
Identification of ATP2B4 Regulatory Element Containing Functional Genetic Variants Associated with Severe Malaria

 , , and
, , and
Abstract
:1. Introduction
2. Results
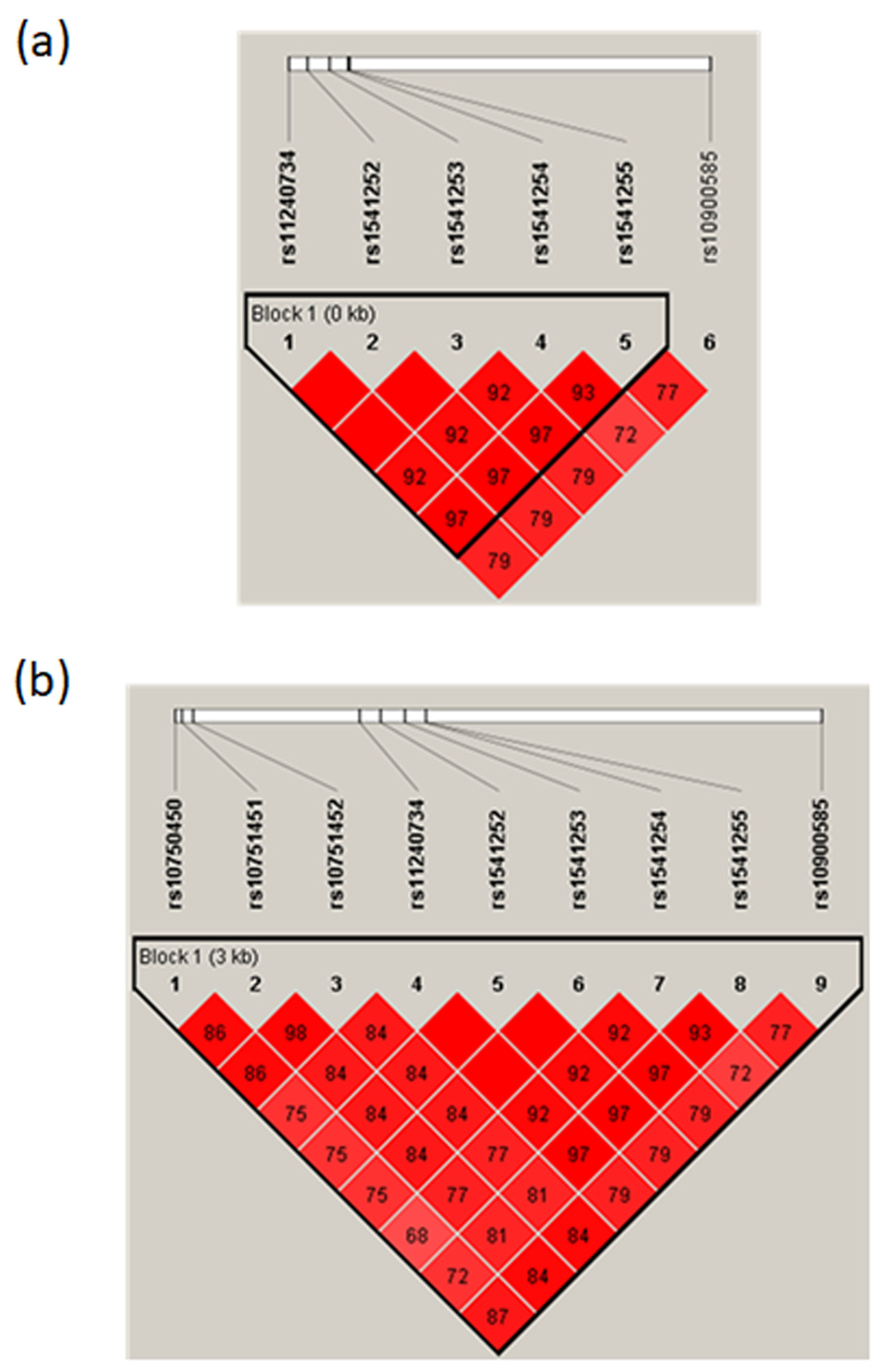
2.1. Prioritization and Annotation of Putative Regulatory SNPs in Linkage Disequilibrium with tagSNPs Associated with Severe Malaria
2.2. Association between the ATP2B4 tagSNP rs10900585 and Severe Malaria in a Senegalese Cohort
2.3. Association of rs11240734, rs1541252, rs1541253, rs1541254, and rs1541255 with Severe Malaria in Senegalese Population
2.4. Association of rs10751450, rs10751451, and rs10751452 with Severe Malaria in Senegalese Population
2.5. Haplotype Analysis of the ATP2B4 SNPs
2.6. Lower Promoter Activity and Higher Enhancer Activity Are Associated with the Risk Haplotype of Severe Malaria
2.7. Genome Editing Confirmed the Regulatory Activity of the Region Containing the SNPs
2.8. K562 Deleted Clones Exhibited Higher Intracellular Calcium Concentration
2.9. PMCA4 Protein Is Not Expressed in K562 Deleted Clones
3. Discussion
4. Materials and Methods
4.1. Study Subjects, Blood Samples, and Phenotypes
4.2. Bioinformatic Prioritization and Functional Annotation of Genetic Variants
4.3. DNA Extraction, DNA Amplification, and Genotyping
4.4. Luciferase Gene Reporter Assay and Site-Directed Mutagenesis:
4.4.1. Promoter Activity
4.4.2. Enhancer Activity
4.5. CRISPR-Cas9 Genome Editing in K562 Cells
4.6. Reverse Transcription-Quantitative PCR
4.7. Calcium Measurement by Flow Cytometry in K562 Clones and Wild-Type Cells
4.8. Detection of PMCA4 by Flow Cytometry in K562 Clones and Wild-Type Cells
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackinnon, M.J.; Mwangi, T.W.; Snow, R.W.; Marsh, K.; Williams, T.N. Heritability of malaria in Africa. PLoS Med. 2005, 2, e340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.; Clegg, J. Genetic variability in response to infection: Malaria and after. Genes Immun. 2002, 3, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damena, D.; Chimusa, E.R. Genome-wide heritability analysis of severe malaria resistance reveals evidence of polygenic inheritance. Hum. Mol. Genet. 2020, 29, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Rihet, P.; Abel, L.; Traore, Y.; Traore-Leroux, T.; Aucan, C.; Fumoux, F. Human malaria: Segregation analysis of blood infection levels in a suburban area and a rural area in Burkina Faso. Genet. Epidemiol. 1998, 15, 435–450. [Google Scholar] [CrossRef]
- Rihet, P.; Traoré, Y.; Abel, L.; Aucan, C.; Traoré-Leroux, T.; Fumoux, F. Malaria in humans: Plasmodium falciparum blood infection levels are linked to chromosome 5q31–q33. Am. J. Hum. Genet. 1998, 63, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Brisebarre, A.; Kumulungui, B.; Sawadogo, S.; Atkinson, A.; Garnier, S.; Fumoux, F.; Rihet, P. A genome scan for Plasmodium falciparum malaria identifies quantitative trait loci on chromosomes 5q31, 6p21. 3, 17p12, and 19p13. Malar. J. 2014, 13, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Flori, L.; Sawadogo, S.; Esnault, C.; Delahaye, N.F.; Fumoux, F.; Rihet, P. Linkage of mild malaria to the major histocompatibility complex in families living in Burkina Faso. Hum. Mol. Genet. 2003, 12, 375–378. [Google Scholar] [CrossRef]
- Jepson, A.; Sisay-Joof, F.; Banya, W.; Hassan-King, M.; Frodsham, A.; Bennett, S.; Hill, A.; Whittle, H. Genetic linkage of mild malaria to the major histocompatibility complex in Gambian children: Study of affected sibling pairs. BMJ 1997, 315, 96–97. [Google Scholar] [CrossRef] [Green Version]
- Flori, L.; Kumulungui, B.; Aucan, C.; Esnault, C.; Traore, A.; Fumoux, F.; Rihet, P. Linkage and association between Plasmodium falciparum blood infection levels and chromosome 5q31–q33. Genes Immun. 2003, 4, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.; Marquet, S.; Bucheton, B.; Hillaire, D.; Cot, M.; Fievet, N.; Dessein, A.J.; Abel, L. Linkage analysis of blood Plasmodium falciparum levels: Interest of the 5q31–q33 chromosome region. Am. J. Trop. Med. Hyg. 1998, 58, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Milet, J.; Nuel, G.; Watier, L.; Courtin, D.; Slaoui, Y.; Senghor, P.; Migot-Nabias, F.; Gaye, O.; Garcia, A. Genome wide linkage study, using a 250K SNP map, of Plasmodium falciparum infection and mild malaria attack in a Senegalese population. PLoS ONE 2010, 5, e11616. [Google Scholar] [CrossRef] [PubMed]
- Sakuntabhai, A.; Ndiaye, R.; Casadémont, I.; Peerapittayamonkol, C.; Rogier, C.; Tortevoye, P.; Tall, A.; Paul, R.; Turbpaiboon, C.; Phimpraphi, W. Genetic determination and linkage mapping of Plasmodium falciparum malaria related traits in Senegal. PLoS ONE 2008, 3, e2000. [Google Scholar] [CrossRef]
- Band, G.; Le, Q.S.; Jostins, L.; Pirinen, M.; Kivinen, K.; Jallow, M.; Sisay-Joof, F.; Bojang, K.; Pinder, M.; Sirugo, G. Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet. 2013, 9, e1003509. [Google Scholar] [CrossRef]
- Jallow, M.; Teo, Y.Y.; Small, K.S.; Rockett, K.A.; Deloukas, P.; Clark, T.G.; Kivinen, K.; Bojang, K.A.; Conway, D.J.; Pinder, M. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat. Genet. 2009, 41, 657–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, M.G.E. A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature 2015, 526, 253–257. [Google Scholar]
- Network, M.G.E. Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania. Nat. Commun. 2019, 10, 5732. [Google Scholar]
- Ravenhall, M.; Campino, S.; Sepúlveda, N.; Manjurano, A.; Nadjm, B.; Mtove, G.; Wangai, H.; Maxwell, C.; Olomi, R.; Reyburn, H. Novel genetic polymorphisms associated with severe malaria and under selective pressure in North-eastern Tanzania. PLoS Genet. 2018, 14, e1007172. [Google Scholar] [CrossRef] [Green Version]
- Timmann, C.; Thye, T.; Vens, M.; Evans, J.; May, J.; Ehmen, C.; Sievertsen, J.; Muntau, B.; Ruge, G.; Loag, W. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature 2012, 489, 443–446. [Google Scholar] [CrossRef]
- Pule, G.; Chimusa, E.; Mnika, K.; Mhandire, K.; Kampira, E.; Dandara, C.; Wonkam, A. Beta-globin gene haplotypes and selected Malaria-associated variants among black Southern African populations. Glob. Health Epidemiol. Genom. 2017, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Leffler, E.M.; Band, G.; Busby, G.B.; Kivinen, K.; Le, Q.S.; Clarke, G.M.; Bojang, K.A.; Conway, D.J.; Jallow, M.; Sisay-Joof, F. Resistance to malaria through structural variation of red blood cell invasion receptors. Science 2017, 356, eaam6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, D.G.; Cofie, J.; Jiang, L.; Hartl, D.L.; Tracy, E.; Kabat, J.; Mendoza, L.H.; Miller, L.H. Glycophorin B is the erythrocyte receptor of Plasmodium falciparum erythrocyte-binding ligand, EBL-1. Proc. Natl. Acad. Sci. USA 2009, 106, 5348–5352. [Google Scholar] [CrossRef] [Green Version]
- Sim, B.; Chitnis, C.; Wasniowska, K.; Hadley, T.; Miller, L. Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science 1994, 264, 1941–1944. [Google Scholar] [CrossRef] [PubMed]
- Lessard, S.; Gatof, E.S.; Beaudoin, M.; Schupp, P.G.; Sher, F.; Ali, A.; Prehar, S.; Kurita, R.; Nakamura, Y.; Baena, E. An erythroid-specific ATP2B4 enhancer mediates red blood cell hydration and malaria susceptibility. J. Clin. Investig. 2017, 127, 3065–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Dayem Ullah, A.Z.; Chelala, C. IW-Scoring: An Integrative Weighted Scoring framework for annotating and prioritizing genetic variations in the noncoding genome. Nucleic Acids Res. 2018, 46, e47. [Google Scholar] [CrossRef] [Green Version]
- Chèneby, J.; Gheorghe, M.; Artufel, M.; Mathelier, A.; Ballester, B. ReMap 2018: An updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 2018, 46, D267–D275. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [Green Version]
- Van Arensbergen, J.; Pagie, L.; FitzPatrick, V.D.; de Haas, M.; Baltissen, M.P.; Comoglio, F.; van der Weide, R.H.; Teunissen, H.; Võsa, U.; Franke, L. High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 2019, 51, 1160. [Google Scholar] [CrossRef]
- Ndila, C.M.; Uyoga, S.; Macharia, A.W.; Nyutu, G.; Peshu, N.; Ojal, J.; Shebe, M.; Awuondo, K.O.; Mturi, N.; Tsofa, B.; et al. Human candidate gene polymorphisms and risk of severe malaria in children in Kilifi, Kenya: A case-control association study. Lancet Haematol. 2018, 5, e333–e345. [Google Scholar] [CrossRef] [Green Version]
- Rockett, K.A.; Clarke, G.M.; Fitzpatrick, K.; Hubbart, C.; Jeffreys, A.E.; Rowlands, K.; Craik, R.; Jallow, M.; Conway, D.J.; Bojang, K.A.; et al. Reappraisal of known malaria resistance loci in a large multicenter study. Nat. Genet. 2014, 46, 1197–1204. [Google Scholar]
- Dao, L.T.; Galindo-Albarrán, A.O.; Castro-Mondragon, J.A.; Andrieu-Soler, C.; Medina-Rivera, A.; Souaid, C.; Charbonnier, G.; Griffon, A.; Vanhille, L.; Stephen, T. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat. Genet. 2017, 49, 1073. [Google Scholar] [CrossRef] [PubMed]
- Dao, L.T.; Spicuglia, S. Transcriptional regulation by promoters with enhancer function. Transcription 2018, 9, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westra, H.-J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zámbó, B.; Várady, G.; Padányi, R.; Szabó, E.; Németh, A.; Langó, T.; Enyedi, Á.; Sarkadi, B. Decreased calcium pump expression in human erythrocytes is connected to a minor haplotype in the ATP2B4 gene. Cell Calcium 2017, 65, 73–79. [Google Scholar] [CrossRef]
- Mozner, O.; Zambo, B.; Sarkadi, B. Modulation of the Human Erythroid Plasma Membrane Calcium Pump (PMCA4b) Expression by Polymorphic Genetic Variants. Membranes 2021, 11, 586. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.-H.; Sipeky, C.; Dong, X.-M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H. Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 2018, 174, 564–575. [Google Scholar] [CrossRef] [Green Version]
- Ying, D.; Li, M.J.; Sham, P.C.; Li, M. A powerful approach reveals numerous expression quantitative trait haplotypes in multiple tissues. Bioinformatics 2018, 34, 3145–3150. [Google Scholar] [CrossRef]
- Corradin, O.; Saiakhova, A.; Akhtar-Zaidi, B.; Myeroff, L.; Willis, J.; Cowper-Sal, R.; Lupien, M.; Markowitz, S.; Scacheri, P.C. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014, 24, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Glessner, J.T.; Zhang, H.; Hou, C.; Wei, Z.; Bradfield, J.P.; Mentch, F.D.; Guo, Y.; Kim, C.; Xia, Q. GWAS of blood cell traits identifies novel associated loci and epistatic interactions in Caucasian and African-American children. Hum. Mol. Genet. 2013, 22, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Villegas-Mendez, A.; Stafford, N.; Haley, M.J.; Pravitasari, N.E.; Baudoin, F.; Ali, A.; Asih, P.B.S.; Siregar, J.E.; Baena, E.; Syafruddin, D.; et al. The plasma membrane calcium ATPase 4 does not influence parasite levels but partially promotes experimental cerebral malaria during murine blood stage malaria. Malar. J. 2021, 20, 297. [Google Scholar] [CrossRef] [PubMed]
- Pance, A.; Ling, B.; Mwikali, K.; Koutsourakis, M.; Agu, C.; Rouhani, F.; Ponstingl, H.; Montandon, R.; Law, F.; Rayner, J. Stem cell technology provides novel tools to understand human variation in Plasmodium falciparum MALARIA. BioRxiv 2021. [Google Scholar] [CrossRef]
- Caride, A.J.; Filoteo, A.G.; Penniston, J.T.; Strehler, E.E. The Plasma Membrane Ca2+ Pump Isoform 4a Differs from Isoform 4b in the Mechanism of Calmodulin Binding and Activation Kinetics IMPLICATIONS FOR Ca2+ SIGNALING. J. Biol. Chem. 2007, 282, 25640–25648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, T.; Kimura, M.; Watanabe, N. Elevated levels of vascular endothelial growth factor (VEGF) and soluble vascular endothelial growth factor receptor (VEGFR)-2 in human malaria. Am. J. Trop. Med. Hyg. 2010, 82, 136–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canavese, M.; Crisanti, A. Vascular endothelial growth factor (VEGF) and lovastatin suppress the inflammatory response to Plasmodium berghei infection and protect against experimental cerebral malaria. Pathog. Glob. Health 2015, 109, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Thiam, A.; Baaklini, S.; Mbengue, B.; Nisar, S.; Diarra, M.; Marquet, S.; Fall, M.M.; Sanka, M.; Thiam, F.; Diallo, R.N. NCR3 polymorphism, haematological parameters, and severe malaria in Senegalese patients. PeerJ 2018, 6, e6048. [Google Scholar] [CrossRef] [Green Version]
- Martorell-Marugan, J.; Toro-Dominguez, D.; Alarcon-Riquelme, M.E.; Carmona-Saez, P. MetaGenyo: A web tool for meta-analysis of genetic association studies. BMC Bioinform. 2017, 18, 563. [Google Scholar] [CrossRef]
- Wallace, B.C.; Dahabreh, I.J.; Trikalinos, T.A.; Lau, J.; Trow, P.; Schmid, C.H. Closing the gap between methodologists and end-users: R as a computational back-end. J. Stat. Softw. 2012, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP rsID | Genomic Coordinate (hg38) | IW Scoring Rank | Number of ChIP-seq Peaks | eQTL Hits |
|---|---|---|---|---|
| rs11240734 | 203682695 | 1 | 54 | 3 hits |
| rs1541252 | 203682798 | 2 | 90 | 3 hits |
| rs10751450 | 203681816 | 3 | 46 | |
| rs2228445 | 203698280 | 4 | 18 | 3 hits |
| rs202111522 | 203682602 | 5 | 43 | |
| rs1541254 | 203683011 | 6 | 104 | |
| rs1541255 | 203683012 | 7 | 103 | |
| rs10594838 | 203685540 | 8 | 2 | |
| rs10625220 | 203686622 | 9 | 15 | |
| rs1541253 | 203682911 | 10 | 149 | |
| rs8176719 | 133257721 | 11 | 7 | 14 hits |
| rs10751451 | 203681849 | 12 | 49 | 3 hits |
| rs10751452 | 203681901 | 13 | 50 |
| SNP | Position a | Minor Allele | MAF b | Risk Genotype | Controls% | Severe Malaria % | OR | 95% CI | p |
|---|---|---|---|---|---|---|---|---|---|
| rs10900585 (T > G) | 203684896 | G | 0.41 | TT | 32.9 | 48.7 | 1.94 | 1.07–3.50 | 0.029 |
| rs11240734 (T > C) | 203682696 | C | 0.40 | TT | 31.6 | 53.8 | 2.52 | 1.39–4.58 | 0.002 |
| rs1541252 (C > T) | 203682799 | T | 0.40 | CC | 31.6 | 53.8 | 2.52 | 1.39–4.58 | 0.002 |
| rs1541253 (C > T) | 203682912 | T | 0.40 | CC | 31.6 | 53.8 | 2.52 | 1.39–4.58 | 0.002 |
| rs1541254 (G > C) | 203683012 | C | 0.43 | GG | 31.6 | 53.8 | 2.52 | 1.39–4.58 | 0.002 |
| rs1541255 (A > G) | 203683013 | G | 0.40 | AA | 31.6 | 53.8 | 2.52 | 1.39–4.58 | 0.002 |
| rs10751450 (C > T) | 203681817 | T | 0.41 | CC | 32.9 | 50.4 | 2.07 | 1.15–3.75 | 0.016 |
| rs10751451 (C > T) | 203681850 | T | 0.40 | CC | 34.2 | 56.4 | 2.49 | 1.38–4.50 | 0.002 |
| rs10751452 (T > C) | 203681902 | C | 0.39 | TT | 35.4 | 55.6 | 2.27 | 1.26–4.09 | 0.006 |
| SM vs. Ctrl | |||||
|---|---|---|---|---|---|
| Major Haplotype | Minor Haplotype | Risk Haplotype | p-Value | OR | 95% CI |
| TCCGA | CTTCG | TCCGA/TCCGA | 0.002 a | 2.52 a | 1.39–4.58 a |
| 0.006 b | 2.38 b | 1.29–4.40 b | |||
| CCTTCCGA | TTCCTTCG | CCTTCCGA/CCTTCCGA | 0.003 a | 2.67 a | 1.41–5.04 a |
| 0.005 b | 2.53 b | 1.32–4.86 b | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nisar, S.; Torres, M.; Thiam, A.; Pouvelle, B.; Rosier, F.; Gallardo, F.; Ka, O.; Mbengue, B.; Diallo, R.N.; Brosseau, L.; et al. Identification of ATP2B4 Regulatory Element Containing Functional Genetic Variants Associated with Severe Malaria. Int. J. Mol. Sci. 2022, 23, 4849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094849
Nisar S, Torres M, Thiam A, Pouvelle B, Rosier F, Gallardo F, Ka O, Mbengue B, Diallo RN, Brosseau L, et al. Identification of ATP2B4 Regulatory Element Containing Functional Genetic Variants Associated with Severe Malaria. International Journal of Molecular Sciences. 2022; 23(9):4849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094849
Chicago/Turabian StyleNisar, Samia, Magali Torres, Alassane Thiam, Bruno Pouvelle, Florian Rosier, Frederic Gallardo, Oumar Ka, Babacar Mbengue, Rokhaya Ndiaye Diallo, Laura Brosseau, and et al. 2022. "Identification of ATP2B4 Regulatory Element Containing Functional Genetic Variants Associated with Severe Malaria" International Journal of Molecular Sciences 23, no. 9: 4849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23094849