
The Aryl Hydrocarbon Receptor (AHR): A Novel Therapeutic Target for Pulmonary Diseases?


Division of Neonatology, Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(3), 1516; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031516
Submission received: 9 July 2021
/
Revised: 30 December 2021
/
Accepted: 13 January 2022
/
Published: 28 January 2022
(This article belongs to the Collection Feature Papers in Molecular Toxicology)
Abstract
:The aryl hydrocarbon receptor (AHR) is a cytoplasmic transcription factor that is well-known for regulating xenobiotic metabolism. Studies in knockout and transgenic mice indicate that the AHR plays a vital role in the development of liver and regulation of reproductive, cardiovascular, hematopoietic, and immune homeostasis. In this focused review on lung diseases associated with acute injury and alveolar development, we reviewed and summarized the current literature on the mechanistic role(s) and therapeutic potential of the AHR in acute lung injury, chronic obstructive pulmonary disease, and bronchopulmonary dysplasia (BPD). Pre-clinical studies indicate that endogenous AHR activation is necessary to protect neonatal and adult lungs against hyperoxia- and cigarette smoke-induced injury. Our goal is to provide insight into the high translational potential of the AHR in the meaningful management of infants and adults with these lung disorders that lack curative therapies.
1. Introduction
The Ah receptor (AHR) was discovered during studies aimed at understanding the metabolism of carcinogenic polycyclic aromatic hydrocarbons (PAHs), such as benzo[a]pyrene (BP) and 3-methylchlanthrene [1,2]. It was found that the aryl hydrocarbon hydroxylase, which later was found to be cytochrome P450 (CYP)1A1 and 1A2, was regulated by the Ah locus and was formally renamed the AHR [3]. More recently, the AHR has been known to be involved in chemical surveillances [4] and in different homeostatic pathways [5,6].
1.1. Regulation of the Aryl Hydrocarbon Receptor (AHR)
The AHR gene consists of 11 exons and is localized to chromosome 7p15 [7] in humans, and chromosome 12 A3 [8] in mice. In both species, the AHR gene’s promoter contains several transcription activation sites in the GC-rich region that lack TATA and CCAAT boxes [9,10]. The basal expression of the AHR is regulated by the zinc-finger transcription factors, such as specificity protein (Sp) 1 and Sp3, that have consensus binding sites in the GC-rich region of the AHR promoter [9,11]. Additional factors that regulate AHR expression include transforming growth factor (TGF)-β [12], nuclear factor erythroid 2–related factor 2 (NRF2) [13], β-catenin [14], and peroxisome proliferator-activated receptor α (PPAR-α) [15]. Interestingly, these factors regulate the AHR gene in a cell-specific manner. For example, TGF-β activation downregulates the AHR gene at the transcriptional level in human A549 lung carcinoma cells [16], whereas, in human HepG2 hepatocarcinoma cells, TGF-β activation increases AHR promoter activity [17]. In addition to these factors, epigenetic factors regulate the AHR gene expression. Histone deacetylase inhibitors increase, whereas histone acetylase inhibitors decrease, AHR promoter activity, indicating that histone acetylation is an important regulator of AHR expression [18]. Likewise, DNA hypermethylation down-regulates AHR expression [19].
1.2. Structure of the AHR
The human AHR protein has a molecular mass of 96 kDa and is composed of 848 amino acids, whereas the mouse AHR protein contains 805 amino acids and has a molecular mass of 90 kDa [20,21]. The AHR is a ligand-activated cytoplasmic transcription factor that belongs to the basic helix-loop-helix (bHLH) family [22]. The highly conserved b and HLH domains are located at the N-terminal of the AHR protein, where the former facilitates the binding of the transcription factor to DNA, and the latter promotes protein-protein interactions. Additionally, AHR contains two PAS domains, PAS-A and PAS-B, which have a homologous sequence to the protein domains found in the Drosophila genes period (Per) and single-minded (Sim) and the human AHR nuclear translocator (ARNT) [23]. The PAS-B domain contains the ligand-binding site [24]. The AHR protein’s C-terminal region contains the transactivation or Q-rich domain that participates in co-activator recruitment and transcriptional activation [25] (Figure 1).
1.3. The AHR Signaling Pathway
There are two pathways of AHR action: the classical pathway and the non-classical pathway.
1.3.1. Classical (Canonical) Pathway
The AHR is expressed practically in all mouse tissues [26], and, in humans, the receptor is highly expressed in the placenta, lungs, thymus, kidney, and liver [27]. The receptor is particularly enriched in lungs and placenta, tissues that participate in oxygen gas exchange [28]. The non-ligand bound AHR is predominantly cytosolic, localized in a core complex comprising two molecules of 90-kDa heat shock protein (Hsp90), the 23-kDa co-chaperone p23, and a single molecule of hepatitis X-associated protein-2 (XAP2), and the Src kinase [29,30]. The Hsp90 and p23 complex protects the receptor from proteolysis and facilitates ligand binding while preventing AHR from binding to the ARNT [31]. The XAP2 binds to the nuclear localization sequence (NLS) and prevents the translocation of non-ligand bound AHR to the nucleus [32]. Ligand-induced AHR activation results in a conformational change of the cytosolic AHR complex and release of XAP2 that exposes the NLS, resulting in translocation of this complex into the nucleus [33,34,35]. In the nucleus, Hsp90, p23, and Src kinase dissociate from the AHR, exposing the PAS domains, which facilitates AHR to dimerize with the ARNT [36]. The AHR/ARNT heterodimer complex then initiates transcription of many phase I (such as cytochrome P450 (CYP)1A) and phase II genes (anti-oxidant enzymes (AOE), such as glutathione S-transferase-α (GST-α), and NAD(P)H quinone reductase-1 (NQO1)), by binding to the xenobiotic responsive element (XRE)/AHR responsive elements (AHRE) motifs that contain the core bases 5′-GCGTG-3′ in the promoter region of these genes [37,38,39,40]. The AHR signaling is terminated upon the elimination of xenobiotics by at least two independent mechanisms, proteasomal degradation and competitive inhibition. AHR undergoes nuclear export, followed by E3 ubiquitin ligase-mediated ubiquitination and subsequent degradation by the 26S proteasome in the cytoplasm [41,42] (Figure 2). Recent evidence demonstrate that activation of autophagy can also degrade AHR protein via p23-dependent mechanisms [43]. In addition, the AHR signaling is terminated by a negative feedback loop via the AHR repressor (AHRR). Since it is structurally similar to AHR, the AHRR competes with the latter to dimerize with ARNT and bind to the XRE [44]. The XRE-bound AHRR recruits co-repressors, such as histone deacetylases, that repress transcription of the target genes [45].
1.3.2. The Non-Classical (Non-Canonical) Pathway
Cross-talk between the AHR and other signaling mechanisms can result in non-canonical pathways of the action of AHR and its ligands [46]. In the nucleus, the AHR has been shown to associate with the hypophosphorylate form of pRB, resulting in growth arrest at the GL/S phase of the cell cycle [46,47]. Other mechanisms entail involvement of the transcription factor c-Maf [13,48], estrogen receptor, NRF2, RelA, and RelB [46].
1.4. AHR and Phase I/II Enzymes
The AHR’s well-established function is to mediate induction of phase I (CYP1A/1B1) and II enzymes that metabolize xenobiotics [49,50]. The phase I enzymes, such as CYP1A/1B1 monooxygenases and NADPH-CYP reductase, act to introduce reactive and polar groups to their xenobiotic substrates, which, in turn, leads to activation or detoxification of the substrates, leading to toxicity or excretion. These substrate modifications include hydroxylation, epoxidation, oxidation, reduction, hydrolysis, cyclization, and decyclization. In phase II reactions, enzymes, such as NQO1, glucuronyl transferases, and GST, conjugate the activated substrates with glutathione, sulfate, glycine, or glucuronic acid to detoxify the substrates and make them more polar so that they can be actively transported. Together, phase I and II enzymes detoxify toxic compounds and metabolites.
The CYP enzymes belong to a superfamily of hemeproteins that are involved in the metabolism of exogenous and endogenous chemicals [51]. The CYP1A enzymes are of particular interest to oxygen toxicity. The CYP1A subfamily has two isoforms, CYP1A1 and 1A2. CYP1A1 is essentially an extrahepatic enzyme that is predominantly present in rodent and human lungs, intestines, placenta, and kidneys. On the other hand, CYP1A2 is expressed mainly in the rodent and human liver and is not, or minimally, expressed in extrahepatic tissues.
In addition, phase II enzymes, such as NQO1 and GST, have been shown to protect cells and tissues against oxidant injury induced by various toxic chemicals [52,53,54] and oxygen [55,56,57]. The protective mechanisms of these enzymes have been attributed to their ability to conjugate and excrete the reactive electrophiles and lipid peroxidation products generated by an oxidant injury [52,56].
1.5. Physiological Roles of the AHR
The AHR is of particular interest to toxicologists, and extensive research has been conducted on its role in the bioactivation of polycyclic and aromatic hydrocarbons, leading to carcinogenesis [58]. Transgenic and knockout mice with AHR deficiencies have provided insight into the potential role(s) that AHR might play in normal physiological homeostasis [59,60]. The very fact that AHR is evolutionarily conserved from invertebrates who lack xenobiotic metabolism suggests that the role of AHR extends beyond xenobiotic metabolism. In fact, the AHR homolog spineless (ss) in Drosophila is necessary for the development of its legs, and distal segments of antennae [61] and AHR deficiency in Caenorhabditis elegans lead to defects in neuronal development [62]. Moreover, studies in knockout and transgenic mice indicate that AHR plays a vital role in the development of liver [63,64] and regulation of reproductive [65], cardiovascular [66,67], renal [68], hematopoietic [69], immune [70], and microbial [71] homeostasis. Additionally, AHR is known to regulate genes involved in proliferation, apoptosis, cell growth and differentiation, and cellular stress response [72].
2. AHR Ligands
Several structurally diverse compounds activate AHR. There are two types of AHR ligands, those coming from exogenous sources, such as diesel exhaust, commercial production, or industrial contamination (e.g., PAHs, PCBs, TCDD, etc.), or diet, or those generated endogenously (e.g., FICZ, indolo-carbazoles, indigoids, etc.) (Table 1).
2.1. Exogenous Ligands
The prototypical exogenous ligand is TCDD [73]. The majority of high affinity ligands are planar, hydrophobic halogenated hydrocarbons (HAHs) (e.g., TCDD, PCBs, dibenzofurans, biphenyls), and PAHs, such as MC, BP, benzanthracenes, benzoflavones, etc.) [73]. The most potent ligands are the ones that are most metabolically stable (e.g., HAHs), with binding affinities in the pM to nM range. The mechanisms of toxicity of HAHs involve the AHR, but PAHs in part mediate their action by inducing CYP1A1, which, in turn, bioactivates the PAHs to DNA-reactive metabolites, resulting in cancers of the lung and other extra-hepatic organs [73].
2.2. Endogenous Ligands
The majority of these compounds are proligands, which are transformed into ligands before they can bind and activate AHR [28]. The tryptophan derivative FICZ is one of the most potent AHR ligands and inducers of CYP1A1 [28].
Several developmental deficits and physiological impairments in AHR-deficient mice indicate the presence of several endogenous AHR ligands, including phytochemicals, microbial bioproducts, and metabolites of indole, tryptophan, heme, and arachidonic acid [99,100]. Additionally, several nonclassical synthetic compounds, such as omeprazole (OM), lansoprazole, thiabendazole, and primaquine, can activate AHR-dependent gene expression indirectly. Although these compounds are not AHR ligands by themselves, they are thought to activate AHR-dependent gene expression, either via metabolic conversion into a ligand or by their ability to affect a cellular pathway that results in AHR activation [101,102,103,104,105]. The prototypical ligands, such as TCDD and MC, are unsuitable for clinical use because of their well-known toxicities. Hence, identifying novel non-toxic AHR ligands, such as OM, is important for developing the AHR as a clinically relevant therapeutic target in oxidant injury- and inflammation-mediated lung disorders. OM, a benzimidazole derivative, is a proton pump inhibitor that inhibits gastric acid secretion both in humans [106] and in animals [107]. It has been widely used in the management of gastric acid disorders in humans [106]. Several in vitro studies suggest that OM activates AHR in human and rat hepatocytes [108,109,110,111], and the mechanistic role of AHR in the induction of CYP1A enzymes by OM in vitro has been extensively studied [112,113,114]. Furthermore, OM activates AHR and attenuates hyperoxic injury in adult mice in vivo [96] and adult human lung H441 cells in vitro [112], which indicates that OM can be used as an AHR agonist to understand AHR biology in hyperoxia-mediated lung disorders. Importantly, these ligands can exert different molecular and cellular responses within the same cell, tissue, or species [115]. The mechanisms of these ligand-specific effects are unclear at this time.
2.3. Selective AHR Modulators
A number of studies have recently showed AHR to be ligands that are selective modulators (sAHRMs) [115]. In addition to binding of 2,3,7,8-teyrachlorodibenzo-p-dioxin (TCDD) and PAHs, the AHR plays an important role in maintaining cellular homeostasis and in pathophysiology of many human diseases, and studies are emerging that the AHR is an important drug target [116]. The AHR binds structurally diverse chemicals, such as pharmaceuticals, phytochemicals, and many endogenous ligands. Thus, the AHR ligands are sAHRMs that display organ, tissue and cell-specific AHR agonist activities, and their functional diversity is very similar to steroid hormone and other nuclear receptors [116].
2.4. Current Barriers/Limitations to Developing AHR Ligands as Therapeutic Agents
The clinical applications of drugs using the AHR as a target have been lacking mainly due to the fact that the AHR was initially identified as the receptor that mediated the toxicity of (TCDD) and other polychlorinated aromatic environmental contaminants [117,118]. However, the discovery of many endogenous ligands, phytochemicals, and therapeutic compounds that activate the AHR suggest that AHR also plays a key role in myriad signaling pathways that regulate the normal physiology of the organism [85,117,119]. In fact, an AHR active drug, e.g., laquinimod, has been in clinical trials for treating multiple sclerosis [120].
3. Roles of the AHR in Lung Inflammation and Oxidative Stress
The recent discovery of the AHR as a crucial regulator of lung immune homeostasis suggests that AHR plays an important role in the modulation of lung inflammation. However, AHR biology in inflammatory lung disease is complex and is context- and disease-dependent. For example, depending on the nature of AHR ligands, the experimental conditions, and the disease model, AHR activation may potentiate or attenuate the lung inflammation [46,121]. Deficient AHR signaling has been reported to affect immune and non-immune cells, such as neutrophils, macrophages, and fibroblasts in the lung, leading to increased lung inflammation upon exposure to tobacco smoke, lipopolysaccharide, and hyperoxia [122,123,124]. Conversely, AHR activation has been shown to decrease airway inflammation in rodent models of asthma by regulating the production and secretion of Th2 cytokines, such as interleukin (IL)-4, IL-5, and IL-15 [125,126]. Interestingly, Wong et al. reported that AHR activation by TCCD increased the expression of the inflammatory cytokines, IL-1β, and monocyte chemoattractant protein-1 (MCP-1), in the mouse lungs, which they attributed to the increased lung infiltration of neutrophils and macrophages [126]. However, we observed that AHR activation by omeprazole (OM) decreased lung inflammation in an adult mouse model of acute hyperoxic lung injury, wherein both the neutrophils infiltration and MCP-1 expression were decreased compared to vehicle-treated animals [96]. These contrasting findings further emphasize the complexity of AHR biology in lung diseases, wherein the outcome is both ligand and context-dependent.
NRF2 is a master regulator of the antioxidant response, but it is also known to regulate the AHR expression transcriptionally. Moreover, the XRE and antioxidant responsive elements (ARE) are adjacent to each other in the promoter region of the genes encoding the antioxidant enzymes, such as NQO1 and GST [127]. Hence, AHR and NRF2 regulate and share a subset of common target genes with antioxidant properties, suggesting that AHR may be an essential regulator of the redox status of the cell. Along those lines, our studies in adult mice and adult human lung cells indicate that AHR deficiency increases, whereas AHR activation by OM decreases, oxidative stress in the lungs [96,112,128]. Additionally, AHR deficiency has shown to increase cardiac ROS levels via the pro-oxidant enzyme, NAD(P)H oxidase [129]. These observations strongly indicate that AHR signaling may be beneficial in inflammation- and oxidant injury-mediated lung disorders.
4. Lung Disorders and AHR
4.1. Acute Lung Injury
Acute respiratory distress syndrome (ARDS) is a life-threatening lung disease that is characterized by acute lung injury (ALI), respiratory failure, bilateral opacities on chest imaging, and a PaO2/FiO2 ratio < 300 mm Hg on at least a positive end-expiratory pressure (PEEP) of 5 or a PaO2/FiO2 ratio < 315 mm Hg without any PEEP requirement [130,131]. Despite improved intensive care management, the treatment of patients with ARDS is mostly supportive, with associated mortality as high as 46% [131]. The recent pandemic due to SARS-CoV-2 infection has, until today, seen numerous deaths (over 511,000) globally, and respiratory illnesses, such as pneumonia and ARDS [132], are the major causes of death. Thus, there is an urgent need for improved therapies for ARDS patients. Oxidative stress from increased reactive oxygen species (ROS) generation is a major contributor to ARDS development [133,134]. Supplemental oxygen, that is traditionally used as a life-saving measure in patients with impaired lung function, in itself, increases ROS generation and exacerbates lung injury [135,136,137]. Hyperoxia-induced acute lung injury in adult mice leads to a phenotype similar to human ARDS [138,139]. ALI is a multi-factorial morbid and fatal lung disorder in humans.
The AHR is expressed in numerous lung cells, including macrophages, club cells, alveolar type II cells, and endothelial cells [140,141,142,143,144,145,146], and plays a significant role in modulating lung function, especially in the context of environmental exposures-induced lung injury. In models of hyperoxic lung injury in adult animals, AHR deficiency potentiates hyperoxia-induced lung inflammation and damage [124,129], whereas AHR activation [124] mitigates these effects of hyperoxia. The molecular mechanisms by which the pulmonary AHR protects against hyperoxic lung injury remains poorly defined; however, CYP1A family of enzymes mediate some of the beneficial effects of the AHR in the context of hyperoxic injury. Hyperoxia for 48 h induces CYP1A1/1A2 in the liver and CYP1A1 in the lung of adult rodents. Interestingly, the induction of CYP1A enzymes in liver and lung decline after continuation of hyperoxia for 60 h [147,148], the time period that coincides with expression of overt respiratory distress in these animals, suggesting that CYP1A induction may protect against hyperoxic lung injury in adult rodents. The protection against hyperoxic lung injury of adult rodents pretreated with beta-naphthoflavone (BNF) [81] or 3-methylcholanthrene (3-MC) [149] has been attributed to the aryl hydrocarbon receptor (AHR)-mediated induction of CYP1A1, an enzyme with high peroxidase activity. It has also been shown that the CYP1A inhibitor 1-aminobenzotriazole potentiates hyperoxic lung injury in rats [124]. Studies have consistently demonstrated that CYP1A enzymes mitigate hyperoxic injury. Genetic or pharmacologic inhibition of CYP1A enzymes potentiates [124,150,151], whereas activation of these enzymes prevents and abrogate [81] hyperoxic injury. Mechanistic studies demonstrate that CYP1A enzymes protect against hyperoxic lung injury by decreasing lipid peroxidation and oxidative DNA damage [151,152]. On the other hand, CYP1B1, which is also regulated by the AHR, appears to play a pro-oxidant role in hyperoxic lung injury, as mice deficient in CYP1B1 are less susceptible to hyperoxic lung injury [152]. Recently, AHR activation was also shown to mitigate lipopolysaccharide (LPS)-mediated acute lung injury in mice by upregulating the immunomodulatory gene, TNF-stimulated gene 6 (TSG-6) [153] (Figure 3).
4.2. Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD), a chronic adult lung disease that affects 300 million people worldwide, includes diseases such as chronic bronchitis and emphysema, and it is predicted by the World Health Organization to be the third most common cause of global deaths, by the end of 2030 [86,154]. The morbidity associated with the disease, including physician visits and hospitalizations, increases with age and is influenced by other comorbid diseases [155,156]. Further, COPD increases the economic and social burden and is predicted to be the seventh leading cause of disability-adjusted life years lost worldwide in 2030 [157]. COPD is a progressive lung disease that is characterized by mucociliary dysfunction and lung inflammation, fibrosis, destruction, and dysfunction and persistent airflow limitation [158]. Cigarette smoking is the most common risk factor for COPD [159]. Occupational exposures to organic and inorganic dusts, chemical agents and fumes, and indoor pollution from biomass cooking and heating are other important risk factors for COPD [154,160]. Additionally, genetics, lung developmental anomalies, and socioeconomic factors play important roles in the development and progression of COPD [159]. All the above mentioned risk factors ultimately cause oxidative stress, inflammation, and aberrant proliferation, death, and senescence of lung cells, leading to parenchymal tissue destruction and the development of COPD [161,162].
The AHR exerts ligand-specific effects on the lungs and can either potentiate or attenuate COPD. For instance, the dioxins and PAHs in tobacco smoke and particulate matter mediate their toxic effects on the lungs through AHR signaling. These xenobiotic ligands induce inflammation, upregulate expression of mucin 5AC and matrix metalloproteinases, and damage ciliated cells, Club cells, and alveolar macrophages, contributing to the pathogenesis of COPD [75,126,163,164,165]. By inflammation and oxidative stress, the major contributors to the COPD pathogenesis [166]. Cigarette smoke (CS) exposure is a major risk factor for the development of COPD [167,168] and is a commonly used insult in animal models to elucidate the molecular mechanism of COPD [169]. Both acute and chronic CS exposure elicits an augmented neutrophilic response in the lungs of AHR-deficient mice than AHR-sufficient mice [122]. The precise molecular mechanisms through which endogenous AHR mediates these effects are unclear, but studies strongly indicate that the NF-κB protein RelB may be partly responsible. The AHR interacts with and modulates the expression of RelB [170,171], which is essential for maintaining immune homeostasis. AHR deficiency potentiates CS-induced RelB degradation, which, in turn, leads to: (1) increased expression of the neutrophil chemokine, intercellular adhesion molecule 1, and neutrophilia [123]; and (2) increased levels of the pro-inflammatory enzyme cyclooxygenase-2 via human antigen R-dependent pathway [75,121,122]. Further, AHR also regulates oxidative stress, the other common risk factor for COPD. AHR-deficient lung cells exhibit more increased reactive oxygen species (ROS) generation and decreased expression of the anti-oxidant enzymes, NQO1, and sulfiredoxin than AHR-sufficient cells, upon exposure to CS [172], suggesting that CS-induced oxidative stress is potentiated in AHR-deficient lungs. These findings collectively indicate that endogenous AHR ligands may protect the lungs against inflammatory and oxidant injuries and provide a mechanistic rationale for developing select AHR agonists as therapeutic agents to prevent and mitigate COPD (Figure 4).
4.3. Bronchopulmonary Dysplasia
Bronchopulmonary dysplasia (BPD) is a chronic lung disease of predominantly preterm infants that is characterized histopathologically by alveolar and pulmonary vascular hypoplasia [173,174,175]. The incidence of BPD remains unchanged despite significant advancement in the medical care of extremely low birth weight infants with respiratory dysfunction [176]. The therapies in the early phases of respiratory dysfunction in premature infants are mostly supportive, and there are no specific interventions known to prevent BPD directly. Furthermore, infants with BPD are more likely to have long-term pulmonary problems, increased re-hospitalizations during the first year of life, and neurodevelopment impairments [177,178,179,180,181,182,183,184]. In addition, BPD increases the economic burden with an estimated cost of BPD infants being twice that of non-BPD infants [185], making it the second most expensive childhood disease after asthma. Inflammatory stimuli, such as infection, hyperoxia, and mechanical ventilation, disrupt growth factor signaling, extracellular matrix assembly, and cell proliferation in the developing lungs and contribute to BPD pathogenesis [186,187,188,189]. Failure to understand the specific molecular mechanisms that contribute to the development of BPD is one of the main reasons for the lack of specific therapies to prevent BPD and its associated economic burden and long-term sequelae.
AHR signaling plays an important role in BPD pathogenesis [190,191]. In humans, placenta expresses the greatest levels of AHR followed by lungs and liver, whereas, in mice, the lungs express the highest levels of AHR followed by the placenta [28]. In human fetal lungs, the AHR is strongly expressed in the epithelial cells of the bronchus, bronchiole, and alveoli, and it is weakly expressed in the endothelial and smooth muscle cells of blood vessels [192]. Evidence indicates that AHR is expressed in the airway and parenchyma of the developing rodent lungs [97,144]; however, the lung cell-specific expression of AHR in rodents is not well characterized. Exposure to chronic hyperoxia activates AHR, as evidenced by increased expression of AHR-regulated phase I and II enzymes, such as CYP1A1 and NQO1, in wild-type (WT) mice but not in AHR dysfunctional (AHRd) mice. Interestingly, the failure of AHR activation in AHRd mice is associated with increased hyperoxia-induced lung inflammation and alveolar simplification. This implies that endogenous AHR signaling protects newborn mice against chronic hyperoxia-induced developmental lung injury [193]. By contrast, AHR activation protects neonatal rodents against hyperoxic lung injury. The AHR agonists, quercetin and β-napthoflavone, up-regulate the anti-oxidant enzymes, reduce oxidative adducts, decrease inflammation, and mitigate hyperoxia-induced neonatal lung injury in mice [82,83,194]. However, the AHR agonist, omeprazole, has differential effects on neonatal hyperoxic lung injury. While omeprazole activates AHR and mitigates hyperoxic lung injury in adult animals [96], prolonged (2-week) omeprazole therapy decreases pulmonary AHR activation and potentiates hyperoxia-induced: alveolar and pulmonary vascular simplification; inflammation; vascular injury; and oxidative stress [97]. In contrast, omeprazole activates AHR, increases surfactant and angiogenic proteins, and improves lung development and function in preterm rabbits exposed to hyperoxia [98]. Differences in the animal species, omeprazole dosage, and the nature and duration of the insult maybe some of the causes for these variable results. Nevertheless, these findings are consistent with the notion that an endogenous AHR response is protective in the context of neonatal hyperoxic lung injury. AHR activation can also potentiate neonatal lung injury in rodents. Maternal exposure to the environmental pollutant, BP, potentiates hyperoxia-induced alveolar hypoplasia in the offspring [195]. Mechanistic studies suggest that BP mediates hyperoxic injury by modulating the CYP1A/1B1 enzymes, leading to increased inflammation and oxidative lipid and DNA damage in the lungs [195]. Collectively, the findings indicate that AHR exerts ligand-specific effects on the developing lungs (Figure 5).
The AHR deficiency also potentiates hyperoxic injury in primary fetal human pulmonary microvascular endothelial cells (HPMECs), the cells which promote alveolarization and facilitate lung development. Silencing AHR signaling in primary fetal HPMEC increases hyperoxia-induced cytotoxicity, ROS generation, and inflammation and decreases the expression of antioxidant enzymes [146]. Interestingly, AHR-deficiency decreases the activation of the alternative NF-κB pathway (RelB) that mediates anti-inflammatory effects in these cells [146]. These results suggest that AHR signaling is also necessary to protect human fetal lung endothelial cells against hyperoxic injury. Gene expression profiling of AHR-sufficient and -deficient HPMEC exposed to hyperoxia indicate that AHR deficiency downregulates genes that mediate organ development and cell proliferation, and it upregulates genes that increase inflammation [145]. These results have important implications for managing BPD, a developmental lung disorder of preterm infants characterized by increased inflammation and interrupted alveolar development.
4.4. AHR Antagonists
Because the AHR is involved in the causation of the above mentioned lung diseases, one approach is to develop drugs and chemicals that target the AHR signaling pathway. The most well-known AHR antagonists are 3′methoxy-4′-nitroflavone (MNF) [94] and resveratrol [95], Recently, AHR activation has been shown to upregulate the expression of mucin SAC (oligomeric mucus/gel-forming (MUC5AC)) in the airway epithelial cell line via formation of ROS [196], which, in turn, contributes to lung diseases, such as COPD [197]. Chiba et al. [196] have shown that the AHR antagonist resveratrol mitigates the production of mucin. Wang et al. [74] have reported that the PAH BP increases dermiaogaphagoides group I (Der f1)-induced allergic lung inflammation via the AHR, and this effect is mitigated by the AHR antagonist CH223191. This AHR antagonist has also been shown to reverse the development of experimental pulmonary hypertension induced by Sugen 5146 in rats [198]. Development of AHR antagonists for human therapeutics is also being considered in the fields of wound healing and cancer [199].
5. Conclusions
The AHR is a versatile transcription factor that is evolutionarily conserved, serving many important physiological and pathological roles beyond its traditionally recognized role in xenobiotic metabolism. Importantly, activation of the AHR can exert opposing effects within the same cell or organ, depending upon the activating ligand and the nature of the insult. In general, endogenous AHR signaling is necessary to protect against both acute lung disease and chronic lung disorders, such as COPD and BPD. Furthermore, while the typical xenobiotic AHR ligands, such as TCDD and BP, can contribute to the development of lung diseases, the atypical AHR ligand, omeprazole, and the natural xenobiotic AHR ligands, quercetin and β-napthoflavone, can protect the lungs against oxidative damage. Despite decades of research, there are several knowledge gaps in the field of AHR biology. One of the most intriguing gaps is the mechanism behind the cell- and tissue-specific effects of the AHR ligands. The biological actions of the same AHR ligand can differ between tissues. There is also a lack of sufficient knowledge of the non-canonical pathways through which the AHR exerts its beneficial or harmful effects. Finally, the role of the negative feedback loop of the AHR pathway, e.g., AHRR, in the pathobiology is unclear. Deciphering these knowledge gaps would advance AHR biology and lay the foundation for selecting and developing the most effective AHR ligands as novel therapies for lung disorders, including ALI, COPD, and BPD.
Author Contributions
B.S. and B.M. participated in literature search, critical review of published literature, and writing and revising the manuscript. C.C. participated in reading the literature, preparing illustrations, and editing and revising the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was in part supported by NIH grants R01HL139594 to BS, and R01HL129794, R01ES029382, P42ES027725, and Cancer Prevention Research Institute of Texas (CPRIT) RP190279 to BM.
Conflicts of Interest
The authors report no conflicts of interest, financial or otherwise in this work.
Abbreviations
| AHR | aryl hydrocarbon receptor |
| AHRE | AHR responsive elements |
| AHRR | AHR repressor |
| ALI | acute lung injury |
| AOE | anti-oxidant enzyme |
| ARDS | acute respiratory distress syndrome |
| ARE | antioxidant responsive elements |
| ARNT | AHR nuclear translocator |
| bHLH | basic helix-loop-helix |
| BNF | beta-naphthoflavone |
| BP | benzo[α]pyrene |
| BPD | bronchopulmonary dysplasia |
| COPD | chronic obstructive pulmonary disease |
| CYP | cytochrome P450 |
| GST-α | glutathione S-transferase-α |
| HPMEC | human pulmonary microvascular endothelial cells |
| Hsp | heat shock protein |
| MC | methylcholanthrene |
| MCP | monocyte chemoattractant protein |
| NLS | nuclear localization sequence |
| NQO1 | NAD(P)H quinone reductase-1 |
| NRF2 | nuclear factor erythroid 2–related factor 2 |
| OM | omeprazole |
| PPARα | peroxisome proliferator-activated receptor α |
| ROS | reactive oxygen species |
| TCDD | 2,3,7,8-Tetrachlorodibenzo-p-dioxin |
| TGF | transforming growth factor |
| XAP2 | hepatitis X-associated protein-2 |
| XRE | xenobiotic responsive element |
References
- Conney, A.H.; Davison, C.; Gastel, R.; Burns, J.J. Adaptive increases in drug-metabolizing enzymes induced by phenobarbital and other drugs. J. Pharmacol. Exp. Ther. 1960, 130, 1–8. [Google Scholar] [PubMed]
- Nebert, D.W.; Gelboin, H.V. The in vivo and in vitro induction of aryl hydrocarbon hydroxylase in mammalian cells of different species, tissues, strains, and developmental and hormonal states. Arch. Biochem. Biophys. 1969, 134, 76–89. [Google Scholar] [CrossRef]
- Nebert, D.W.; Negishi, M.; Lang, M.A.; Hjelmeland, L.M.; Eisen, H.J. The Ah locus, a multigene family necessary for survival in a chemically adverse environment: Comparison with the immune system. Adv. Genet. 1982, 21, 1–52. [Google Scholar] [PubMed]
- Lai, A.; Baumgartner, J.; Schauer, J.J.; Rudich, Y.; Pardo, M. Cytotoxicity and chemical composition of women’s personal PM2.5 exposures from rural China. Environ. Sci. Atmos. 2021, 1, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Barouki, R.; Coumoul, X. Aryl hydrocarbon receptor and its diverse ligands and functions: An exposome receptor. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 383–404. [Google Scholar] [CrossRef] [PubMed]
- Sahebnasagh, A.; Hashemi, J.; Khoshi, A.; Saghafi, F.; Avan, R.; Faramarzi, F.; Azimi, S.; Habtemariam, S.; Sureda, A.; Khayatkashani, M.; et al. Aromatic hydrocarbon receptors in mitochondrial biogenesis and function. Mitochondrion 2021, 61, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Micka, J.; Milatovich, A.; Menon, A.; Grabowski, G.A.; Puga, A.; Nebert, D.W. Human Ah receptor (AHR) gene: Localization to 7p15 and suggestive correlation of polymorphism with CYP1A1 inducibility. Pharmacogenetics 1997, 7, 95–101. [Google Scholar] [CrossRef]
- Poland, A.; Glover, E.; Taylor, B.A. The murine Ah locus: A new allele and mapping to chromosome 12. Mol. Pharmacol. 1987, 32, 471–478. [Google Scholar]
- Eguchi, H.; Hayashi, S.; Watanabe, J.; Gotoh, O.; Kawajiri, K. Molecular cloning of the human AH receptor gene promoter. Biochem. Biophys. Res. Commun. 1994, 203, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Garrison, P.M.; Denison, M.S. Analysis of the murine AhR gene promoter. J. Biochem. Mol. Toxicol. 2000, 14, 1–10. [Google Scholar] [CrossRef]
- Fitzgerald, C.T.; Nebert, D.W.; Puga, A. Regulation of mouse Ah receptor (Ahr) gene basal expression by members of the Sp family of transcription factors. DNA Cell Biol. 1998, 17, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Dohr, O.; Abel, J. Transforming growth factor-beta1 coregulates mRNA expression of aryl hydrocarbon receptor and cell-cycle-regulating genes in human cancer cell lines. Biochem. Biophys. Res. Commun. 1997, 241, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 modulates aryl hydrocarbon receptor signaling: Influence on adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [Green Version]
- Chesire, D.R.; Dunn, T.A.; Ewing, C.M.; Luo, J.; Isaacs, W.B. Identification of aryl hydrocarbon receptor as a putative Wnt/beta-catenin pathway target gene in prostate cancer cells. Cancer Res. 2004, 64, 2523–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villard, P.H.; Caverni, S.; Baanannou, A.; Khalil, A.; Martin, P.G.; Penel, C.; Pineau, T.; Seree, E.; Barra, Y. PPARalpha transcriptionally induces AhR expression in Caco-2, but represses AhR pro-inflammatory effects. Biochem. Biophys. Res. Commun. 2007, 364, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Dohr, O.; Sinning, R.; Vogel, C.; Munzel, P.; Abel, J. Effect of transforming growth factor-beta1 on expression of aryl hydrocarbon receptor and genes of Ah gene battery: Clues for independent down-regulation in A549 cells. Mol. Pharmacol. 1997, 51, 703–710. [Google Scholar] [CrossRef]
- Wolff, S.; Harper, P.A.; Wong, J.M.; Mostert, V.; Wang, Y.; Abel, J. Cell-specific regulation of human aryl hydrocarbon receptor expression by transforming growth factor-beta(1). Mol. Pharmacol. 2001, 59, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Garrison, P.M.; Rogers, J.M.; Brackney, W.R.; Denison, M.S. Effects of histone deacetylase inhibitors on the Ah receptor gene promoter. Arch. Biochem. Biophys. 2000, 374, 161–171. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Carvajal-Gonzalez, J.M.; Herranz, M.; Ballestar, E.; Fraga, M.F.; Ropero, S.; Esteller, M.; Fernandez-Salguero, P.M. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis 2006, 27, 1099–1104. [Google Scholar] [CrossRef]
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [Green Version]
- Dolwick, K.M.; Schmidt, J.V.; Carver, L.A.; Swanson, H.I.; Bradfield, C.A. Cloning and expression of a human Ah receptor cDNA. Mol. Pharmacol. 1993, 44, 911–917. [Google Scholar] [PubMed]
- Hankinson, O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Sogawa, K.; Watanabe, N.; Chujoh, Y.; Matsushita, N.; Gotoh, O.; Funae, Y.; Fujii-Kuriyama, Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 1992, 184, 246–253. [Google Scholar] [CrossRef]
- Coumailleau, P.; Poellinger, L.; Gustafsson, J.A.; Whitelaw, M.L. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J. Biol. Chem. 1995, 270, 25291–25300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.B.; Ramadoss, P.; Reen, R.K.; Vanden Heuvel, J.P.; Perdew, G.H. The Q-rich subdomain of the human Ah receptor transactivation domain is required for dioxin-mediated transcriptional activity. J. Biol. Chem. 2001, 276, 42302–42310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, B.D.; Birnbaum, L.S.; Perdew, G.H. Developmental expression of two members of a new class of transcription factors: I. Expression of aryl hydrocarbon receptor in the C57BL/6N mouse embryo. Dev. Dyn. 1995, 204, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Tirona, R.G.; Kim, R.B. Nuclear receptors and drug disposition gene regulation. J. Pharm. Sci. 2005, 94, 1169–1186. [Google Scholar] [CrossRef]
- Avilla, M.N.; Malecki, K.M.C.; Hahn, M.E.; Wilson, R.H.; Bradfield, C.A. The Ah Receptor: Adaptive Metabolism, Ligand Diversity, and the Xenokine Model. Chem. Res. Toxicol. 2020, 33, 860–879. [Google Scholar] [CrossRef]
- Denis, M.; Cuthill, S.; Wikstrom, A.C.; Poellinger, L.; Gustafsson, J.A. Association of the dioxin receptor with the Mr 90,000 heat shock protein: A structural kinship with the glucocorticoid receptor. Biochem. Biophys. Res. Commun. 1988, 155, 801–807. [Google Scholar] [CrossRef]
- Carver, L.A.; Bradfield, C.A. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J. Biol. Chem. 1997, 272, 11452–11456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskas, A.; Poellinger, L.; Pongratz, I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (Aryl hydrocarbon) receptor. J. Biol. Chem. 1999, 274, 13519–13524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrulis, J.R.; Kusnadi, A.; Ramadoss, P.; Hollingshead, B.; Perdew, G.H. The hsp90 Co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J. Biol. Chem. 2003, 278, 2677–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollenz, R.S.; Sattler, C.A.; Poland, A. The aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localizations in Hepa 1c1c7 cells by immunofluorescence microscopy. Mol. Pharmacol. 1994, 45, 428–438. [Google Scholar] [PubMed]
- Hord, N.G.; Perdew, G.H. Physicochemical and immunocytochemical analysis of the aryl hydrocarbon receptor nuclear translocator: Characterization of two monoclonal antibodies to the aryl hydrocarbon receptor nuclear translocator. Mol. Pharmacol. 1994, 46, 618–626. [Google Scholar] [PubMed]
- Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probst, M.R.; Reisz-Porszasz, S.; Agbunag, R.V.; Ong, M.S.; Hankinson, O. Role of the aryl hydrocarbon receptor nuclear translocator protein in aryl hydrocarbon (dioxin) receptor action. Mol. Pharmacol. 1993, 44, 511–518. [Google Scholar] [PubMed]
- Emi, Y.; Ikushiro, S.; Iyanagi, T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J. Biol. Chem. 1996, 271, 3952–3958. [Google Scholar] [CrossRef] [Green Version]
- Favreau, L.V.; Pickett, C.B. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J. Biol. Chem. 1991, 266, 4556–4561. [Google Scholar] [CrossRef]
- Fujisawa-Sehara, A.; Sogawa, K.; Yamane, M.; Fujii-Kuriyama, Y. Characterization of xenobiotic responsive elements upstream from the drug-metabolizing cytochrome P-450c gene: A similarity to glucocorticoid regulatory elements. Nucleic Acids Res. 1987, 15, 4179–4191. [Google Scholar] [CrossRef] [Green Version]
- Rushmore, T.H.; King, R.G.; Paulson, K.E.; Pickett, C.B. Regulation of glutathione S-transferase Ya subunit gene expression: Identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc. Natl. Acad. Sci. USA 1990, 87, 3826–3830. [Google Scholar] [CrossRef] [Green Version]
- Pollenz, R.S.; Santostefano, M.J.; Klett, E.; Richardson, V.M.; Necela, B.; Birnbaum, L.S. Female Sprague-Dawley rats exposed to a single oral dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin exhibit sustained depletion of aryl hydrocarbon receptor protein in liver, spleen, thymus, and lung. Toxicol. Sci. Off. J. Soc. Toxicol. 1998, 42, 117–128. [Google Scholar]
- Davarinos, N.A.; Pollenz, R.S. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J. Biol. Chem. 1999, 274, 28708–28715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Chan, W.K. Selective Autophagy Maintains the Aryl Hydrocarbon Receptor Levels in HeLa Cells: A Mechanism That Is Dependent on the p23 Co-Chaperone. Int. J. Mol. Sci. 2020, 21, 3449. [Google Scholar] [CrossRef]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haarmann-Stemmann, T.; Bothe, H.; Kohli, A.; Sydlik, U.; Abel, J.; Fritsche, E. Analysis of the transcriptional regulation and molecular function of the aryl hydrocarbon receptor repressor in human cell lines. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 2262–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, C.; Rannug, A.; Stockinger, B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009, 30, 447–454. [Google Scholar] [CrossRef]
- Levine-Fridman, A.; Chen, L.; Elferink, C.J. Cytochrome P4501A1 promotes G1 phase cell cycle progression by controlling aryl hydrocarbon receptor activity. Mol. Pharmacol. 2004, 65, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Schrenk, D. Impact of dioxin-type induction of drug-metabolizing enzymes on the metabolism of endo- and xenobiotics. Biochem. Pharmacol. 1998, 55, 1155–1162. [Google Scholar]
- Rowlands, J.C.; Gustafsson, J.A. Aryl hydrocarbon receptor-mediated signal transduction. Crit. Rev. Toxicol. 1997, 27, 109–134. [Google Scholar] [CrossRef]
- Guengerich, F.P. Enzymatic oxidation of xenobiotic chemicals. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 97–153. [Google Scholar] [CrossRef]
- Johansson, K.; Jarvliden, J.; Gogvadze, V.; Morgenstern, R. Multiple roles of microsomal glutathione transferase 1 in cellular protection: A mechanistic study. Free Radic. Biol. Med. 2010, 49, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J. Molecular mechanisms of quinone cytotoxicity. Chem. Biol. Interact. 1991, 80, 1–41. [Google Scholar] [CrossRef]
- Rahman, Q.; Abidi, P.; Afaq, F.; Schiffmann, D.; Mossman, B.T.; Kamp, D.W.; Athar, M. Glutathione redox system in oxidative lung injury. Crit. Rev. Toxicol. 1999, 29, 543–568. [Google Scholar] [CrossRef] [PubMed]
- McGrath-Morrow, S.; Lauer, T.; Yee, M.; Neptune, E.; Podowski, M.; Thimmulappa, R.K.; O’Reilly, M.; Biswal, S. Nrf2 increases survival and attenuates alveolar growth inhibition in neonatal mice exposed to hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L565–L573. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef]
- Das, A.; Kole, L.; Wang, L.; Barrios, R.; Moorthy, B.; Jaiswal, A.K. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic. Biol. Med. 2006, 40, 1843–1856. [Google Scholar] [CrossRef]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef] [Green Version]
- Bock, K.W.; Kohle, C. The mammalian aryl hydrocarbon (Ah) receptor: From mediator of dioxin toxicity toward physiological functions in skin and liver. Biol. Chem. 2009, 390, 1225–1235. [Google Scholar] [CrossRef]
- Fujii-Kuriyama, Y.; Kawajiri, K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Duncan, D.M.; Burgess, E.A.; Duncan, I. Control of distal antennal identity and tarsal development in Drosophila by spineless-aristapedia, a homolog of the mammalian dioxin receptor. Genes Dev. 1998, 12, 1290–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Powell-Coffman, J.A. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev. Biol. 2004, 270, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Abbott, B.D.; Schmid, J.E.; Pitt, J.A.; Buckalew, A.R.; Wood, C.R.; Held, G.A.; Diliberto, J.J. Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol. Appl. Pharmacol. 1999, 155, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, A.; Atallah-Yunes, N.; Smith, F.C.; You, X.; Chase, S.E.; Silverstone, A.E.; Vikstrom, K.L. A role for the aryl hydrocarbon receptor in cardiac physiology and function as demonstrated by AhR knockout mice. Cardiovasc. Toxicol. 2003, 3, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.K.; Goens, M.B.; Kanagy, N.L.; Walker, M.K. Cardiac hypertrophy in aryl hydrocarbon receptor null mice is correlated with elevated angiotensin II, endothelin-1, and mean arterial blood pressure. Toxicol. Appl. Pharmacol. 2003, 193, 177–187. [Google Scholar] [CrossRef]
- Harrill, J.A.; Hukkanen, R.R.; Lawson, M.; Martin, G.; Gilger, B.; Soldatow, V.; Lecluyse, E.L.; Budinsky, R.A.; Rowlands, J.C.; Thomas, R.S. Knockout of the aryl hydrocarbon receptor results in distinct hepatic and renal phenotypes in rats and mice. Toxicol. Appl. Pharmacol. 2013, 272, 503–518. [Google Scholar] [CrossRef]
- Gasiewicz, T.A.; Singh, K.P.; Casado, F.L. The aryl hydrocarbon receptor has an important role in the regulation of hematopoiesis: Implications for benzene-induced hematopoietic toxicity. Chem.-Biol. Interact. 2010, 184, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef]
- Bock, K.W. Aryl hydrocarbon receptor (AHR) functions: Balancing opposing processes including inflammatory reactions. Biochem. Pharmacol. 2020, 178, 114093. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Roe, A.L.; Dieter, M.Z.; Solis, W.A.; Yang, Y.; Dalton, T.P. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem. Pharmacol. 2000, 59, 65–85. [Google Scholar] [CrossRef]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Liu, X.; Tu, W.; Do, D.C.; Yu, H.; Yang, L.; Zhou, Y.; Xu, D.; Huang, S.K.; Yang, P.; et al. Benzo(a)pyrene facilitates dermatophagoides group 1 (Der f 1)-induced epithelial cytokine release through aryl hydrocarbon receptor in asthma. Allergy 2019, 74, 1675–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, J.A.; Zago, M.; Nair, P.; Li, P.Z.; Bourbeau, J.; Tan, W.C.; Hamid, Q.; Eidelman, D.H.; Benedetti, A.L.; Baglole, C.J. Decreased expression of the NF-κB family member RelB in lung fibroblasts from Smokers with and without COPD potentiates cigarette smoke-induced COX-2 expression. Respir. Res. 2015, 16, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baginski, T.K.; Dabbagh, K.; Satjawatcharaphong, C.; Swinney, D.C. Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. Am. J. Respir. Cell Mol. Biol. 2006, 35, 165–174. [Google Scholar] [CrossRef]
- Yang, L.; Wang, W.C.; Lung, S.C.; Sun, Z.; Chen, C.; Chen, J.K.; Zou, Q.; Lin, Y.H.; Lin, C.H. Polycyclic aromatic hydrocarbons are associated with increased risk of chronic obstructive pulmonary disease during haze events in China. Sci. Total Environ. 2017, 574, 1649–1658. [Google Scholar] [CrossRef]
- Islam, J.; Shree, A.; Afzal, S.M.; Vafa, A.; Sultana, S. Protective effect of Diosmin against benzo(a)pyrene-induced lung injury in Swiss Albino Mice. Environ. Toxicol. 2020, 35, 747–757. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alrumaihi, F.; Alsahli, M.A.; Alhommrani, M.F.; Khan, A.; Rahmani, A.H. Curcumin, an active constituent of turmeric spice: Implication in the prevention of lung injury induced by benzo(a)pyrene (BaP) in rats. Molecules 2020, 25, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolterbeek, A.P.; Schoevers, E.J.; Rutten, A.A.; Feron, V.J. A critical appraisal of intratracheal instillation of benzo[a]pyrene to Syrian golden hamsters as a model in respiratory tract carcinogenesis. Cancer Lett. 1995, 89, 107–116. [Google Scholar] [CrossRef]
- Sinha, A.; Muthiah, K.; Jiang, W.; Couroucli, X.; Barrios, R.; Moorthy, B. Attenuation of hyperoxic lung injury by the CYP1A inducer beta-naphthoflavone. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 87, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Couroucli, X.I.; Liang, Y.H.; Jiang, W.; Wang, L.; Barrios, R.; Yang, P.; Moorthy, B. Prenatal administration of the cytochrome P4501A inducer, Β-naphthoflavone (BNF), attenuates hyperoxic lung injury in newborn mice: Implications for bronchopulmonary dysplasia (BPD) in premature infants. Toxicol. Appl. Pharmacol. 2011, 256, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K.; Maturu, P.; Liang, Y.W.; Jiang, W.; Wang, L.; Moorthy, B.; Couroucli, X.I. β-Naphthoflavone treatment attenuates neonatal hyperoxic lung injury in wild type and Cyp1a2-knockout mice. Toxicol. Appl. Pharmacol. 2018, 339, 133–142. [Google Scholar] [CrossRef]
- Maturu, P.; Wei-Liang, Y.; Androutsopoulos, V.P.; Jiang, W.; Wang, L.; Tsatsakis, A.M.; Couroucli, X.I. Quercetin attenuates the hyperoxic lung injury in neonatal mice: Implications for Bronchopulmonary dysplasia (BPD). Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 114, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Passi, M.; Shahid, S.; Chockalingam, S.; Sundar, I.K.; Packirisamy, G. Conventional and Nanotechnology Based Approaches to Combat Chronic Obstructive Pulmonary Disease: Implications for Chronic Airway Diseases. Int. J. Nanomed. 2020, 15, 3803–3826. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Suda, T.; Furuhashi, K.; Suzuki, M.; Fujie, M.; Hahimoto, D.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer 2010, 67, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Tang, X.; Cui, Y.; Xiong, X.; Song, J.; Wang, C.; Zhang, Y. 6-Formylindolo[3,2-b]carbazole alleviates lipopolysaccharide-induced acute lung injury via suppressing endoplasmic reticulum stress. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2021, 33, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Weems, J.M.; Cutler, N.S.; Moore, C.; Nichols, W.K.; Martin, D.; Makin, E.; Lamb, J.G.; Yost, G.S. 3-Methylindole is mutagenic and a possible pulmonary carcinogen. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 112, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Li, X.; Hu, F.; Li, Y.; Yang, Y.; Yan, J.; Kuang, C.; Yang, Q. Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in Lewis lung cancer (LLC) tumor-bearing mice. J. Med. Chem. 2013, 56, 8321–8331. [Google Scholar] [CrossRef] [PubMed]
- Dera, A.A.; Rajagopalan, P.; Al Fayi, M.; Ahmed, I.; Chandramoorthy, H.C. Indirubin-3-monoxime and thymoquinone exhibit synergistic efficacy as therapeutic combination in in-vitro and in-vivo models of Lung cancer. Arch. Pharmacal Res. 2020, 43, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Li, H.; Li, S. Indirubin improves antioxidant and anti-inflammatory functions in lipopolysaccharide-challenged mice. Oncotarget 2017, 8, 36658–36663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, G.A.; Murray, I.A.; Krishnegowda, G.; Amin, S.; Perdew, G.H. Perdew GH. Selective aryl hydrocarbon receptor modulator-mediated repression of CD55 expression induced by cytokine exposure. J. Pharmacol. Exp. Ther. 2012, 342, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dertinger, S.D.; Lantum, H.B.; Silverstone, A.E.; Gasiewicz, T.A. Effect of 3′-methoxy-4′-nitroflavone on benzo[a]pyrene toxicity. Aryl hydrocarbon receptor-dependent and -independent mechanisms. Biochem. Pharmacol. 2000, 60, 189–196. [Google Scholar] [CrossRef]
- Revel, A.; Raanani, H.; Younglai, E.; Xu, J.; Rogers, I.; Han, R.; Savouret, J.F.; Casper, R.F. Resveratrol, a natural aryl hydrocarbon receptor antagonist, protects lung from DNA damage and apoptosis caused by benzo[a]pyrene. J. Appl. Toxicol. JAT 2003, 23, 255–261. [Google Scholar] [CrossRef]
- Shivanna, B.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic lung injury in mice via aryl hydrocarbon receptor activation and is associated with increased expression of cytochrome P4501A enzymes. J. Pharmacol. Exp. Ther. 2011, 339, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, B.; Zhang, S.; Patel, A.; Jiang, W.; Wang, L.; Welty, S.E.; Moorthy, B. Omeprazole Attenuates Pulmonary Aryl hydrocarbon Receptor Activation and Potentiates Hyperoxia-Induced Developmental Lung Injury in Newborn Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 148, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.; Jimenez, J.; Nagatomo, T.; Toelen, J.; Brady, P.; Salaets, T.; Lesage, F.; Vanoirbeek, J.; Deprest, J. Proton-pump inhibitor omeprazole attenuates hyperoxia induced lung injury. J. Transl. Med. 2016, 14, 247. [Google Scholar] [CrossRef] [Green Version]
- Furue, M.; Hashimoto-Hachiya, A.; Tsuji, G. Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2019, 20, 5424. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Daujat, M.; Peryt, B.; Lesca, P.; Fourtanier, G.; Domergue, J.; Maurel, P. Omeprazole, an inducer of human CYP1A1 and 1A2, is not a ligand for the Ah receptor. Biochem. Biophys. Res. Commun. 1992, 188, 820–825. [Google Scholar] [CrossRef]
- Lesca, P.; Peryt, B.; Larrieu, G.; Alvinerie, M.; Galtier, P.; Daujat, M.; Maurel, P.; Hoogenboom, L. Evidence for the ligand-independent activation of the AH receptor. Biochem. Biophys. Res. Commun. 1995, 209, 474–482. [Google Scholar] [CrossRef]
- Daujat, M.; Charrasse, S.; Fabre, I.; Lesca, P.; Jounaidi, Y.; Larroque, C.; Poellinger, L.; Maurel, P. Induction of CYP1A1 gene by benzimidazole derivatives during Caco-2 cell differentiation. Evidence for an aryl-hydrocarbon receptor-mediated mechanism. Eur. J. Biochem. FEBS 1996, 237, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Aix, L.; Rey-Grobellet, X.; Larrieu, G.; Lesca, P.; Galtier, P. Thiabendazole is an inducer of cytochrome P4501A1 in cultured rabbit hepatocytes. Biochem. Biophys. Res. Commun. 1994, 202, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, F.; de Sousa, G.; Duchene, P.; Rahmani, R. Cytochrome P450 Induction and Cytotoxic effects of Antimalarials in Rat Hepatocytes. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 1998, 12, 545–549. [Google Scholar] [CrossRef]
- Li, X.Q.; Andersson, T.B.; Ahlstrom, M.; Weidolf, L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 821–827. [Google Scholar] [CrossRef]
- Larsson, H.; Carlsson, E.; Ryberg, B.; Fryklund, J.; Wallmark, B. Rat parietal cell function after prolonged inhibition of gastric acid secretion. Am. J. Physiol. 1988, 254, G33-9. [Google Scholar] [CrossRef]
- Quattrochi, L.C.; Tukey, R.H. Nuclear uptake of the Ah (dioxin) receptor in response to omeprazole: Transcriptional activation of the human CYP1A1 gene. Mol. Pharm. 1993, 43, 504–508. [Google Scholar]
- Yoshinari, K.; Ueda, R.; Kusano, K.; Yoshimura, T.; Nagata, K.; Yamazoe, Y. Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem. Pharm. 2008, 76, 139–145. [Google Scholar] [CrossRef]
- Backlund, M.; Ingelman-Sundberg, M. Regulation of aryl hydrocarbon receptor signal transduction by protein tyrosine kinases. Cell. Signal. 2005, 17, 39–48. [Google Scholar] [CrossRef]
- Murray, I.A.; Perdew, G.H. Omeprazole stimulates the induction of human insulin-like growth factor binding protein-1 through aryl hydrocarbon receptor activation. J. Pharmacol. Exp. Ther. 2008, 324, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivanna, B.; Chu, C.; Welty, S.E.; Jiang, W.; Wang, L.; Couroucli, X.I.; Moorthy, B. Omeprazole attenuates hyperoxic injury in H441 cells via the aryl hydrocarbon receptor. Free Radic. Biol. Med. 2011, 51, 1910–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiizaki, K.; Ohsako, S.; Kawanishi, M.; Yagi, T. Identification of amino acid residues in the ligand-binding domain of the aryl hydrocarbon receptor causing the species-specific response to omeprazole: Possible determinants for binding putative endogenous ligands. Mol. Pharmacol. 2014, 85, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Backlund, M.; Johansson, I.; Mkrtchian, S.; Ingelman-Sundberg, M. Signal transduction-mediated activation of the aryl hydrocarbon receptor in rat hepatoma H4IIE cells. J. Biol. Chem. 1997, 272, 31755–31763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safe, S.; Jin, U.H.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl hydrocarbon receptor (AHR) ligands as selective AHR modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 124, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors - Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Haggiag, S.; Ruggieri, S.; Gasperini, C. Efficacy and safety of laquinimod in multiple sclerosis: Current status. Ther. Adv. Neurol. Disord. 2013, 6, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Baglole, C.J.; Maggirwar, S.B.; Gasiewicz, T.A.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J. Biol. Chem. 2008, 283, 28944–28957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatcher, T.H.; Maggirwar, S.B.; Baglole, C.J.; Lakatos, H.F.; Gasiewicz, T.A.; Phipps, R.P.; Sime, P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am. J. Pathol. 2007, 170, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luebke, R.W.; Copeland, C.B.; Daniels, M.; Lambert, A.L.; Gilmour, M.I. Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol. Sci. Off. J. Soc. Toxicol. 2001, 62, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, B.; Parker, K.M.; Smith, C.V.; Bend, J.R.; Welty, S.E. Potentiation of oxygen-induced lung injury in rats by the mechanism-based cytochrome P-450 inhibitor, 1-aminobenzotriazole. J. Pharmacol. Exp. Ther. 2000, 292, 553–560. [Google Scholar] [PubMed]
- Moon, D.O.; Kim, M.O.; Lee, H.J.; Choi, Y.H.; Park, Y.M.; Heo, M.S.; Kim, G.Y. Curcumin attenuates ovalbumin-induced airway inflammation by regulating nitric oxide. Biochem. Biophys. Res. Commun. 2008, 375, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.S.; Vogel, C.F.; Kokosinski, K.; Matsumura, F. Arylhydrocarbon receptor activation in NCI-H441 cells and C57BL/6 mice: Possible mechanisms for lung dysfunction. Am. J. Respir. Cell Mol. Biol. 2010, 42, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Welty, S.E.; Couroucli, X.I.; Barrios, R.; Kondraganti, S.R.; Muthiah, K.; Yu, L.; Avery, S.E.; Moorthy, B. Disruption of the Ah receptor gene alters the susceptibility of mice to oxygen-mediated regulation of pulmonary and hepatic cytochromes P4501A expression and exacerbates hyperoxic lung injury. J. Pharmacol. Exp. Ther. 2004, 310, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.K.; Peterson, S.L.; Timmins, G.S.; Walker, M.K. Endothelin-1-mediated increase in reactive oxygen species and NADPH Oxidase activity in hearts of aryl hydrocarbon receptor (AhR) null mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2005, 88, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [PubMed]
- Riviello, E.D.; Kiviri, W.; Twagirumugabe, T.; Mueller, A.; Banner-Goodspeed, V.M.; Officer, L.; Novack, V.; Mutumwinka, M.; Talmor, D.S.; Fowler, R.A. Hospital Incidence and Outcomes of the Acute Respiratory Distress Syndrome Using the Kigali Modification of the Berlin Definition. Am. J. Respir. Crit. Care Med. 2016, 193, 52–59. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet. Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Chabot, F.; Mitchell, J.A.; Gutteridge, J.M.; Evans, T.W. Reactive oxygen species in acute lung injury. Eur. Respir. J. 1998, 11, 745–757. [Google Scholar]
- Tasaka, S.; Amaya, F.; Hashimoto, S.; Ishizaka, A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid. Redox Signal. 2008, 10, 739–753. [Google Scholar] [CrossRef]
- Crapo, J.D. Morphologic changes in pulmonary oxygen toxicity. Annu. Rev. Physiol. 1986, 48, 721–731. [Google Scholar] [CrossRef]
- Crapo, J.D.; Barry, B.E.; Foscue, H.A.; Shelburne, J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am. Rev. Respir. Dis. 1980, 122, 123–143. [Google Scholar]
- Crapo, J.D.; Hayatdavoudi, G.; Knapp, M.J.; Fracica, P.J.; Wolfe, W.G.; Piantadosi, C.A. Progressive alveolar septal injury in primates exposed to 60% oxygen for 14 days. Am. J. Physiol. 1994, 267, L797–L806. [Google Scholar] [CrossRef]
- Barazzone, C.; Horowitz, S.; Donati, Y.R.; Rodriguez, I.; Piguet, P.F. Oxygen toxicity in mouse lung: Pathways to cell death. Am. J. Respir. Cell Mol. Biol. 1998, 19, 573–581. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Metrailler-Ruchonnet, I.; Schappi, M.; Donati, Y.; Matthay, M.A.; Krause, K.H.; Barazzone Argiroffo, C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981. [Google Scholar] [CrossRef]
- Saint-Georges, F.; Abbas, I.; Billet, S.; Verdin, A.; Gosset, P.; Mulliez, P.; Shirali, P.; Garçon, G. Gene expression induction of volatile organic compound and/or polycyclic aromatic hydrocarbon-metabolizing enzymes in isolated human alveolar macrophages in response to airborne particulate matter (PM2.5). Toxicology 2008, 244, 220–230. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Wong, P.; Kuzmicky, P.; Kado, N.; Matsumura, F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ. Health Perspect. 2005, 113, 1536–1541. [Google Scholar] [CrossRef]
- Martin, J.; Dinsdale, D.; White, I.N. Characterization of Clara and type II cells isolated from rat lung by fluorescence-activated flow cytometry. Biochem. J. 1993, 295, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Oyama, T.; Sugio, K.; Uramoto, H.; Iwata, T.; Onitsuka, T.; Isse, T.; Nozoe, T.; Kagawa, N.; Yasumoto, K.; Kawamoto, T. Increased cytochrome P450 and aryl hydrocarbon receptor in bronchial epithelium of heavy smokers with non-small cell lung carcinoma carries a poor prognosis. Front. Biosci. A J. Virtual Libr. 2007, 12, 4497–4503. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.K.; Vogel, C.F.; Baek, J.; Kodani, S.D.; Uppal, R.S.; Bein, K.J.; Anderson, D.S.; Van Winkle, L.S. Combustion derived ultrafine particles induce cytochrome P-450 expression in specific lung compartments in the developing neonatal and adult rat. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L665–L677. [Google Scholar] [CrossRef] [Green Version]
- Shivanna, B.; Maity, S.; Zhang, S.; Patel, A.; Jiang, W.; Wang, L.; Welty, S.E.; Belmont, J.; Coarfa, C.; Moorthy, B. Gene Expression Profiling Identifies Cell Proliferation and Inflammation as the Predominant Pathways Regulated by Aryl Hydrocarbon Receptor in Primary Human Fetal Lung Cells Exposed to Hyperoxia. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 152, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Patel, A.; Chu, C.; Jiang, W.; Wang, L.; Welty, S.E.; Moorthy, B.; Shivanna, B. Aryl hydrocarbon receptor is necessary to protect fetal human pulmonary microvascular endothelial cells against hyperoxic injury: Mechanistic roles of antioxidant enzymes and RelB. Toxicol. Appl. Pharmacol. 2015, 286, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Couroucli, X.I.; Welty, S.E.; Geske, R.S.; Moorthy, B. Regulation of pulmonary and hepatic cytochrome P4501A expression in the rat by hyperoxia: Implications for hyperoxic lung injury. Mol. Pharmacol. 2002, 61, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, B.; Nguyen, U.T.; Gupta, S.; Stewart, K.D.; Welty, S.E.; Smith, C.V. Induction and decline of hepatic cytochromes P4501A1 and 1A2 in rats exposed to hyperoxia are not paralleled by changes in glutathione S-transferase-alpha. Toxicol. Lett. 1997, 90, 67–75. [Google Scholar] [CrossRef]
- Mansour, H.; Levacher, M.; Azoulay-Dupuis, E.; Moreau, J.; Marquetty, C.; Gougerot-Pocidalo, M.A. Genetic differences in response to pulmonary cytochrome P-450 inducers and oxygen toxicity. J. Appl. Physiol. 1988, 64, 1376–1381. [Google Scholar] [CrossRef]
- Lingappan, K.; Jiang, W.; Wang, L.; Wang, G.; Couroucli, X.I.; Shivanna, B.; Welty, S.E.; Barrios, R.; Khan, M.F.; Nebert, D.W.; et al. Mice Deficient in the Gene for Cytochrome P450 (CYP)1A1 are More Susceptible than Wild-Type to Hyperoxic Lung Injury: Evidence for Protective Role of CYP1A1 Against Oxidative Stress. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 141, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lingappan, K.; Jiang, W.; Couroucli, X.I.; Welty, S.E.; Shivanna, B.; Barrios, R.; Wang, G.; Firoze Khan, M.; Gonzalez, F.J.; et al. Disruption of cytochrome P4501A2 in mice leads to increased susceptibility to hyperoxic lung injury. Free Radic. Biol. Med. 2015, 82, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Veith, A.C.; Bou Aram, B.; Jiang, W.; Wang, L.; Zhou, G.; Jefcoate, C.R.; Couroucli, X.I.; Lingappan, K.; Moorthy, B. Mice Lacking the Cytochrome P450 1B1 Gene Are Less Susceptible to Hyperoxic Lung Injury Than Wild Type. Toxicol. Sci. Off. J. Soc. Toxicol. 2018, 165, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cao, K.; Liu, K.; Xue, Y.; Roberts, A.I.; Li, F.; Han, Y.; Rabson, A.B.; Wang, Y.; Shi, Y. Kynurenic acid, an IDO metabolite, controls TSG-6-mediated immunosuppression of human mesenchymal stem cells. Cell Death Differ. 2018, 25, 1209–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvuna, L.; Sood, A. Epidemiology of Chronic Obstructive Pulmonary Disease. Clin. Chest Med. 2020, 41, 315–327. [Google Scholar] [CrossRef]
- Eisner, M.D.; Anthonisen, N.; Coultas, D.; Kuenzli, N.; Perez-Padilla, R.; Postma, D.; Romieu, I.; Silverman, E.K.; Balmes, J.R. An official American Thoracic Society public policy statement: Novel risk factors and the global burden of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 693–718. [Google Scholar] [CrossRef]
- Eisner, M.D.; Iribarren, C.; Blanc, P.D.; Yelin, E.H.; Ackerson, L.; Byl, N.; Omachi, T.A.; Sidney, S.; Katz, P.P. Development of disability in chronic obstructive pulmonary disease: Beyond lung function. Thorax 2011, 66, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [Green Version]
- Vestbo, J.; Hurd, S.S.; Rodriguez-Roisin, R. The 2011 revision of the global strategy for the diagnosis, management and prevention of COPD (GOLD)--why and what? Clin. Respir. J. 2012, 6, 208–214. [Google Scholar] [CrossRef]
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef]
- Pathak, U.; Gupta, N.C.; Suri, J.C. Risk of COPD due to indoor air pollution from biomass cooking fuel: A systematic review and meta-analysis. Int. J. Environ. Health Res. 2020, 30, 75–88. [Google Scholar] [CrossRef]
- Yao, H.; Rahman, I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol. Appl. Pharmacol. 2011, 254, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Eeden, S.F.; Sin, D.D. Oxidative stress in chronic obstructive pulmonary disease: A lung and systemic process. Can. Respir. J. 2013, 20, 27–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.J.; Hsu, Y.L.; Wang, T.N.; Wu, L.Y.; Lien, C.T.; Hung, C.H.; Kuo, P.L.; Huang, M.S. Aryl hydrocarbon receptor (AhR) agonists increase airway epithelial matrix metalloproteinase activity. J. Mol. Med. 2014, 92, 615–628. [Google Scholar] [CrossRef]
- Guerrina, N.; Traboulsi, H.; Eidelman, D.H.; Baglole, C.J. The Aryl Hydrocarbon Receptor and the Maintenance of Lung Health. Int. J. Mol. Sci. 2018, 19, 3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley, A.J.; Tetley, T.D. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2007, 2, 409–428. [Google Scholar]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [Green Version]
- De Cunto, G.; Cavarra, E.; Bartalesi, B.; Lucattelli, M.; Lungarella, G. Innate Immunity and Cell Surface Receptors in the Pathogenesis of COPD: Insights from Mouse Smoking Models. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1143–1154. [Google Scholar] [CrossRef]
- Rogers, S.; de Souza, A.R.; Zago, M.; Iu, M.; Guerrina, N.; Gomez, A.; Matthews, J.; Baglole, C.J. Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure. Sci. Rep. 2017, 7, 40539. [Google Scholar] [CrossRef] [Green Version]
- de Souza, A.R.; Zago, M.; Eidelman, D.H.; Hamid, Q.; Baglole, C.J. Aryl hydrocarbon receptor (AhR) attenuation of subchronic cigarette smoke-induced pulmonary neutrophilia is associated with retention of nuclear RelB and suppression of intercellular adhesion molecule-1 (ICAM-1). Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 140, 204–223. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Matsumura, F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem. Pharmacol. 2009, 77, 734–745. [Google Scholar] [CrossRef] [Green Version]
- Iu, M.; Zago, M.; Rico de Souza, A.; Bouttier, M.; Pareek, S.; White, J.H.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. RelB attenuates cigarette smoke extract-induced apoptosis in association with transcriptional regulation of the aryl hydrocarbon receptor. Free Radic. Biol. Med. 2017, 108, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Sarill, M.; Zago, M.; Sheridan, J.A.; Nair, P.; Matthews, J.; Gomez, A.; Roussel, L.; Rousseau, S.; Hamid, Q.; Eidelman, D.H.; et al. The aryl hydrocarbon receptor suppresses cigarette-smoke-induced oxidative stress in association with dioxin response element (DRE)-independent regulation of sulfiredoxin 1. Free Radic. Biol. Med. 2015, 89, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H. Animal Models, Learning Lessons to Prevent and Treat Neonatal Chronic Lung Disease. Front. Med. 2015, 2, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobe, A.H.; Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 163, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.N.; Siddiqui, N.H.; Stocker, J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum. Pathol. 1998, 29, 710–717. [Google Scholar] [CrossRef]
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Hall, G.L.; Wilson, A.C. Lung function following very preterm birth in the era of ’new’ bronchopulmonary dysplasia. Respirology 2015, 20, 535–540. [Google Scholar] [CrossRef]
- Islam, J.Y.; Keller, R.L.; Aschner, J.L.; Hartert, T.V.; Moore, P.E. Understanding the Short- and Long-Term Respiratory Outcomes of Prematurity and Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2015, 192, 134–156. [Google Scholar] [CrossRef] [Green Version]
- Gou, X.; Yang, L.; Pan, L.; Xiao, D. Association between bronchopulmonary dysplasia and cerebral palsy in children: A meta-analysis. BMJ Open 2018, 8, e020735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, S.J.; Turkovic, L.; Wilson, A.C.; Verheggen, M.; Logie, K.M.; Pillow, J.J.; Hall, G.L. Lung function trajectories throughout childhood in survivors of very preterm birth: A longitudinal cohort study. Lancet. Child Adolesc. Health 2018, 2, 350–359. [Google Scholar] [CrossRef]
- Twilhaar, E.S.; Wade, R.M.; de Kieviet, J.F.; van Goudoever, J.B.; van Elburg, R.M.; Oosterlaan, J. Cognitive Outcomes of Children Born Extremely or Very Preterm Since the 1990s and Associated Risk Factors: A Meta-analysis and Meta-regression. JAMA Pediatrics 2018, 172, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Haggie, S.; Robinson, P.; Selvadurai, H.; Fitzgerald, D.A. Bronchopulmonary dysplasia: A review of the pulmonary sequelae in the post-surfactant era. J. Paediatr. Child Health 2020, 56, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.R.; Beckmann, J.; Ni, Y.; Bolton, C.E.; McEniery, C.M.; Cockcroft, J.R.; Marlow, N. Respiratory and Cardiovascular Outcomes in Survivors of Extremely Preterm Birth at 19 Years. Am. J. Respir. Crit. Care Med. 2020, 202, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.L.Y.; Doyle, L.W. An update on pulmonary and neurodevelopmental outcomes of bronchopulmonary dysplasia. Semin. Perinatol. 2018, 42, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Lapcharoensap, W.; Bennett, M.V.; Xu, X.; Lee, H.C.; Dukhovny, D. Hospitalization costs associated with bronchopulmonary dysplasia in the first year of life. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2019, 40, 130–137. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Abman, S.H. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Annu. Rev. Physiol. 2005, 67, 623–661. [Google Scholar] [CrossRef] [Green Version]
- Thebaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Rong, M.; Platteau, A.; Hehre, D.; Smith, H.; Ruiz, P.; Whitsett, J.; Bancalari, E.; Wu, S. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: Implication in the pathogenesis of severe bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L330–L340. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Zhou, Z.; Yee, M.; Chu, C.Y.; Lopez, A.M.; Lunger, V.A.; Solleti, S.K.; Resseguie, E.; Buczynski, B.; Mariani, T.J.; et al. The genome-wide transcriptional response to neonatal hyperoxia identifies Ahr as a key regulator. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L516–L523. [Google Scholar] [CrossRef]
- Shivanna, B.; Zhang, W.; Jiang, W.; Welty, S.E.; Couroucli, X.I.; Wang, L.; Moorthy, B. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol. Appl. Pharmacol. 2013, 267, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.Z.; Wang, K.; Fang, R.; Zheng, J. Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2010, 58, 679–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Maltepe, E.; Lu, M.M.; Simon, C.; Bradfield, C.A. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha and Ah receptor mRNAs in the developing mouse. Mech. Dev. 1998, 73, 117–123. [Google Scholar] [CrossRef]
- Maturu, P.; Wei-Liang, Y.; Jiang, W.; Wang, L.; Lingappan, K.; Barrios, R.; Liang, Y.; Moorthy, B.; Couroucli, X.I. Newborn Mice Lacking the Gene for Cyp1a1 Are More Susceptible to Oxygen-Mediated Lung Injury, and Are Rescued by Postnatal β-Naphthoflavone Administration: Implications for Bronchopulmonary Dysplasia in Premature Infants. Toxicol. Sci. Off. J. Soc. Toxicol. 2017, 157, 260–271. [Google Scholar] [CrossRef]
- Thakur, V.S.; Liang, Y.W.; Lingappan, K.; Jiang, W.; Wang, L.; Barrios, R.; Zhou, G.; Guntupalli, B.; Shivanna, B.; Maturu, P.; et al. Increased susceptibility to hyperoxic lung injury and alveolar simplification in newborn rats by prenatal administration of benzo[a]pyrene. Toxicol. Lett. 2014, 230, 322–332. [Google Scholar] [CrossRef]
- Chiba, T.; Uchi, H.; Tsuji, G.; Gondo, H.; Moroi, Y.; Furue, M. Arylhydrocarbon receptor (AhR) activation in airway epithelial cells induces MUC5AC via reactive oxygen species (ROS) production. Pulm. Pharmacol. Ther. 2011, 24, 133–140. [Google Scholar] [CrossRef]
- Chiba, T.; Uchi, H.; Yasukawa, F.; Furue, M. Role of the arylhydrocarbon receptor in lung disease. Int. Arch. Allergy Immunol. 2011, 155 (Suppl. 1), 129–134. [Google Scholar] [CrossRef]
- Dean, A.; Gregorc, T.; Docherty, C.K.; Harvey, K.Y.; Nilsen, M.; Morrell, N.W.; MacLean, M.R. Role of the aryl hydrocarbon receptor in sugen 5416-induced experimental pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2018, 58, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Murray, I.A.; Perdew, G.H. How Ah receptor ligand specificity became imprortant in understanding its physiological function. Int. J. Mol. Sci. 2020, 21, 9614. [Google Scholar] [CrossRef]
Figure 1.
Structure of the AHR. bHLH: basic helix-loop-helix; PAS: PER-ARNT-SIM.

Figure 2.
The AHR signaling pathway. Prior to ligand binding, the AHR (Aryl hydrocarbon receptor) is located in the cytoplasm as a complex comprising menu proteins, including Hsp90 (Heat shock protein 90), XAP2: (Hepatitis B virus X-associated protein 2), p23, and Src Kinase. Upon entry of the ligand-AHR into the nucleus, the associated proteins are dissociated, and the ligand-AHR complex binds to the ARNT (Aryl hydrocarbon receptor nuclear translocator,), which, in turn, binds to the AHRE (Aryl hydrocarbon receptor responsive elements) on the CYP1A1 promoter, leading to transcriptional activation of CYP1A1 and other phase II genes.
Figure 2.
The AHR signaling pathway. Prior to ligand binding, the AHR (Aryl hydrocarbon receptor) is located in the cytoplasm as a complex comprising menu proteins, including Hsp90 (Heat shock protein 90), XAP2: (Hepatitis B virus X-associated protein 2), p23, and Src Kinase. Upon entry of the ligand-AHR into the nucleus, the associated proteins are dissociated, and the ligand-AHR complex binds to the ARNT (Aryl hydrocarbon receptor nuclear translocator,), which, in turn, binds to the AHRE (Aryl hydrocarbon receptor responsive elements) on the CYP1A1 promoter, leading to transcriptional activation of CYP1A1 and other phase II genes.

Figure 3.
AHR modulates ALI in vivo via CYP1A enzymes. AHR is expressed in lungs and liver. In hyperoxic lung injury animal models, AHR deficiency potentiates the symptoms, which may be associated with AHR-regulated genes, such as CYP1A1/2, in these tissues. AHR-regulated genes may also alleviate LPS-induced lung injury. The CYP1A enzymes attenuate lung injury by detoxifying lipid hydroperoxides, such as F2-isoprostanes [129,152,153].
Figure 3.
AHR modulates ALI in vivo via CYP1A enzymes. AHR is expressed in lungs and liver. In hyperoxic lung injury animal models, AHR deficiency potentiates the symptoms, which may be associated with AHR-regulated genes, such as CYP1A1/2, in these tissues. AHR-regulated genes may also alleviate LPS-induced lung injury. The CYP1A enzymes attenuate lung injury by detoxifying lipid hydroperoxides, such as F2-isoprostanes [129,152,153].

Figure 4.
The role of AHR in the pathogenesis of COPD. Cigarette smoke and occupational pollutants may cause COPD due to abnormal immune homeostasis in the lung, which is attenuated by activation of AHR. It suggests AHR agonists may prevent or treat COPD.
Figure 4.
The role of AHR in the pathogenesis of COPD. Cigarette smoke and occupational pollutants may cause COPD due to abnormal immune homeostasis in the lung, which is attenuated by activation of AHR. It suggests AHR agonists may prevent or treat COPD.

Figure 5.
The role of the AHR in development of experimental BPD. Hyperoxia is one of major factors that contribute to the development of BPD. Some AHR ligands alleviate the hyperoxia-induced BPD, which may be associated with the activation of many AHR-regulated genes, such as phase I and II enzymes. However, other AHR ligands, such as environmental pollutants, potentiate the hyperoxia-induced BPD.
Figure 5.
The role of the AHR in development of experimental BPD. Hyperoxia is one of major factors that contribute to the development of BPD. Some AHR ligands alleviate the hyperoxia-induced BPD, which may be associated with the activation of many AHR-regulated genes, such as phase I and II enzymes. However, other AHR ligands, such as environmental pollutants, potentiate the hyperoxia-induced BPD.


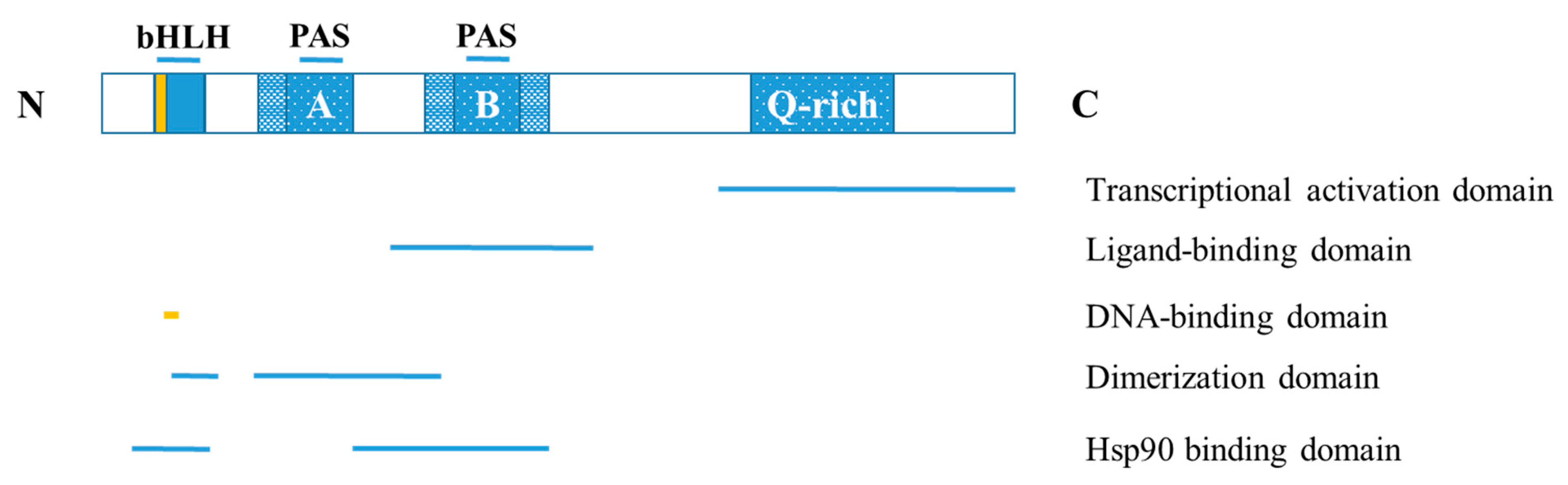{kind=link}
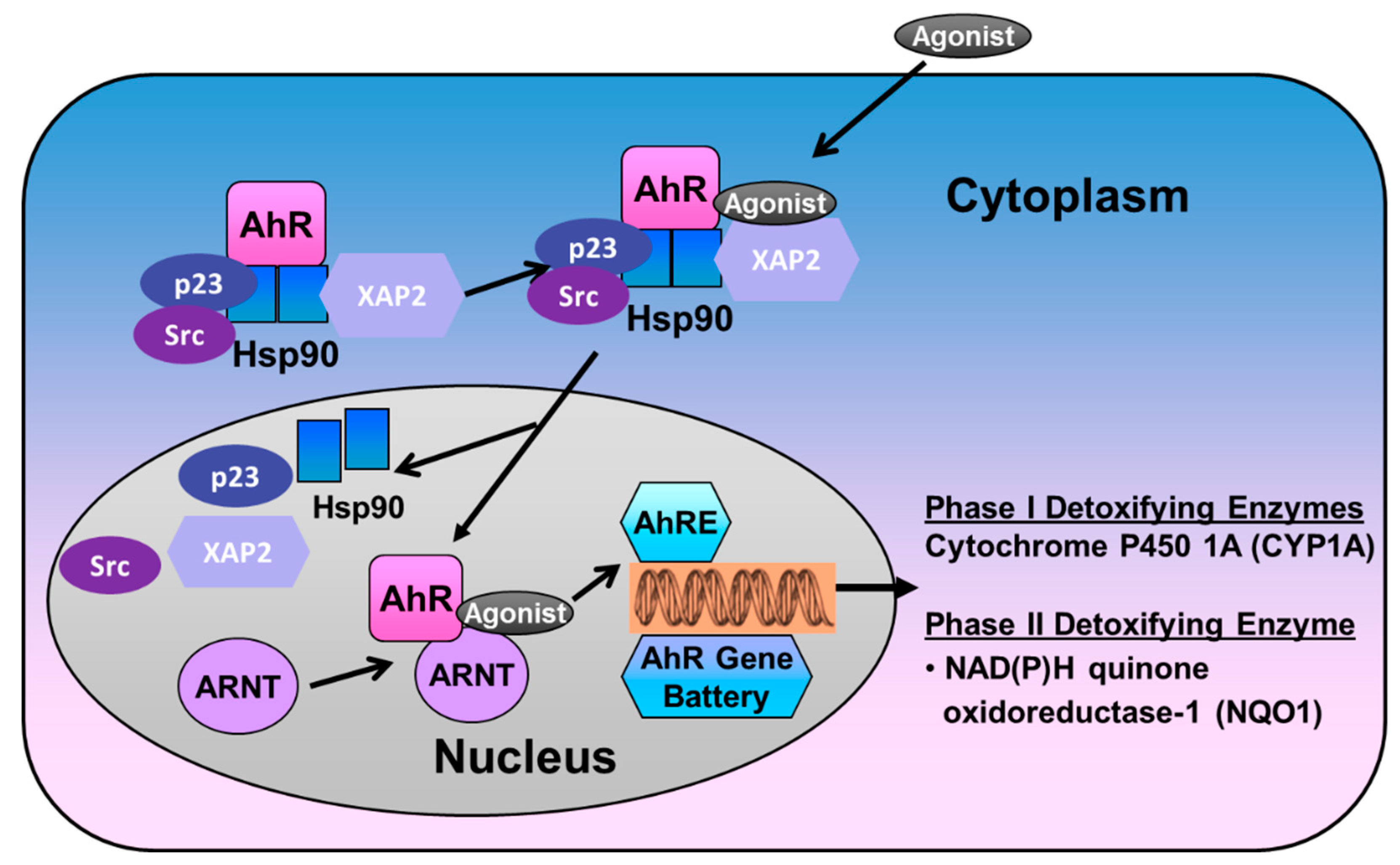{kind=link}
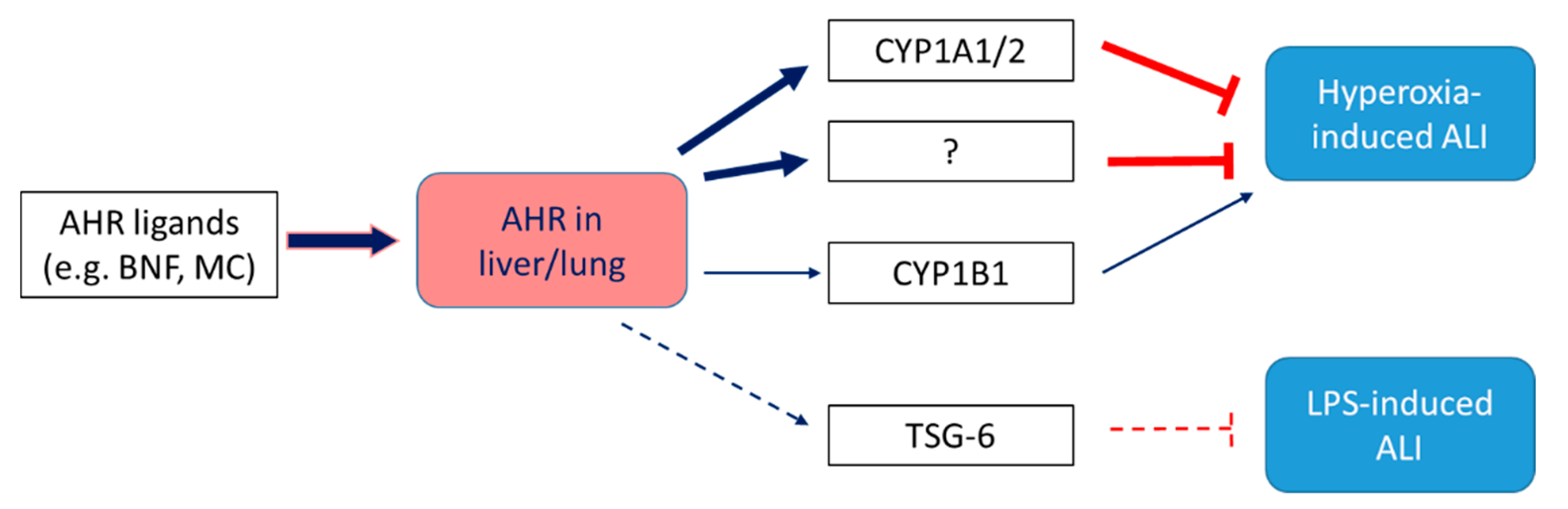{kind=link}
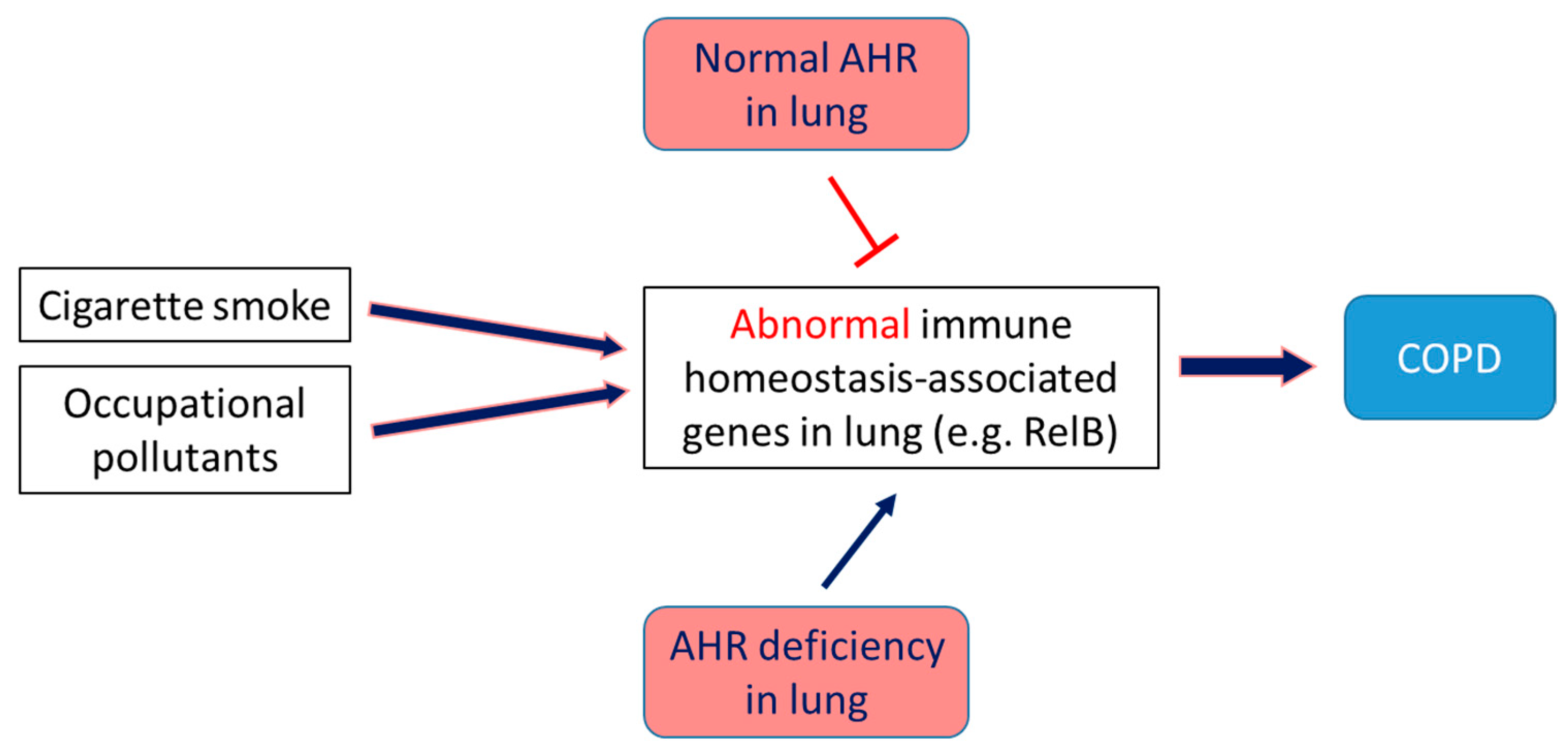{kind=link}
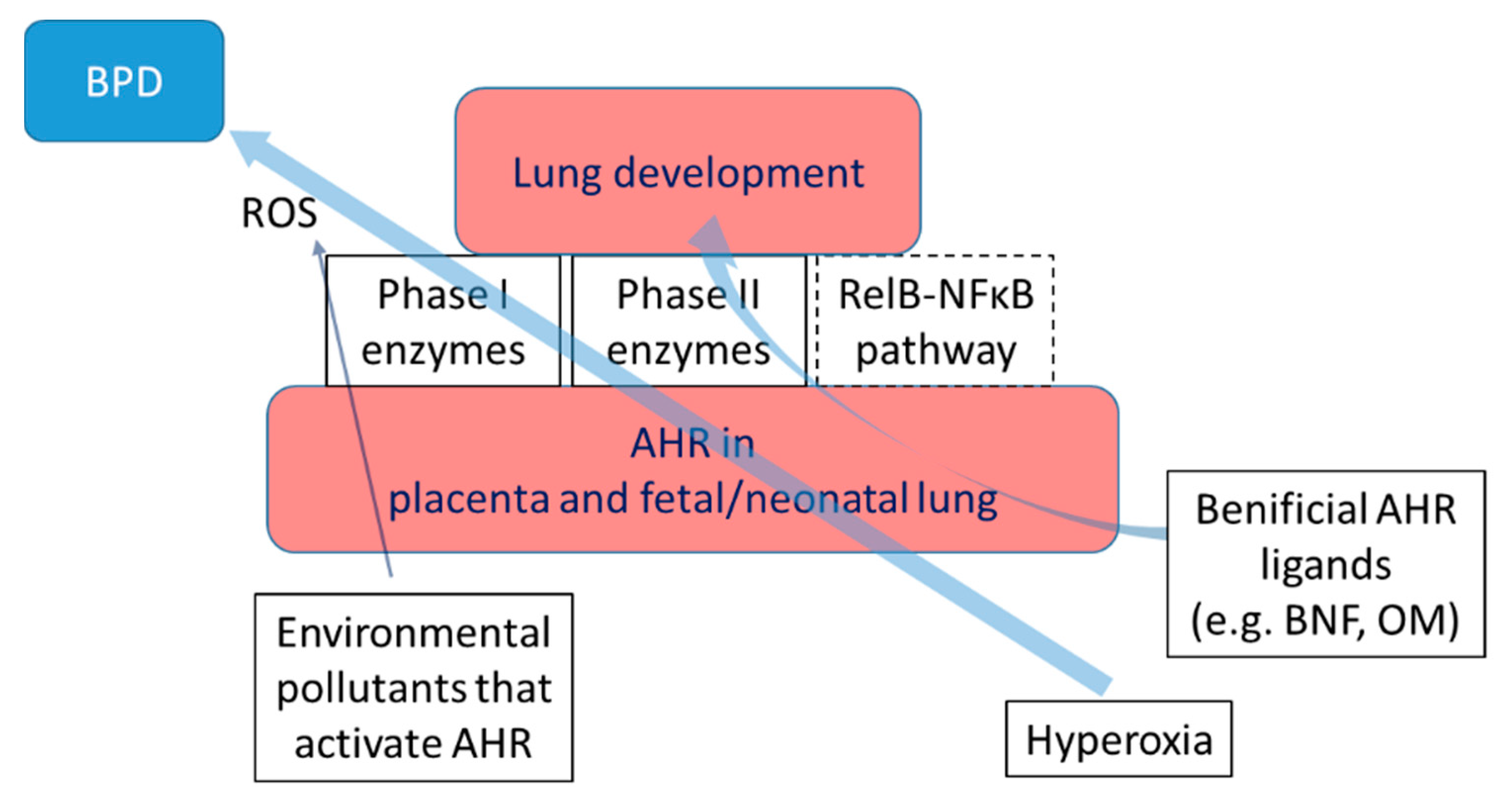{kind=link}
Table 1.
List of major agonists (exogenous and endogenous ligands) and antagonists of the AHR. The table also describes the major target organs and the diseases that are modulated by the AHR.
Table 1.
List of major agonists (exogenous and endogenous ligands) and antagonists of the AHR. The table also describes the major target organs and the diseases that are modulated by the AHR.
| Source | Examples | Target Organ/Disease |
|---|---|---|
| Exogenous | Halogenated aromatic hydrocarbons | Lung cancer [28,73] |
| Dibenzofurans | Lung toxicity not confirmed | |
| Biphenyls | Lung toxicity not confirmed | |
| Polycyclic aromatic hydrocarbons | Lung cancer [28,73], asthma [74], COPD [75], chronic bronchitis [76,77] | |
| 3-Methylcholanthrene | No severe lung toxicity | |
| Benzo[a]pyrene | Lung inflammation [78,79], respiratory tract cancer [80] | |
| Benzanthracenes | No immediate severe lung toxicity | |
| Benzoflavones | Non-toxic | |
| Dietary Endogenous | Flavonoids | BPD/ARDS [81,82,83] |
| Quercetin | BPD [84] | |
| Indole-3-carbinol | COPD, asthma, ARDS, BPD | |
| 3,3′-Diindolylmethane | Lung cancer chemoprevention [85] | |
| Indolo[3,2-b]carbazole | No pulmonary therapeutic application reported | |
| Tryptophan metabolites | Kynurenic acid | ALI [86] |
| Kynurenine | Lung cancer [87] | |
| Tryptamine | No pulmonary therapeutic application reported | |
| 6-Formylindolo[3,2-b]carbazole | LPS-induced ALI [88] | |
| Indoxyl sulfate | No immediate severe lung toxicity | |
| Microbiota | 3-Methylindole | May cause lung cancer [89] |
| Tryptanthrin | Lung cancer [90] | |
| 1,4-Dihydroxy-2-naphthoic acid | No pulmonary therapeutic application reported | |
| Indole-3-aldehyde | No immediate severe lung toxicity | |
| Indole-3-acetate | No pulmonary therapeutic application reported | |
| Phenazines | No pulmonary therapeutic application reported | |
| Indirubin | Lung cancer [91], anti-inflammatory [92] | |
| Malassezin | No pulmonary therapeutic application reported | |
| Xenobiotic | 3,4-Dimethoxy-a-naphthoflavone | Lung cancer [93] |
| MNF | Lung cancer, COPD, asthma [94] | |
| CH-223191 | Lung cancer, COPD, asthma [74] | |
| Dietary | Resveratrol | Lung cancer, asthma COPD [95] |
| AHR Active Pharmaceuticals | Tranilast | COPD, Asthma [85] |
| Leflunomide | BPD, ARDS [85] | |
| Omeprazole | BPD, ARDS [85,96,97,98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shivanna, B.; Chu, C.; Moorthy, B. The Aryl Hydrocarbon Receptor (AHR): A Novel Therapeutic Target for Pulmonary Diseases? Int. J. Mol. Sci. 2022, 23, 1516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031516
AMA Style
Shivanna B, Chu C, Moorthy B. The Aryl Hydrocarbon Receptor (AHR): A Novel Therapeutic Target for Pulmonary Diseases? International Journal of Molecular Sciences. 2022; 23(3):1516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031516
Chicago/Turabian StyleShivanna, Binoy, Chun Chu, and Bhagavatula Moorthy. 2022. "The Aryl Hydrocarbon Receptor (AHR): A Novel Therapeutic Target for Pulmonary Diseases?" International Journal of Molecular Sciences 23, no. 3: 1516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031516
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.