
Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease



Abstract
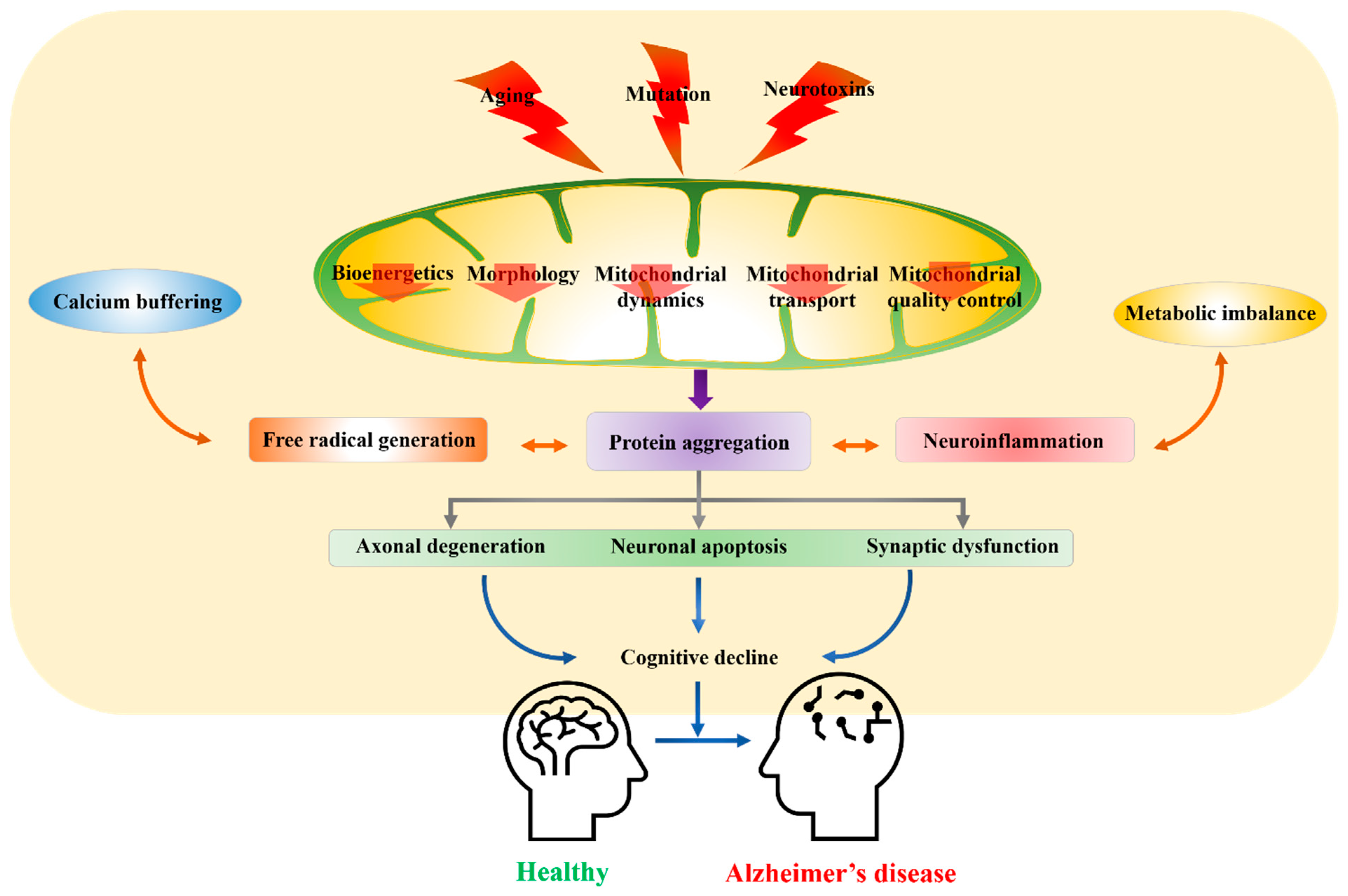
:1. Introduction
2. Factors Debilitating Mitochondrial Function
2.1. Aging
2.2. Genetically Induced Mitochondrial Dysfunction
2.3. Environmental Toxins and Mitochondrial Dysfunction
2.4. Metabolic Syndrome (MetS)-Induced Mitochondrial Dysfunction
3. Current Etiological Hypothesis of AD Involving Mitochondrial Dysfunction
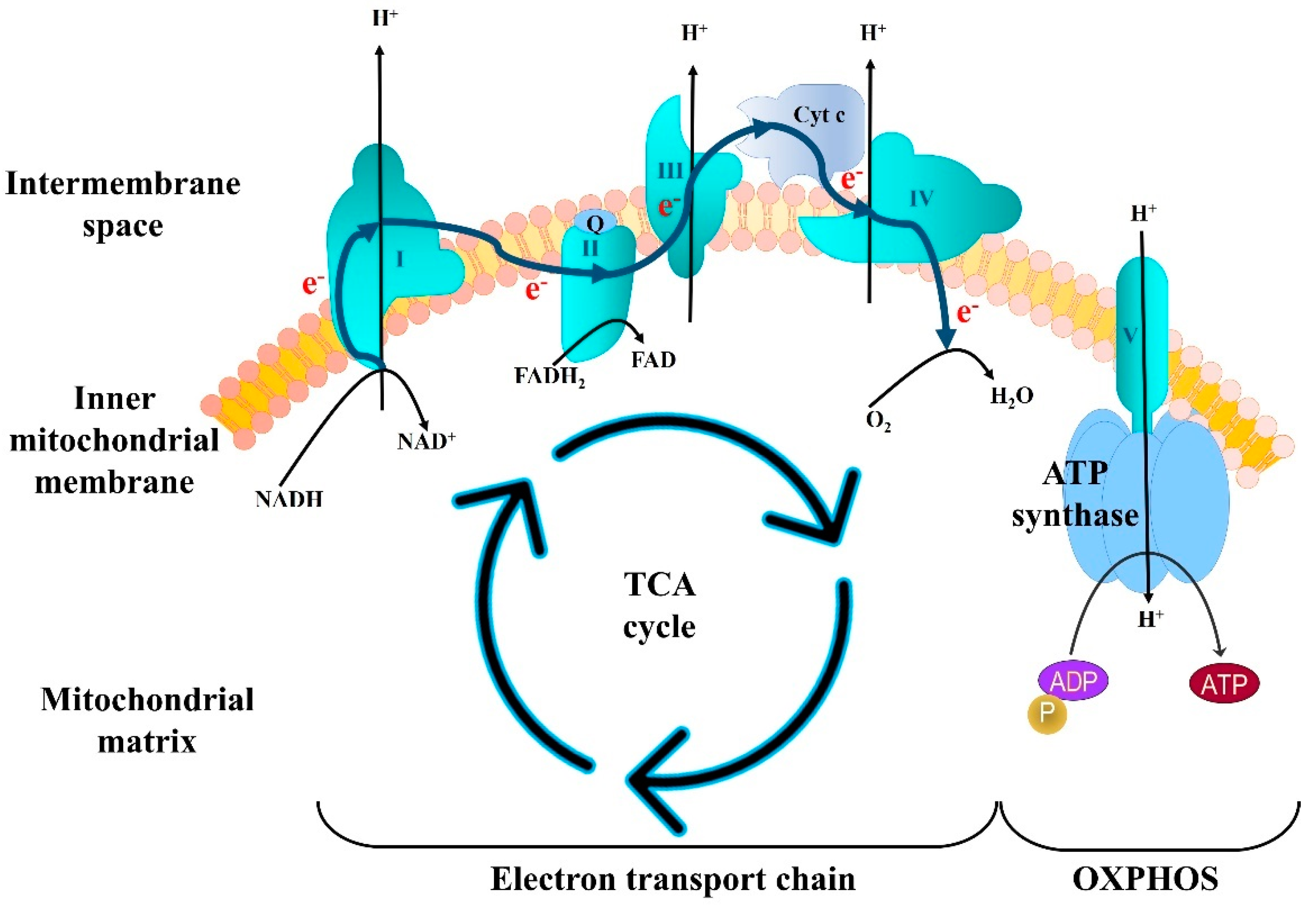
3.1. Deficits in Mitochondrial Bioenergetics
3.2. Deficits in Mitochondrial Dynamics
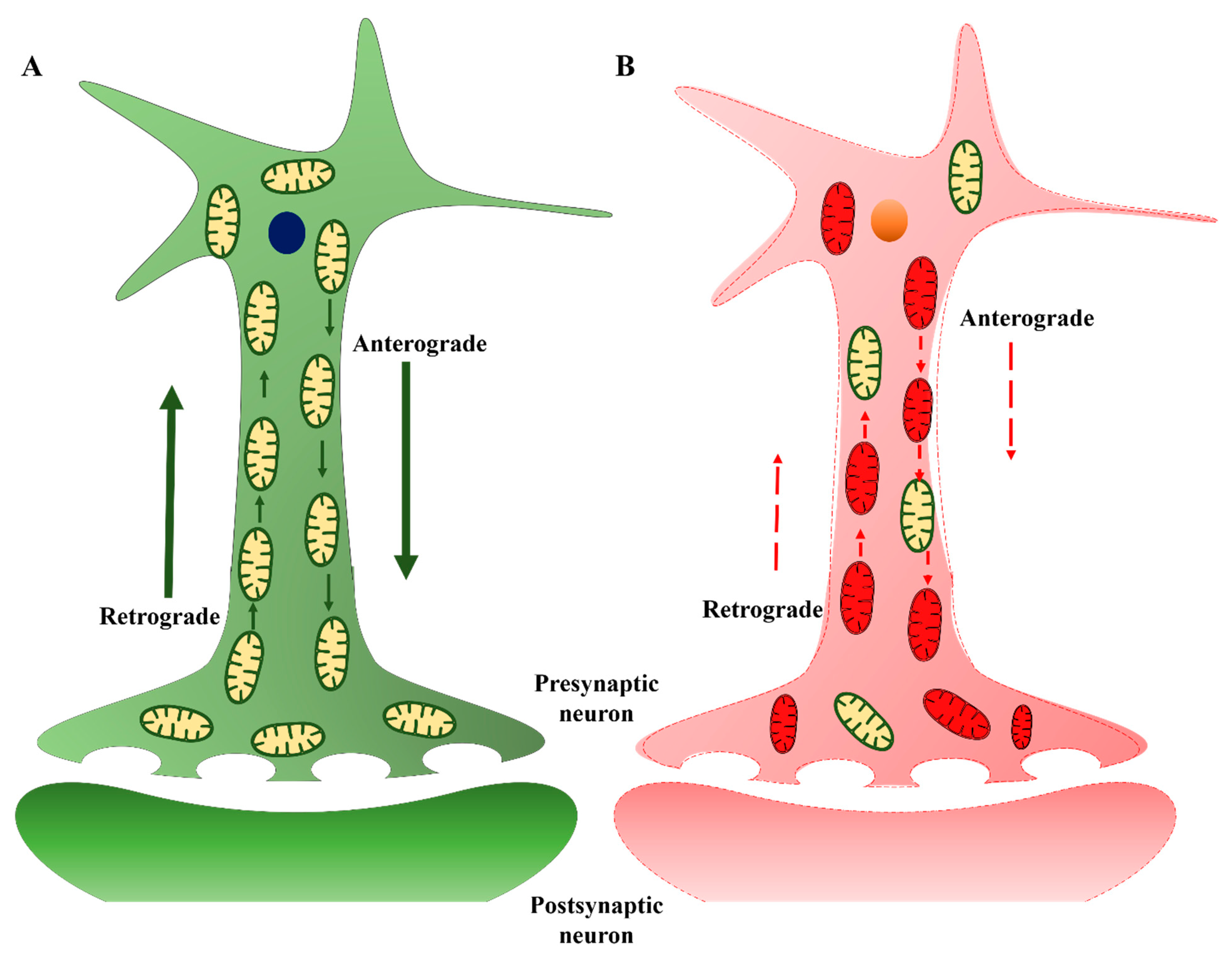
3.3. Mitochondrial Transport
3.4. Mitochondrial Quality Control (mtQC)-Associated Deficits
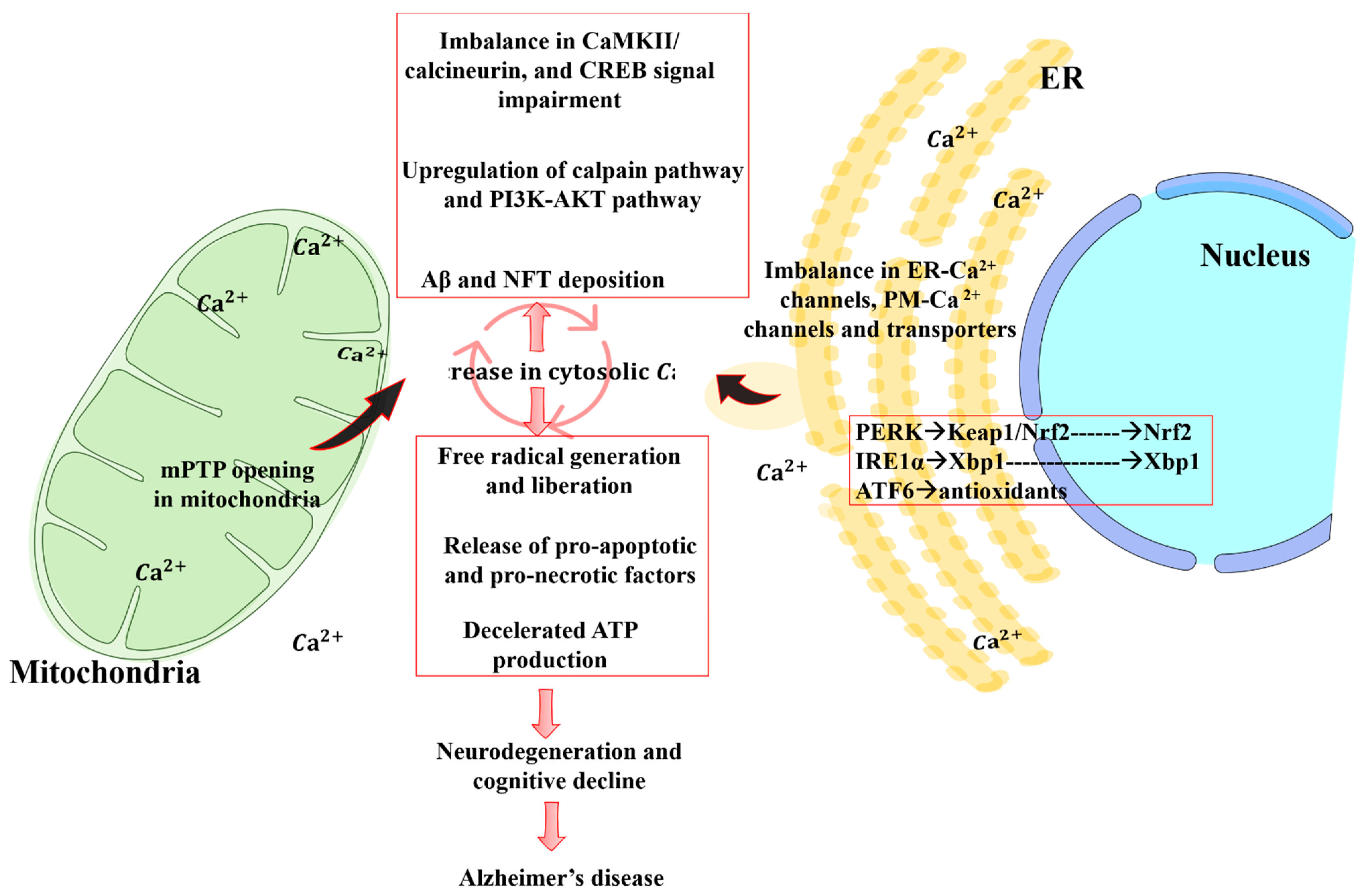
4. Crosstalk between Mitochondrial Dysfunction and Calcium Signaling
5. Free Radical Generation and Mitochondrial Dysfunction
6. Therapeutic Interventions
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C. Age, Neuropathology, and Dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Chételat, G.; Dickson, D.; Fagan, A.M.; Frisoni, G.B.; Jagust, W.; Mormino, E.C.; Petersen, R.C.; Sperling, R.A.; et al. Suspected non-Alzheimer disease pathophysiology-concept and controversy. Nat. Rev. Neurol. 2016, 12, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dourlen, P.; Kilinc, D.; Malmanche, N.; Chapuis, J.; Lambert, J.C. The new genetic landscape of Alzheimer’s disease: From amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019, 138, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Price, J.L.; McKeel, D.W.; Buckles, V.D.; Roe, C.M.; Xiong, C.; Grundman, M.; Hansen, L.A.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W.; et al. Neuropathology of nondemented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 2009, 30, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Chételat, G.; La Joie, R.; Villain, N.; Perrotin, A.; De La Sayette, V.; Eustache, F.; Vandenberghe, R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. Neuroimage Clin. 2013, 2, 356–365. [Google Scholar] [CrossRef]
- Bryan, K.J.; Lee, H.; Perry, G.; Smith, M.A.; Casadesus, G. Transgenic Mouse Models of Alzheimer’s Disease: Behavioral Testing and Considerations; CRC Press: Boca Raton, FL, USA, 2009; ISBN 9781420052343. [Google Scholar]
- Sowade, R.F.; Jahn, T.R. Seed-induced acceleration of amyloid -β Mediated neurotoxicity in vivo. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Onstead, L.; Randle, S.; Price, R.; Smithson, L.; Zwizinski, C.; Dickson, D.W.; Golde, T.; McGowan, E. Aβ40 inhibits amyloid deposition in vivo. J. Neurosci. 2007, 27, 627–633. [Google Scholar] [CrossRef]
- Kim, J.; Chakrabarty, P.; Hanna, A.; March, A.; Dickson, D.W.; Borchelt, D.R.; Golde, T.; Janus, C. Normal cognition in transgenic BRI2-Aβ mice. Mol. Neurodegener. 2013, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Lannfelt, L.; Möller, C.; Basun, H.; Osswald, G.; Sehlin, D.; Satlin, A.; Logovinsky, V.; Gellerfors, P. Perspectives on future Alzheimer therapies: Amyloid-β protofibrils-A new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Lindwall, G.; David Cole, R. The Purification of Tau Protein and the Occurrence of Two Phosphorylation States of Tau in Brain. J. Biol. Chem. 1984, 259, 12241–12245. [Google Scholar] [CrossRef]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, S.; Afonin, A.; Evsyukov, I.; Bondarenko, A. Alzheimer’s disease: As it was in the beginning. Rev. Neurosci. 2017, 28, 825–843. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Albensi, B.C. Dysfunction of mitochondria: Implications for Alzheimer’s disease. In International Review of Neurobiology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 145, pp. 13–27. ISBN 9780128172247. [Google Scholar] [CrossRef]
- Chakravorty, A.; Teresa Jetto, C.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Int. Rev. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, M.; Faas, G.C.; Saggau, P.; Craigen, W.J.; Sweatt, J.D. Mitochondrial regulation of synaptic plasticity in the hippocampus. J. Biol. Chem. 2003. [Google Scholar] [CrossRef] [Green Version]
- Werth, J.L.; Thayer, S.A. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 1994. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, A.; Zarb, C.; Caruana, M.; Ostermeier, U.; Ghio, S.; Högen, T.; Schmidt, F.; Giese, A.; Vassallo, N. Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2532–2543. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Mancuso, M.; Calsolaro, V.; Orsucci, D.; Carlesi, C.; Choub, A.; Piazza, S.; Siciliano, G. Mitochondria, cognitive impairment, and Alzheimer’s disease. Res. Int. J. Alzheimer’s Dis. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys Acta 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abyadeh, M.; Gupta, V.; Chitranshi, N.; Gupta, V.; Wu, Y.; Saks, D.; Wander Wall, R.; Fitzhenry, M.J.; Basavarajappa, D.; You, Y.; et al. Mitochondrial Dysfunction in Alzheimer’s Disease—A Proteomics Perspective. Expert Rev. Proteom. 2021. [Google Scholar] [CrossRef]
- Eckert, A.; Pagani, L. Amyloid-beta interaction with mitochondria. Int. J. Alzheimers Dis. 2011, 2011, 925050. [Google Scholar]
- Wang, J.; Chen, G.J. Mitochondria as a therapeutic target in Alzheimer’s disease. Genes Dis. 2016, 3, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. biomedicines Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kühlbrandt, W. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 15301–15306. [Google Scholar] [CrossRef] [Green Version]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010, 47, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Harada, C.N.; Natelson Love, M.C.; Triebel, K.L. Normal cognitive aging. Clin. Geriatr. Med. 2013, 29, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar]
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging Europe PMC Funders Group. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnane, A.W.; Ozawa, T.; Marzuki, S.; Tanaka, M. Mitochondrial DNA Mutations As An Important Contributor to Ageing and Degenerative Diseases. Lancet 1989, 333, 642–645. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Luong, T.; Neely, T.R.; Robinson, D.; Wands, J.R. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab. Investig. 2000, 80, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Imbimbo, B.P.; Lombard, J.; Pomara, N. Pathophysiology of Alzheimer’s disease. Neuroimaging Clin. N. Am. 2005, 15, 727–753. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Pathogenesis of Alzheimer’s disease. Clin. Interv. Aging 2007, 2, 347–359. [Google Scholar]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar]
- Cantuti-Castelvetri, I.; Lin, M.T.; Zheng, K.; Keller-McGandy, C.E.; Betensky, R.A.; Johns, D.R.; Beal, M.F.; Standaert, D.G.; Simon, D.K. Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol. Aging 2005, 26, 1343–1355. [Google Scholar] [CrossRef]
- Finsterer, J. Cognitive decline as a manifestation of mitochondrial disorders (mitochondrial dementia). J. Neurol. Sci. 2008, 272, 20–33. [Google Scholar] [CrossRef]
- Tanaka, D.; Nakada, K.; Takao, K.; Ogasawara, E.; Kasahara, A.; Sato, A.; Yonekawa, H.; Miyakawa, T.; Hayashi, J.I. Normal mitochondrial respiratory function is essential for spatial remote memory in mice. Mol. Brain 2008, 1. [Google Scholar] [CrossRef] [Green Version]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Wang, J.; Xiong, S.; Xie, C.; Markesbery, W.R.; Lovell, M.A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 2005, 93, 953–962. [Google Scholar] [CrossRef]
- Inczedy-Farkas, G.; Trampush, J.W.; Perczel Forintos, D.; Beech, D.; Andrejkovics, M.; Varga, Z.; Remenyi, V.; Bereznai, B.; Gal, A.; Molnar, M.J. Mitochondrial DNA mutations and cognition: A case-series report. Arch. Clin. Neuropsychol. 2014, 29, 315–321. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leung, M.C.K.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Karri, V.; Schuhmacher, M.; Kumar, V. Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Environ. Toxicol. Pharmacol. 2016, 48, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Miah, M.R.; Aschner, M. Metals and Neurodegeneration. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.Y.; Lee, Y.J.; Lee, S.Y.; Lee, Y.M.; Lee, H.H.; Choi, I.S.; Oh, K.W.; Han, S.B.; Nam, S.Y.; Hong, J.T. Attenuation of scopolamine-induced cognitive dysfunction by obovatol. Arch. Pharm. Res. 2012, 35, 1279–1286. [Google Scholar] [CrossRef]
- El-Khadragy, M.; Al-Olayan, E.; Moneim, A. Neuroprotective Effects of Citrus reticulata in Scopolamine-Induced Dementia Oxidative Stress in Rats. Cns Neurol. Disord. Drug Targets 2014, 13, 684–690. [Google Scholar] [CrossRef] [Green Version]
- Wong-Guerra, M.; Jiménez-Martin, J.; Pardo-Andreu, G.L.; Fonseca-Fonseca, L.A.; Souza, D.O.; de Assis, A.M.; Ramirez-Sanchez, J.; del Valle, R.M.S.; Nuñez-Figueredo, Y. Mitochondrial involvement in memory impairment induced by scopolamine in rats. Neurol. Res. 2017, 39, 649–659. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Santos, M.S.; Casadesus, G.; Lamanna, J.C.; Perry, G.; Smith, M.A.; Moreira, P.I. Mitochondrial Abnormalities in a Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2013, 10, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Pahuja, M.; Gupta, Y.K. Streptozotocin-induced sporadic Alzheimer’s Disease: Selection of appropriate dose. J. Alzheimer’s Dis. 2013, 33, 17–21. [Google Scholar] [CrossRef]
- Paidi, R.; Nthenge-Ngumbau, D.; Singh, R.; Kankanala, T.; Mehta, H.; Mohanakumar, K. Mitochondrial Deficits Accompany Cognitive Decline Following Single Bilateral Intracerebroventricular Streptozotocin. Curr. Alzheimer Res. 2015, 12, 785–795. [Google Scholar] [CrossRef]
- Lee, E.B.; Mattson, M.P. The neuropathology of obesity: Insights from human disease. Acta Neuropathol. 2014, 127, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Jha, N.K.; Kumar, D.; Ambasta, R.K.; Kumar, P. Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in Neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Williams, J.; Smith, F.; Bhatti, J.S.; Kumar, S.; Vijayan, M.; Kandimalla, R.; Kuruva, C.S.; Wang, R.; Manczak, M.; et al. MicroRNAs, Aging, Cellular Senescence, and Alzheimer’s Disease. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; Volume 146, pp. 127–171. [Google Scholar] [CrossRef]
- Jayaraman, A.; Pike, C.J. Alzheimer’s disease and type 2 diabetes: Multiple mechanisms contribute to interactions topical collection on pathogenesis of type 2 diabetes and insulin Resistance. Curr. Diab. Rep. 2014, 14. [Google Scholar] [CrossRef] [Green Version]
- Efremov, R.G.; Sazanov, L.A. Structure of the membrane domain of respiratory complex i. Nature 2011, 476, 414–421. [Google Scholar] [CrossRef]
- Tang, J.X.; Thompson, K.; Taylor, R.W.; Oláhová, M. Mitochondrial OXPHOS Biogenesis: Co-Regulation of Protein Synthesis, Import, and Assembly Pathways. Int. J. Mol. Sci. 2020, 21, 3820. [Google Scholar] [CrossRef]
- Fessel, J. A vaccine to prevent initial loss of cognition and eventual Alzheimer’s disease in elderly persons. Alzheimers Demat. (N. Y.) 2021. [Google Scholar] [CrossRef]
- Lindeboom, J.; Weinstein, H. Neuropsychology of cognitive ageing, minimal cognitive impairment, Alzheimer’s disease, and vascular cognitive impairment. Eur. J. Pharmacol. 2004, 490, 83–86. [Google Scholar] [CrossRef]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-Hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2008, 30, 107–120. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Fujii, S. Extracellular ATP modulates synaptic plasticity induced by activation of metabotropic glutamate receptors in the hippocampus. Biomed. Res. 2015, 36, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, C.; Bloch, M.H.; Williams, K. Glutamate abnormalities in obsessive compulsive disorder: Neurobiology, pathophysiology, and treatment. Pharmacol. Ther. 2011, 132, 314–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FiŠar, Z.; Hroudová, J.; Hansíková, H.; Spá|ilová, J.; Lelková, P.; Wenchich, L.; Jirák, R.; Zvová, M.; Zeman, J.; Martásek, P.; et al. Mitochondrial Respiration in the Platelets of Patients with Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 930–941. [Google Scholar] [CrossRef]
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Proteinopathies and OXP HOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H. Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer’s disease. Cns Spectr. 2009, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s diseases. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 507–513. [Google Scholar] [CrossRef]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Keller, J.N.; Begley, J.G. Evidence for synaptic apoptosis. Exp. Neurol. 1998, 153, 35–48. [Google Scholar] [CrossRef]
- Cao, L.L.; Guan, P.P.; Liang, Y.Y.; Huang, X.S.; Wang, P. Calcium ions stimulate the hyperphosphorylation of tau by activating microsomal prostaglandin E synthase 1. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S.I. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; et al. Increased Autophagic Degradation of Mitochondria in Alzheimer Disease. Autophagy 2007, 3, 614–615. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Rugarli, E.I.; Langer, T. Focus Review Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef] [Green Version]
- Voos, W. Mitochondrial protein homeostasis: The cooperative roles of chaperones and proteases. Res. Microbiol. 2009, 160, 718–725. [Google Scholar] [CrossRef]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Chen, Y.; Liu, X.; Wang, S.; Lv, Y.; Wu, D.; Wang, Q.; Luo, M.; Deng, H. Downregulation of HSP60 disrupts mitochondrial proteostasis to promote tumorigenesis and progression in clear cell renal cell carcinoma. Oncotarget 2016, 7, 38822–38834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.P.; Morse, K.M.; Metcalfe, H.M.; Skalska, J.; Andreeff, M.; et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 2012, 119, 3321–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deepa, S.S.; Bhaskaran, S.; Ranjit, R.; Qaisar, R.; Nair, B.C.; Liu, Y.; Walsh, M.E.; Fok, W.C.; Van Remmen, H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic. Biol. Med. 2016, 91, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Sauer, R.T.; Baker, T.A. AAA+ Proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; Rugarli, E.I. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levytskyy, R.M.; Germany, E.M.; Khalimonchuk, O. Mitochondrial Quality Control Proteases in Neuronal Welfare. J. Neuroimmune Pharmacol. 2016, 11, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 2011, 22. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.H. Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Ivannikov, M.V.; Sugimori, M.; Llinás, R.R. Calcium clearance and its energy requirements in cerebellar neurons. Cell Calcium 2010, 47, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IPreceptors, mitochondria, and Ca2+ signaling: Implications for aging. J. Aging Res. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Frazier, H.N.; Maimaiti, S.; Anderson, K.L.; Brewer, L.D.; Gant, J.C.; Porter, N.M.; Thibault, O. Calcium’s role as nuanced modulator of cellular physiology in the brain. Biochem. Biophys. Res. Commun. 2017, 483, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Investig. 2014, 124, 1552–1567. [Google Scholar] [CrossRef] [Green Version]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; De Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Butterfield, D.A. β-amyloid-associated free radical oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Chem. Res. Toxicol. 1997, 10, 495–506. [Google Scholar] [CrossRef] [PubMed]
- De Grey, A.D.N.J. A proposed refinement of the mitochondrial free radical theory of aging. BioEssays 1997, 19, 161–166. [Google Scholar] [CrossRef]
- Dragicevic, N.; Mamcarz, M.; Zhu, Y.; Buzzeo, R.; Tan, J.; Arendash, G.W.; Bradshaw, P.C. Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimer’s Dis. 2010, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Cochemé, H.M.; Kelso, G.F.; James, A.M.; Ross, M.F.; Trnka, J.; Mahendiran, T.; Asin-Cayuela, J.; Blaikie, F.H.; Manas, A.R.B.; Porteous, C.M.; et al. Mitochondrial targeting of quinones: Therapeutic implications. Mitochondrion 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- Mcmanus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant mitoq prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Han, D.; Xu, D.; Li, X.; Sun, L. CREB Protects against Temporal Lobe Epilepsy Associated with Cognitive Impairment by Controlling Oxidative Neuronal Damage. Neurodegener. Dis. 2020, 19, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuro Mol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Echtay, K.S.; Blaikie, F.H.; Asin-Cayuelat, J.; Cochemé, H.M.; Green, K.; Buckingham, J.A.; Taylort, E.R.; Hurrellt, F.; Hughes, G.; et al. Superoxide activates uncoupling proteins by generating carbon-centered radicals and initiating lipid peroxidation: Studies using a mitochondria- targeted spin trap derived from α-phenyl-N-tert-butylnitrone. J. Biol. Chem. 2003, 278, 48534–48545. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Li, M. Mitochondria-targeted antioxidant mitotempo protects mitochondrial function against amyloid beta toxicity in primary cultured mouse neurons. Biochem. Biophys. Res. Commun. 2016, 478, 174–180. [Google Scholar] [CrossRef]
- Senin, U.; Parnetti, L.; Barbagallo-Sangiorgi, G.; Bartorelli, L.; Bocola, V.; Capurso, A.; Cuzzupoli, M.; Denaro, M.; Marigliano, V.; Tammaro, A.E.; et al. Idebenone in senile dementia of Alzheimer type: A multicentre study. Arch. Gerontol. Geriatr. 1992, 15, 249–260. [Google Scholar] [CrossRef]
- Weyer, G.; Babej-Dölle, R.M.; Hadler, D.; Hofmann, S.; Herrmann, W.M. A controlled study of 2 doses of idebenone in the treatment of alzheimer’s disease. Neuropsychobiology 1997, 36, 73–82. [Google Scholar] [CrossRef]
- Gutzmann, H.; Hadler, D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer’s disease: Update on a 2-year double-blind multicentre study. J. Neural Transm. Suppl. 1998, 54, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Santos, M.S.; Oliveira, C. Involvement of oxidative stress on the impairment of energy metabolism induced by Aβ peptides on PC12 cells: Protection by antioxidants. Neurobiol. Dis. 1999, 6, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Nabeshima, T. Protectiye effects of idebenone and α-tocopherol on β-amyloid-(1-42)-induced learning and memory deficits in rats: Implication of oxidative stress in β-amyloid-induced neurotoxicity in vivo. Eur. J. Neurosci. 1999, 11, 83–90. [Google Scholar] [CrossRef]
- Zhao, K.; Luo, G.; Giannelli, S.; Szeto, H.H. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-β toxicity in Alzheimer’s disease neurons. J. Alzheimer’s Dis. 2010, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, H.H.; Birk, A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X.L.; Reddy, A.P. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.; Gallo, G. To mdivi-1 or not to mdivi-1: Is that the question? Dev. Neurobiol. 2017, 77, 1260–1268. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Hemachandra Reddy, P. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 2019, 28, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Kuruva, C.S.; Manczak, M.; Yin, X.; Ogunmokun, G.; Reddy, A.P.; Reddy, P.H. Aqua-soluble DDQ reduces the levels of Drp1 and Aβ and inhibits abnormal interactions between Aβ and Drp1 and protects Alzheimer’s disease neurons from Aβ- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 2017, 26, 3375–3395. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.J.; Kirchhausen, T.; Murthy, V.M. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 17955–17960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.H.; Barylko, B.; Leitz, J.; Liu, X.; Kavalali, E.T. Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J. Neurosci. 2010, 30, 13631376. [Google Scholar] [CrossRef]
- Dessolin, J.; Schuler, M.; Quinart, A.; De Giorgi, F.; Ghosez, L.; Ichas, F. Selective targeting of synthetic antioxidants to mitochondria: Towards a mitochondrial medicine for neurodegenerative diseases? Eur. J. Pharmacol. 2002, 447, 155–161. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somayajulu, M.; McCarthy, S.; Hung, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Role of mitochondria in neuronal cell death induced by oxidative stress; Neuroprotection by Coenzyme Q10. Neurobiol. Dis. 2005, 18, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zou, L.Y.; Cao, C.M.; Yang, E.S. Coenzyme Q10 protects SHSY5Y neuronal cells from beta amyloid toxicity and oxygen-glucose deprivation by inhibiting the opening of the mitochondrial permeability transition pore. BioFactors 2005, 25, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Preformed β-amyloid fibrils are destabilized by coenzyme Q 10 in vitro. Biochem. Biophys. Res. Commun. 2005, 330, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Noh, Y.H.; Kim, K.Y.; Shim, M.S.; Choi, S.H.; Choi, S.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A.; Ju, W.K. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Andres, R.H.; Ducray, A.D.; Schlattner, U.; Wallimann, T.; Widmer, H.R. Functions and effects of creatine in the central nervous system. Brain Res. Bull. 2008, 76, 329–343. [Google Scholar] [CrossRef]
- Kontush, A.; Schekatolina, S. Vitamin E in neurodegenerative disorders: Alzheimer’s disease. N. Y. Acad. Sci. 2004, 1031, 249–262. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Hagl, S.; Heinrich, M.; Kocher, A.; Schiborr, C.; Frank, J.; Eckert, G.P. Curcumin Micelles Improve Mitochondrial Function in a Mouse Model of Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2014, 1, 80–83. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Sun, J.; Han, Y.; Gong, W.; Li, Y.; Feng, Y.; Wang, H.; Yang, M.; Li, Z.; et al. Neuronal mitochondria-targeted delivery of curcumin by biomimetic engineered nanosystems in Alzheimer’s disease mice. Acta Biomater. 2020, 108, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Kumar, P.; Padi, S.S.V.; Naidu, P.S.; Kumar, A. Effect of resveratrol on 3-nitropropionic acid-induced biochemical and behavioural changes: Possible neuroprotective mechanisms. Behav. Pharmacol. 2006, 17, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Wallimann, T.W. Protective effect of the energy precursor creatine against toxicity of glutamate and β-amyloid in rat hippocampal neurons. J. Neurochem. 2000, 74, 1968–1978. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Tan, K.N.; Borges, K. Sulforaphane is anticonvulsant and improves mitochondrial function. J. Neurochem. 2015, 135, 932–942. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, J.G.; Marosi, K.; Halpern, J.; Mattson, M.P. DNP, mitochondrial uncoupling, and neuroprotection: A little dab’ll do ya. Alzheimer’s Dement. Alzheimers Dement 2017, 13, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of alzheimer’s disease. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Cha, M.Y.; Kim, D.; Kim, D.K.; Soh, M.; Shin, K.; Hyeon, T.; Mook-Jung, I. Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimeŕs Disease. ACS Nano 2016, 10, 2860–2870. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Agent | Mode of Action | Mechanistic Pathways and Effects | References |
|---|---|---|---|
| Mitochondrial Targeted Synthetic Analogs/Peptides | |||
| MitoQ | Enhances electron transport chain (ETC) activity | Free radical scavenger, prevents mPTP opening (anti-apoptotic), enhances CREB signaling, and improves mitochondrial health | [123,124,125,126,127,128] |
| MitoVitE | Inhibits lipid peroxidation | Free radical scavenger, prevents apoptosis by inhibiting mPTP opening, cytochrome c release, and caspase-3 activity | [125,129,130] |
| MitoPBN | Inhibits lipid peroxidation | Anti-apoptotic | [131] |
| MitoTEMPO | Inhibits lipid peroxidation | Eliminates mitochondrial superoxide, inhibits lipid peroxidation, and maintains mtDNA fidelity and copy number | [132] |
| Idebenone | Enhances ETC activity | Free radical scavenger, protects mitochondrial complex and ETC | [133,134,135,136,137] |
| SS-31 | Inhibits lipid peroxidation. Activates PGC1α | Free radical scavenger, maintains mitochondrial transport, prevents mitochondrial depolarization by enhancing mitochondrial processing peptidase expression, and prevents apoptosis by inhibiting cytochrome c release | [138,139,140,141] |
| Mdivi-1 | Drp1 inhibitor | Decreases mitochondrial fission, reduces ROS generation, and enhances mitochondrial biogenesis | [142,143] |
| DDQ | Inhibits Aβ and Drp1 binding | Decreases fission, increases fusion, and increases PGC1α, Nrfl, Nrf2, and TFAM | [144] |
| P110 | Drp1 inhibitor | Decreases fission and ROS, enhances MMP, and prevents apoptosis | [145] |
| Dynasore | Drp1 and mTORC1 inhibitor | Inhibits mitochondrial fission and enhances biogenesis and mitophagy | [146,147] |
| TEMPOL | Superoxide dismutase mimetic | Protects against MMP depolarization | [148] |
| EUK-134 | Superoxide dismutase mimetic | Protects against MMP depolarization | [148] |
| Naturally Present | |||
| Co-Q10 | Enhances ETC activity | Mitigates free radicals and enhances mitochondrial biogenesis | [149,150,151,152,153] |
| Creatine | Maintains energy reserve capacity | Mitigates ROS and enhances ATP reserves | [154] |
| Vitamin E | Antioxidant | Free radical scavenger, decreases abnormal protein nitration | [155] |
| Vitamin C | Antioxidant | Free radical scavenger | [156] |
| Glutathione | Targets glutathione to the mitochondrion | Free radical scavenger and anti-apoptotic | [130] |
| Natural Compounds and Their Synthetic Derivatives | |||
| Curcumin | Antioxidant | Mitophagy modulator, | [157,158] |
| Resveratrol | SIRT1 activator | regulates mitochondrial biogenesis, acts as an antioxidant, and anti-inflammatory | [159,160,161] |
| Sulforaphane | Nrf2 activator | Reduces ROS and maintains redox homeostasis, upregulates cytoprotective genes, and reduces inflammation | [162,163] |
| Bezafibrate | PGC1α activator | Increases mitochondrial biogenesis and ATP production | [164] |
| NAD+ precursors | Enhances NAD+ signaling | Enhances mitophagy and increases ROS resistance | [165,166] |
| DNP | Activates CREB and PGClα | Mitigates ROS, stimulates autophagy | [167] |
| Rapamycin | mTOR inhibitor | Enhances mitophagy | [168] |
| N-acetyl-L-cysteine | Precursor of GSH | Free radical scavenger, protects against mPTP opening, anti-apoptotic | [125] |
| Nanoparticles | |||
| Triphenylphosphonium conjugated-Ceria (CeO2) nanoparticles | Reduces superoxides | Protects mitochondrial morphology from oxidative damage | [169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094850
Sharma C, Kim S, Nam Y, Jung UJ, Kim SR. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(9):4850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094850
Chicago/Turabian StyleSharma, Chanchal, Sehwan Kim, Youngpyo Nam, Un Ju Jung, and Sang Ryong Kim. 2021. "Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 9: 4850. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094850