genome Biol. Evol. 2017, 9, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Litwin, S. Examining The Theory of Error Catastrophe. J. Virol. 2006, 80, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, J.J.; Sanjuán, R.; Wilke, C.O. Theory of Lethal Mutagenesis for Viruses. J. Virol. 2007, 81, 2930–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dapp, M.J.; Clouser, C.L.; Patterson, S.; Mansky, L.M. 5-Azacytidine Can Induce Lethal Mutagenesis in Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 11950–11958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.S.; Brabant, W.; Styrchak, S.; Gall, A.; Daifuku, R. KP-1212/1461, a nucleoside designed for the treatment of HIV by viral mutagenesis. Antivir. Res. 2005, 67, 1–9. [Google Scholar] [CrossRef]
- Loeb, L.A.; Essigmann, J.M.; Kazazi, F.; Zhang, J.; Rose, K.D.; Mullins, J.I. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA 1999, 96, 1492–1497. [Google Scholar] [CrossRef] [Green Version]
- Chiang, R.Z.-H.; Gan, S.K.-E.; Su, C.T.-T. A computational study for rational HIV-1 non-nucleoside reverse transcriptase inhibitor selection and the discovery of novel allosteric pockets for inhibitor design. Biosci. Rep. 2018, 38, BSR20171113. [Google Scholar] [CrossRef] [Green Version]
- Su, C.T.-T.; Ling, W.-L.; Lua, W.-H.; Haw, Y.-X.; Gan, S.K.-E. Structural analyses of 2015-updated drug-resistant mutations in HIV-1 protease: An implication of protease inhibitor cross-resistance. BMC Bioinform. 2016, 17, 500. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.T.; Verma, C.S.; Lane, D.P.; Gan, S.K.E. A comparison and optimization of methods and factors affecting the transformation of Escherichia coli. Biosci. Rep. 2013, 33, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Ling, W.-L.; Lua, W.-H.; Poh, J.-J.; Yeo, J.Y.; Lane, D.P.; Gan, S.K.-E. Effect of VH–VL Families in Pertuzumab and Trastuzumab Recombinant Production, Her2 and FcγIIA Binding. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lua, W.-H.; Ling, W.-L.; Yeo, J.Y.; Poh, J.-J.; Lane, D.P.S.; Gan, S.K.-E. The effects of Antibody Engineering CH and CL in Trastuzumab and Pertuzumab Recombinant Models: Impact on antibody production and antigen-binding. Sci. Rep. 2018, 8, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lua, W.-H.; Su, C.T.-T.; Yeo, J.Y.; Poh, J.-J.; Ling, W.-L.; Phua, S.-X.; Gan, S.K.-E. Role of the IgE variable heavy chain in FcεRIα and superantigen binding in allergy and immunotherapy. J. Allergy Clin. Immunol. 2019, 144, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
- Sim, J.-Z.; Nguyen, P.-V.; Lee, H.-K.; Gan, S.K.-E. Republication—GelApp: Mobile gel electrophoresis analyser. Sci. Phone Apps Mob. Devices 2019, 5, 4. [Google Scholar] [CrossRef]
- Poh, J.J.; Gan, S.K.E. The Determination of Factors involved in Column-Based Nucleic Acid Extraction and Purification. J. Bioprocess. Biotech. 2014, 4. [Google Scholar] [CrossRef]
- Koh, W.-S.D.; Chan, K.-F.; Wu, W.; Gan, S.K.-E. Yet Another Quick Assembly, Analysis and Trimming Tool (YAQAAT): A server for the automated assembly and analysis of Sanger sequencing data. J. Biomol. Tech. in press.
- Sim, J.Z.; Nguyen, P.V.; Zang, Y.; Gan, S.K.E. DNA2App: Mobile sequence analyser. Sci. Phone Apps Mob. Devices 2016, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- LosAlamos. HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/index (accessed on 9 March 2020).
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. Ccp4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bradley, P.; Greisen, P.; Liu, Y.; Mulligan, V.K.; Kim, D.E.; Baker, D.; DiMaio, F. Simultaneous Optimization of Biomolecular Energy Functions on Features from Small Molecules and Macromolecules. J. Chem. Theory Comput. 2016, 12, 6201–6212. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 Gene | No. of Clones | Nucleotide Length | Total No. of Bases | Excluding ‘A to-G’ Hypermutations | Including ‘A to G’ Hypermutations | ||
|---|---|---|---|---|---|---|---|
| No. of Mutations | Mutation Rate (Mutations/bp) | No. of Mutations | Mutation Rate (Mutations/bp) | ||||
| Gag | 801 | 1485 | 1,189,485 | 40 | 3.36 × 10−5 | 56 | 4.71 × 10−5 |
| protease | 640 | 285 | 182,400 | 11 | 6.03 × 10−5 | - | - |
| RT p66 | 571 | 1680 | 959,280 | 41 | 4.27 × 10−5 | 68 | 7.09 × 10−5 |
| Codon mutated RT p66 | 700 | 1617 | 1,131,900 | 74 | 6.53 × 10−5 | 213 | 1.88 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, J.Y.; Koh, D.W.-S.; Yap, P.; Goh, G.-R.; Gan, S.K.-E. Spontaneous Mutations in HIV-1 Gag, Protease, RT p66 in the First Replication Cycle and How They Appear: Insights from an In Vitro Assay on Mutation Rates and Types. Int. J. Mol. Sci. 2021, 22, 370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010370
Yeo JY, Koh DW-S, Yap P, Goh G-R, Gan SK-E. Spontaneous Mutations in HIV-1 Gag, Protease, RT p66 in the First Replication Cycle and How They Appear: Insights from an In Vitro Assay on Mutation Rates and Types. International Journal of Molecular Sciences. 2021; 22(1):370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010370
Chicago/Turabian StyleYeo, Joshua Yi, Darius Wen-Shuo Koh, Ping Yap, Ghin-Ray Goh, and Samuel Ken-En Gan. 2021. "Spontaneous Mutations in HIV-1 Gag, Protease, RT p66 in the First Replication Cycle and How They Appear: Insights from an In Vitro Assay on Mutation Rates and Types" International Journal of Molecular Sciences 22, no. 1: 370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010370