Pax9 and Gbx2 Interact in the Pharyngeal Endoderm to Control Cardiovascular Development

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Breeding
2.3. Imaging
2.4. Identifying Conserved Binding Sites in the GBX2 Locus
2.5. Luciferase Assay
2.6. Flow Cytometry
2.7. Quantitative Real-Time RT-PCR (qPCR)
2.8. Statistical Analysis
3. Results
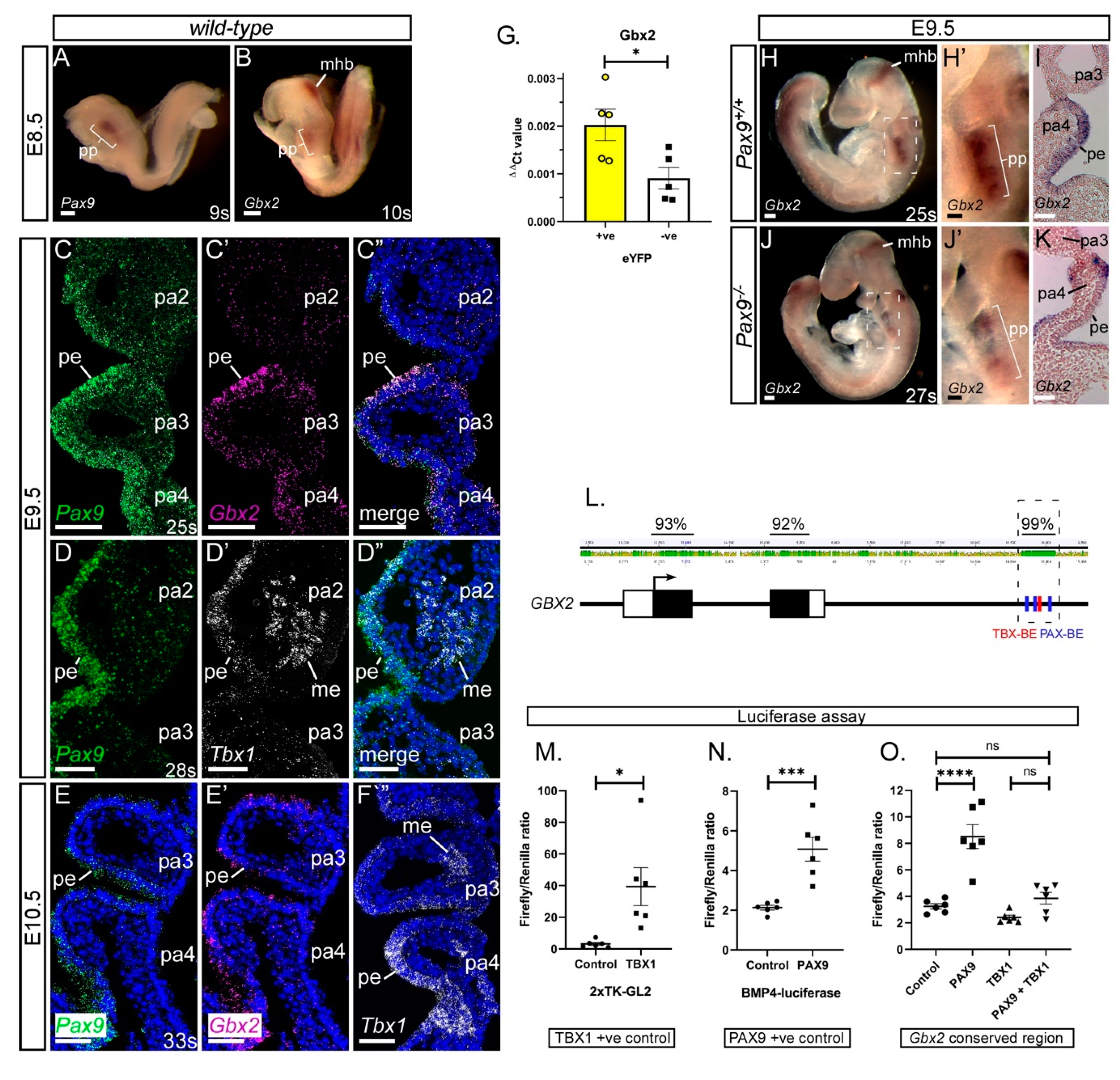
3.1. Gbx2 Expression in the Pharyngeal Endoderm
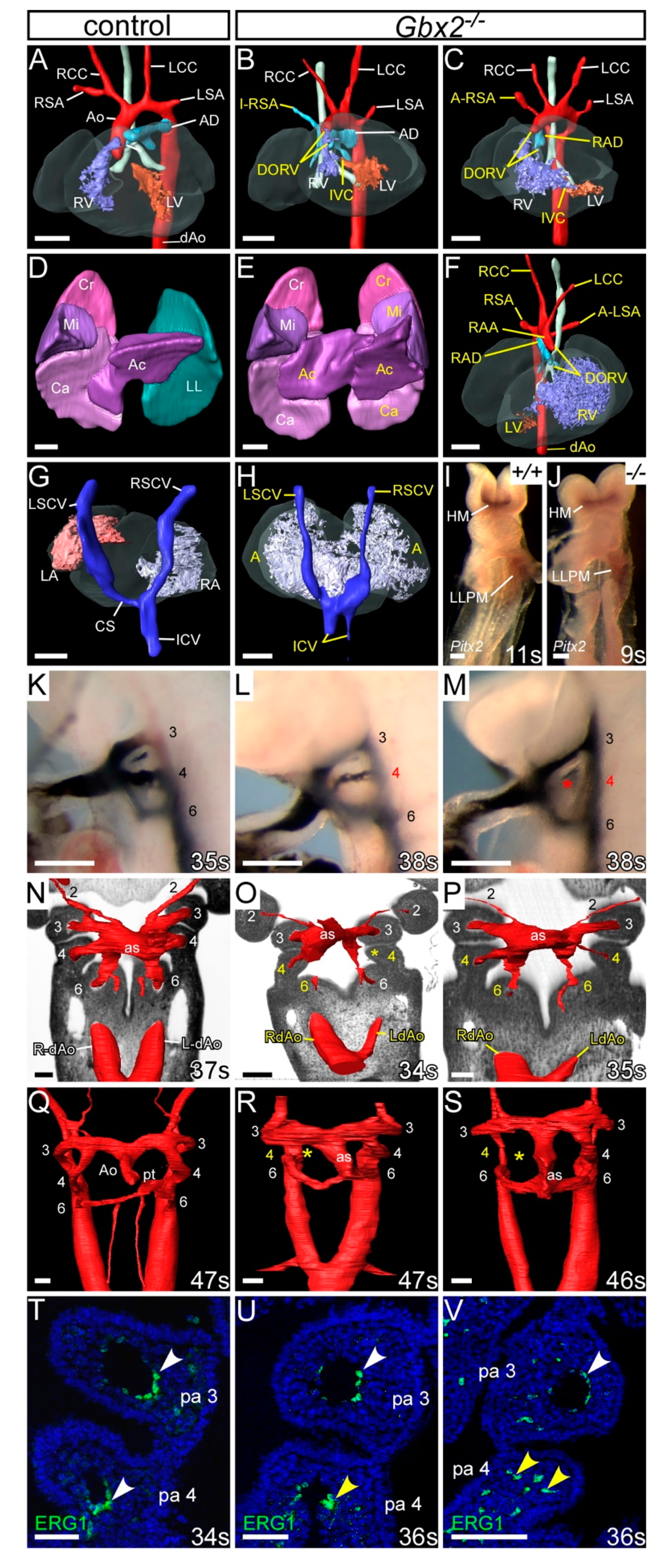
3.2. Gbx2-Null Cardiovascular Defects
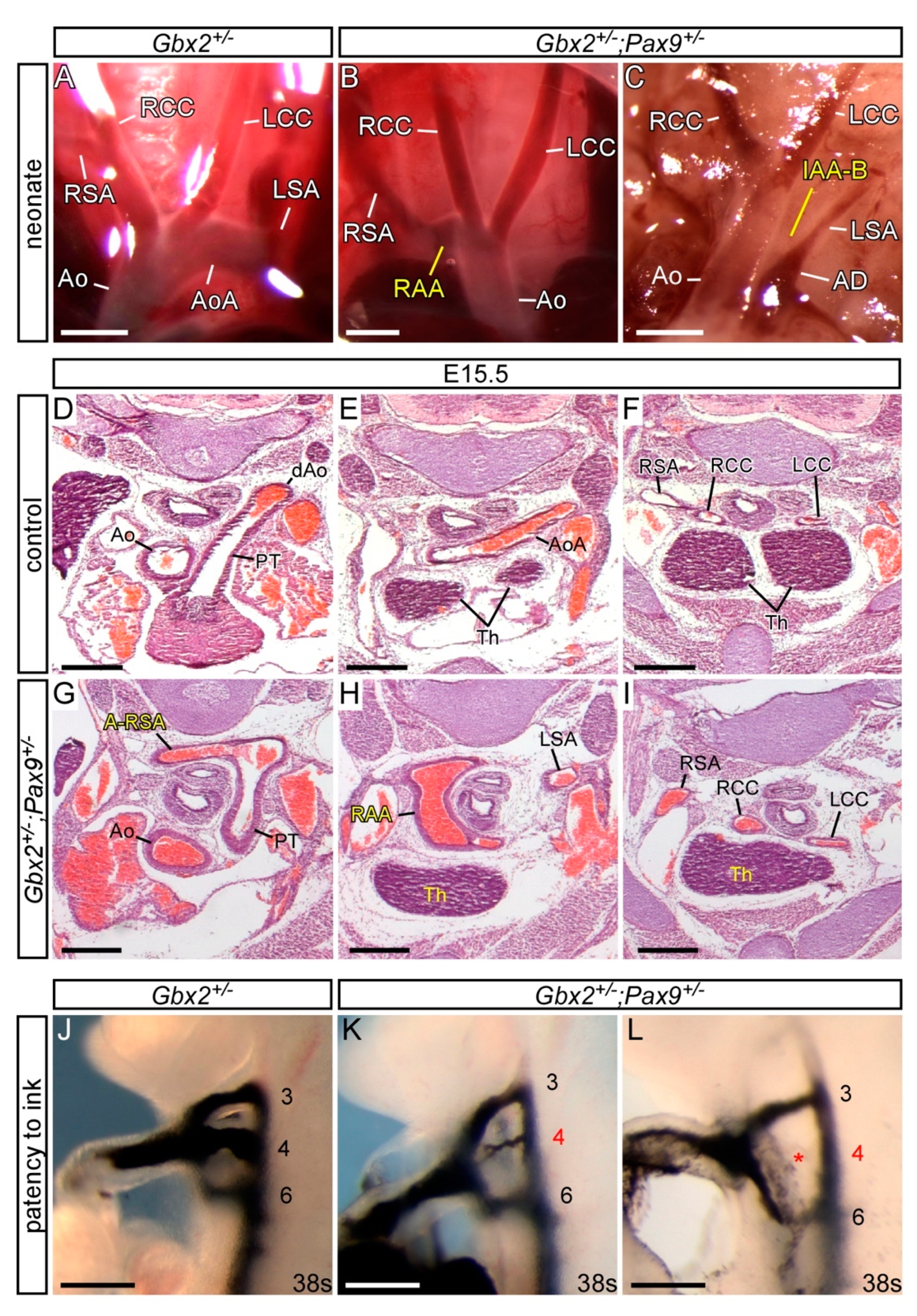
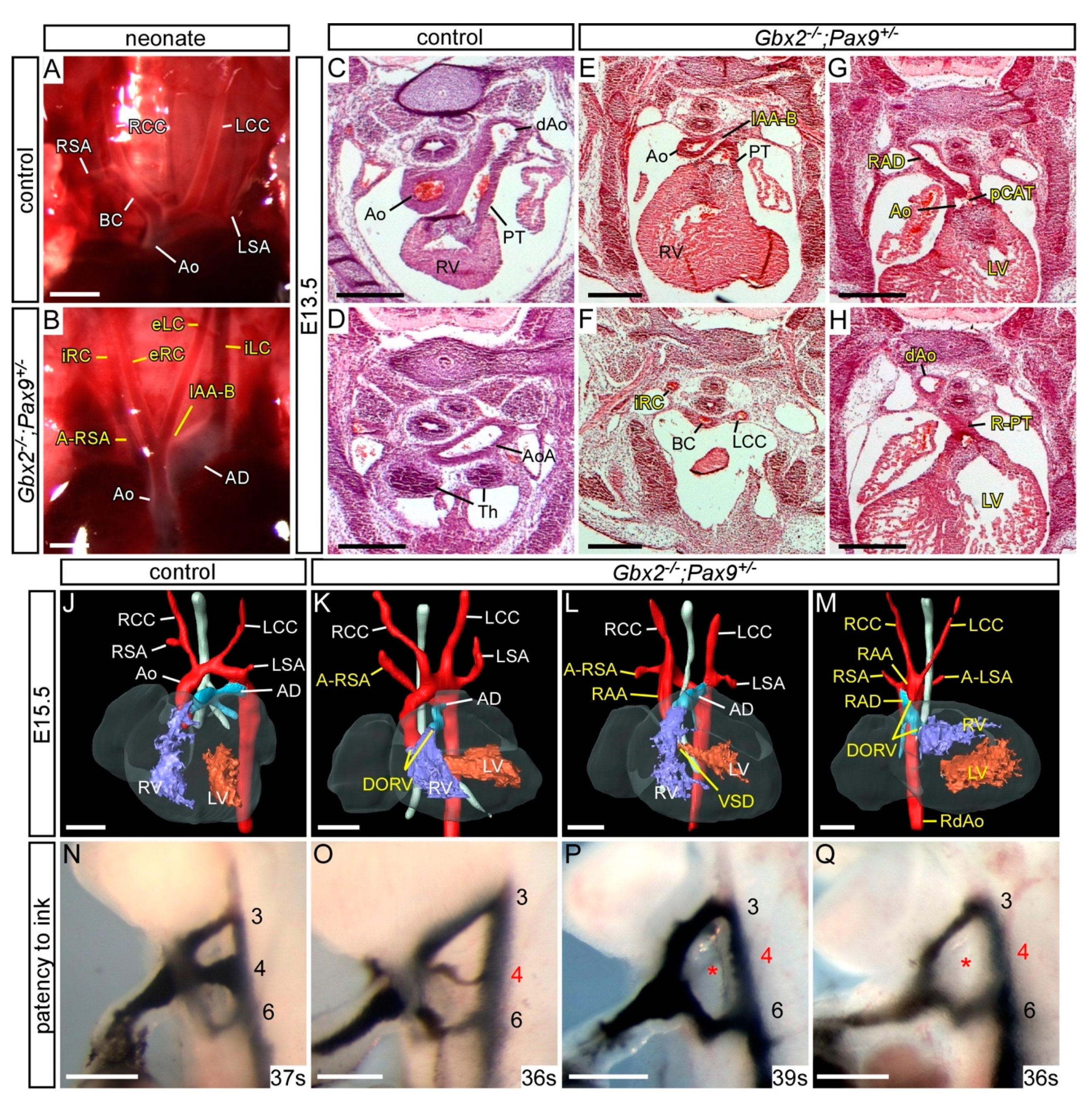
3.3. Genetic Interaction between Gbx2 and Pax9
3.4. Gbx2 and Pax9 Interact in the Pharyngeal Endoderm for Cardiovascular Development
3.5. Exploring a Genetic Interaction between Gbx2, Pax9 and Tbx1
4. Discussion
4.1. Gbx2-Null Mice
4.2. Genetic Interaction with Pax9
4.3. Gbx2 and Pax9 Interact in the Pharyngeal Endoderm
4.4. The Tbx1;Pax9 Double Heterozygous Phenotype is not Modulated by Gbx2 Haploinsufficiency
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andersen, T.A.; Troelsen Kde, L.; Larsen, L.A. Of mice and men: Molecular genetics of congenital heart disease. Cell. Mol. Life Sci. 2014, 71, 1327–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudjemline, Y.; Fermont, L.; Le Bidois, J.; Lyonnet, S.; Sidi, D.; Bonnet, D. Prevalence of 22q11 deletion in fetuses with conotruncal cardiac defects: A 6-year prospective study. J. Pediatr. 2001, 138, 520–524. [Google Scholar] [CrossRef]
- Papangeli, I.; Scambler, P. The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Furutani, Y.; Hamada, H.; Sasaki, T.; Asakawa, S.; Minoshima, S.; Ichida, F.; Joo, K.; Kimura, M.; Imamura, S.; et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003, 362, 1366–1373. [Google Scholar] [CrossRef]
- Zweier, C.; Sticht, H.; Aydin-Yaylagul, I.; Campbell, C.E.; Rauch, A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am. J. Hum. Genet. 2007, 80, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.G. The second heart field. Curr. Top. Dev. Biol. 2012, 100, 33–65. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.H.; Moorman, A.F.; Brown, N.A.; Bamforth, S.D.; Chaudhry, B.; Henderson, D.J.; Mohun, T.J. Normal and abnormal development of the heart. In Pediatric and Congenital Cardiology, Cardiac Surgery and Intensive Care; da Cruz, E., Ivy, D., Jaggers, J., Eds.; Springer: London, UK, 2014; pp. 151–177. [Google Scholar]
- Waldo, K.; Miyagawa-Tomita, S.; Kumiski, D.; Kirby, M.L. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: Aortic sac to ventricular septal closure. Dev. Biol. 1998, 196, 129–144. [Google Scholar] [CrossRef]
- Bajolle, F.; Zaffran, S.; Kelly, R.G.; Hadchouel, J.; Bonnet, D.; Brown, N.A.; Buckingham, M.E. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ. Res. 2006, 98, 421–428. [Google Scholar] [CrossRef]
- Anderson, R.H.; Wessels, A.; Vettukattil, J.J. Morphology and Morphogenesis of Atrioventricular Septal Defect With Common Atrioventricular Junction. World J. Pediatr. Congenit. Heart Surg. 2010, 1, 59–67. [Google Scholar] [CrossRef]
- Wang, X.; Chen, D.; Chen, K.; Jubran, A.; Ramirez, A.; Astrof, S. Endothelium in the pharyngeal arches 3, 4 and 6 is derived from the second heart field. Dev. Biol. 2017, 421, 108–117. [Google Scholar] [CrossRef]
- Graham, A.; Smith, A. Patterning the pharyngeal arches. Bioessays 2001, 23, 54–61. [Google Scholar] [CrossRef]
- Chapman, D.L.; Garvey, N.; Hancock, S.; Alexiou, M.; Agulnik, S.I.; Gibson-Brown, J.J.; Cebra-Thomas, J.; Bollag, R.J.; Silver, L.M.; Papaioannou, V.E. Expression of the T-box family genes,Tbx1-Tbx5, during early mouse development. Dev. Dyn. 1996, 206, 379–390. [Google Scholar] [CrossRef]
- Veitch, E.; Begbie, J.; Schilling, T.F.; Smith, M.M.; Graham, A. Pharyngeal arch patterning in the absence of neural crest. Curr. Biol. 1999, 9, 1481–1484. [Google Scholar] [CrossRef] [Green Version]
- Piotrowski, T.; Nusslein-Volhard, C. The endoderm plays an important role in patterning the segmented pharyngeal region in zebrafish (Danio rerio). Dev. Biol. 2000, 225, 339–356. [Google Scholar] [CrossRef] [Green Version]
- McCauley, D.W.; Bronner-Fraser, M. Neural crest contributions to the lamprey head. Development 2003, 130, 2317–2327. [Google Scholar] [CrossRef] [Green Version]
- Graham, A.; Okabe, M.; Quinlan, R. The role of the endoderm in the development and evolution of the pharyngeal arches. J. Anat. 2005, 207, 479–487. [Google Scholar] [CrossRef]
- Edlund, R.K.; Ohyama, T.; Kantarci, H.; Riley, B.B.; Groves, A.K. Foxi transcription factors promote pharyngeal arch development by regulating formation of FGF signaling centers. Dev. Biol. 2014, 390, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hasten, E.; Morrow, B.E. Tbx1 and Foxi3 genetically interact in the pharyngeal pouch endoderm in a mouse model for 22q11.2 deletion syndrome. PLoS Genet. 2019, 15, e1008301. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.; Kasah, S.; Mansour, S.L.; Morrow, B.; Basson, M.A. Endoderm-specific deletion of Tbx1 reveals an FGF-independent role for Tbx1 in pharyngeal apparatus morphogenesis. Dev. Dyn. 2014, 243, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Hiruma, T.; Nakajima, Y.; Nakamura, H. Development of pharyngeal arch arteries in early mouse embryo. J. Anat. 2002, 201, 15–29. [Google Scholar] [CrossRef]
- Bamforth, S.D.; Chaudhry, B.; Bennett, M.; Wilson, R.; Mohun, T.J.; Van Mierop, L.H.; Henderson, D.J.; Anderson, R.H. Clarification of the identity of the mammalian fifth pharyngeal arch artery. Clin. Anat. 2013, 26, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suntratonpipat, S.; Bamforth, S.D.; Johnson, A.L.; Noga, M.; Anderson, R.H.; Smallhorn, J.; Tham, E. Childhood presentation of interrupted aortic arch with persistent carotid ducts. World J. Pediatr. Congenit. Heart Surg. 2015, 6, 335–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivins, S.; van Lammerts Beuren, K.; Roberts, C.; James, C.; Lindsay, E.; Baldini, A.; Ataliotis, P.; Scambler, P.J. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev. Biol. 2005, 285, 554–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, H.M.; Stothard, C.A.; Shaikh Qureshi, W.M.; Kousa, A.I.; Briones-Leon, J.A.; Khasawneh, R.R.; O’Loughlin, C.; Sanders, R.; Mazzotta, S.; Dodds, R.; et al. Pax9 is required for cardiovascular development and interacts with Tbx1 in the pharyngeal endoderm to control 4th pharyngeal arch artery morphogenesis. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Neubuser, A.; Koseki, H.; Balling, R. Characterization and developmental expression of Pax9, a paired-box-containing gene related to Pax1. Dev. Biol. 1995, 170, 701–716. [Google Scholar] [CrossRef] [Green Version]
- Peters, H.; Neubuser, A.; Kratochwil, K.; Balling, R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998, 12, 2735–2747. [Google Scholar] [CrossRef] [Green Version]
- Calmont, A.; Ivins, S.; Van Bueren, K.L.; Papangeli, I.; Kyriakopoulou, V.; Andrews, W.D.; Martin, J.F.; Moon, A.M.; Illingworth, E.A.; Basson, M.A.; et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 2009, 136, 3173–3183. [Google Scholar] [CrossRef] [Green Version]
- Byrd, N.A.; Meyers, E.N. Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev. Biol. 2005, 284, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.H.; Joyner, A.L. Otx2 and Gbx2 are required for refinement and not induction of mid-hindbrain gene expression. Development 2001, 128, 4979–4991. [Google Scholar]
- Bouillet, P.; Chazaud, C.; Oulad-Abdelghani, M.; Dollé, P.; Chambon, P. Sequence and expression pattern of the Stra7 (Gbx-2) homeobox-containing gene induced by retinoic acid in P19 embryonal carcinoma cells. Dev. Dyn. 1995, 204, 372–382. [Google Scholar] [CrossRef]
- Li, J.Y.; Lao, Z.; Joyner, A.L. Changing requirements for Gbx2 in development of the cerebellum and maintenance of the mid/hindbrain organizer. Neuron 2002, 36, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Lewis, P.; Pevny, L.; McMahon, A.P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 2002, 119, S97–S101. [Google Scholar] [CrossRef]
- Jerome, L.A.; Papaioannou, V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001, 27, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Geyer, S.H.; Mohun, T.J.; Weninger, W.J. Visualizing Vertebrate Embryos with Episcopic 3D Imaging Techniques. Sci. World J. 2009, 9, 1423–1437. [Google Scholar] [CrossRef]
- Degenhardt, K.; Wright, A.C.; Horng, D.; Padmanabhan, A.; Epstein, J.A. Rapid 3D phenotyping of cardiovascular development in mouse embryos by micro-CT with iodine staining. Circ. Cardiovasc. Imaging 2010, 3, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Bamforth, S.D.; Schneider, J.E.; Bhattacharya, S. High-throughput analysis of mouse embryos by magnetic resonance imaging. Cold Spring Harb. Protoc. 2012, 2012, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.E.; Bose, J.; Bamforth, S.D.; Gruber, A.D.; Broadbent, C.; Clarke, K.; Neubauer, S.; Lengeling, A.; Bhattacharya, S. Identification of cardiac malformations in mice lacking Ptdsr using a novel high-throughput magnetic resonance imaging technique. BMC Dev. Biol. 2004, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Kapadia, H.; Feng, J.Q.; Raghow, R.; Peters, H.; D’Souza, R.N. Functional Consequences of Interactions between Pax9 and Msx1 Genes in Normal and Abnormal Tooth Development. J. Biol. Chem. 2006, 281, 18363–18369. [Google Scholar] [CrossRef]
- Wassarman, K.M.; Lewandoski, M.; Campbell, K.; Joyner, A.L.; Rubenstein, J.L.; Martinez, S.; Martin, G.R. Specification of the anterior hindbrain and establishment of a normal mid/hindbrain organizer is dependent on Gbx2 gene function. Development 1997, 124, 2923–2934. [Google Scholar]
- Caprio, C.; Baldini, A. p53 suppression partially rescues the mutant phenotype in mouse models of DiGeorge syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 13385–13390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, J.; Norris, D.P.; Robertson, E.J. Nodal activity in the node governs left-right asymmetry. Genes Dev. 2002, 16, 2339–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Praagh, R.; Van Praagh, S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am. J. Cardiol. 1965, 16, 406–425. [Google Scholar] [CrossRef]
- Olson, E.N.; Arnold, H.H.; Rigby, P.W.J.; Wold, B.J. Know Your Neighbors: Three Phenotypes in Null Mutants of the Myogenic bHLH Gene MRF4. Cell 1996, 85, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, E.A.; Baldini, A. Recovery from arterial growth delay reduces penetrance of cardiovascular defects in mice deleted for the DiGeorge syndrome region. Hum. Mol. Genet. 2001, 10, 997–1002. [Google Scholar] [CrossRef] [Green Version]
- Guris, D.L.; Duester, G.; Papaioannou, V.E.; Imamoto, A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell. 2006, 10, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Randall, V.; McCue, K.; Roberts, C.; Kyriakopoulou, V.; Beddow, S.; Barrett, A.N.; Vitelli, F.; Prescott, K.; Shaw-Smith, C.; Devriendt, K.; et al. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J. Clin. Investig. 2009, 119, 3301–3310. [Google Scholar] [CrossRef] [Green Version]
- Ryckebusch, L.; Bertrand, N.; Mesbah, K.; Bajolle, F.; Niederreither, K.; Kelly, R.G.; Zaffran, S. Decreased Levels of Embryonic Retinoic Acid Synthesis Accelerate Recovery From Arterial Growth Delay in a Mouse Model of DiGeorge Syndrome. Circ. Res. 2010, 106, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, H.; Hamada, H. The left-right axis in the mouse: From origin to morphology. Development 2006, 133, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Cox, C.J.; Espinoza, H.M.; McWilliams, B.; Chappell, K.; Morton, L.; Hjalt, T.A.; Semina, E.V.; Amendt, B.A. Differential regulation of gene expression by PITX2 isoforms. J. Biol. Chem. 2002, 277, 25001–25010. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Liu, W.; Lu, M.F.; Brown, N.A.; Martin, J.F. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development 2001, 128, 2039–2048. [Google Scholar] [PubMed]
- Franco, D.; Campione, M. The role of Pitx2 during cardiac development. Linking left-right signaling and congenital heart diseases. Trends Cardiovasc. Med. 2003, 13, 157–163. [Google Scholar] [CrossRef]
- Liu, C.; Liu, W.; Palie, J.; Lu, M.F.; Brown, N.A.; Martin, J.F. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development 2002, 129, 5081–5091. [Google Scholar] [PubMed]
- Ai, D.; Liu, W.; Ma, L.; Dong, F.; Lu, M.F.; Wang, D.; Verzi, M.P.; Cai, C.; Gage, P.J.; Evans, S.; et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev. Biol. 2006, 296, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Snarr, B.S.; O’Neal, J.L.; Chintalapudi, M.R.; Wirrig, E.E.; Phelps, A.L.; Kubalak, S.W.; Wessels, A. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ. Res. 2007, 101, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Unolt, M.; Versacci, P.; Anaclerio, S.; Lambiase, C.; Calcagni, G.; Trezzi, M.; Carotti, A.; Crowley, T.B.; Zackai, E.H.; Goldmuntz, E.; et al. Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am. J. Med. Genet. Part A 2018, 176, 2087–2098. [Google Scholar] [CrossRef]
- Merscher, S.; Funke, B.; Epstein, J.A.; Heyer, J.; Puech, A.; Lu, M.M.; Xavier, R.J.; Demay, M.B.; Russell, R.G.; Factor, S.; et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 2001, 104, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Morishima, M.; Wylie, J.N.; Schwartz, R.J.; Bruneau, B.G.; Lindsay, E.A.; Baldini, A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 2004, 131, 3217–3227. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Ogden, L.A.; Talbot, A.; Evans, S.; Cai, C.L.; Black, B.L.; Frank, D.U.; Moon, A.M. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 2006, 133, 2419–2433. [Google Scholar] [CrossRef] [Green Version]
- Conway, S.J.; Henderson, D.J.; Copp, A.J. Pax3 is required for cardiac neural crest migration in the mouse: Evidence from the splotch (Sp2H) mutant. Development 1997, 124, 505–514. [Google Scholar] [PubMed]
- Keyte, A.; Hutson, M.R. The neural crest in cardiac congenital anomalies. Differentiation 2012, 84, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, L.; Chaudhry, B.; Hildreth, V.; Webb, S.; Henderson, D.J. Dual role for neural crest cells during outflow tract septation in the neural crest-deficient mutant Splotch(2H). J. Anat. 2009, 214, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.J.; et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Baldini, A. In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum. Mol. Genet. 2008, 17, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Dupays, L.; Mohun, T. Spatiotemporal regulation of enhancers during cardiogenesis. Cell. Mol. Life Sci. CMLS 2017, 74, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
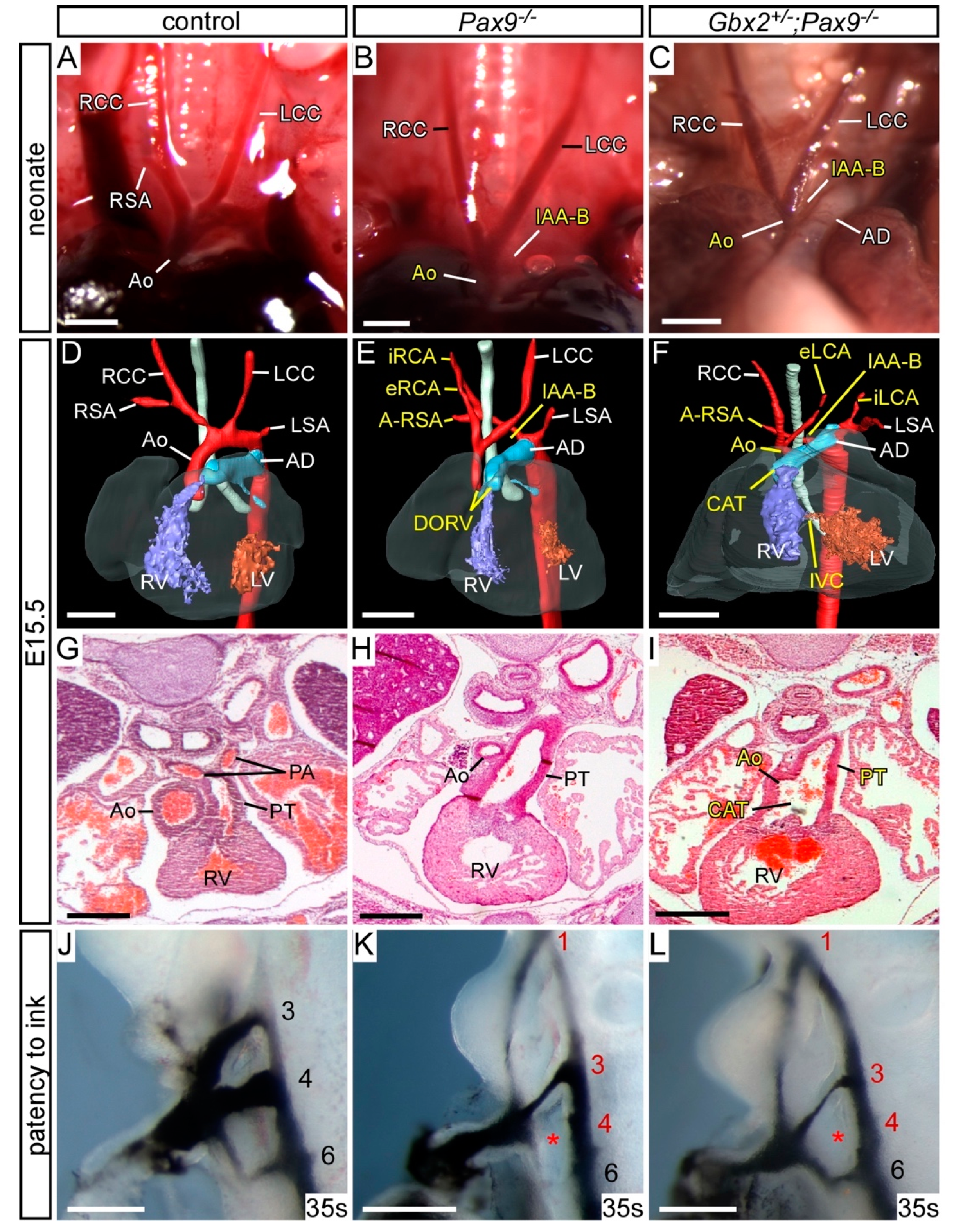{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | n | Abnormal | VSD | AVSD | DORV + IVC | CAT | RAA/RAD +/or A-SA a | IAA-B +/− A-RSA | Absent CC b | L-R Defect c |
|---|---|---|---|---|---|---|---|---|---|---|
| Gbx2−/− | 25 | 16 (64%) | 2 (8%) | 0 | 10 (40%) | 0 | 13 (52%) | 0 | 0 | 9 (36%) |
| Gbx2+/−; Pax9+/− | 28 | 6 (21%) | 0 | 0 | 0 | 0 | 4 (14%) | 2 (7%) | 0 | 0 |
| Gbx2−/−; Pax9+/− | 14 | 14 (100%) * | 3 (21%) | 0 | 7 (50%) | 1 (7%) | 8 (57%) | 5 ** (36%) | 2 (16%) | 6 (43%) |
| Pax9−/− | 9 | 9 (100%) | 2 (22%) | 0 | 3 (44%) | 0 | 0 | 8 (89%) | 5 (56%) | 0 |
| Gbx2+/−; Pax9−/− | 9 | 9 (100%) | 2 (22%) | 0 | 5 (56%) | 2 (22%) | 1 (11%) | 8 (89%) | 6 (67%) | 0 |
| Gbx2−/−; Pax9−/− | 2 | 2 (100%) | 0 | 1 (50%) | 0 | 1 (50%) | 1 (50%) | 1 (50%) | 1 (50%) | 0 |
| Gbx2−/flox; Pax9Cre | 14 | 3 (21%) | 0 | 0 | 0 | 0 | 3 (21%) | 0 | 1 (7%) | 0 |
| Bilateral Defects | ||||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | n | PAA | Abnormal (%) | Unilateral Defect | Bilateral Defect | Present | Hypo/Int/Abs | Absent |
| Gbx2+/− | 44 | 4 | 3 (7%) | 3 (7%) | 0 | - | - | - |
| Gbx2+/−; Pax9+/− | 63 | 4 | 20 (32%) | 13 (21%) | 7 (11%) | - | 3 | 4 |
| Gbx2−/− | 17 | 4 | 12 (71%) | 7 (41%) | 5 (29%) | - | 3 | 2 |
| Gbx2−/−; Pax9+/− | 7 | 4 | 7 (100%) | 1 (14%) | 6 (86%) * | - | 2 | 4 |
| Pax9−/− a | 22 | 1 | 13 (59%) | 1 (5%) | 12 (55%) | 11 | 1 | - |
| 2 | 8 (36%) | 3 (14%) | 5 (23%) | 4 | 1 | - | ||
| 3 | 17 (77%) | 3 (14%) | 14 (64%) | - | 10 | 4 | ||
| 4 | 22 (100%) | 1 (5%) | 21 (95%) | - | 3 | 18 | ||
| Gbx2+/−; Pax9−/− | 6 | 1 | 6 (100%) | 0 | 6 (100%) | 6 | - | - |
| 2 | 2 (33%) | 2 (33%) | 0 | - | - | - | ||
| 3 | 6 (100%) | 0 | 6 (100%) | - | 1 | 5 | ||
| 4 | 6 (100%) | 0 | 6 (100%) | - | - | 6 | ||
| Gbx2−/−; Pax9−/− | 1 | 4 | 1 (100%) | 0 | 1 (100%) | - | - | 1 |
| Gbx2+/flox; Pax9Cre | 19 | 4 | 2 (10%) | 1 (5%) | 1 (5%) | - | - | 1 |
| Gbx2−/flox; Pax9Cre | 14 | 4 | 7 (50%) * | 5 (36%) | 2 (14%) | - | 1 | 1 |
| Genotype | n | VSD | RAA +/or A-RSA | IAA-B +/− A-RSA | 4th PAA Defect |
|---|---|---|---|---|---|
| Tbx1+/− a | 21 | 2 (10%) | 13 (62%) | 1 (5%) | 14 (67%) |
| Tbx1+/−; Pax9+/− b | 24 | 5 (21%) | 9 (38%) | 15 (63%) ** | 24 (100%) |
| Tbx1+/−; Gbx2+/− | 10 | 3(30%) | 2(20%) | 1(10%) | 3(30%) |
| Tbx1+/−; Gbx2+/−; Pax9+/− | 8 | 3 (38%) | 4 (50%) | 4 (50%) | 8 (100%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stothard, C.A.; Mazzotta, S.; Vyas, A.; Schneider, J.E.; Mohun, T.J.; Henderson, D.J.; Phillips, H.M.; Bamforth, S.D. Pax9 and Gbx2 Interact in the Pharyngeal Endoderm to Control Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 20. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7020020
Stothard CA, Mazzotta S, Vyas A, Schneider JE, Mohun TJ, Henderson DJ, Phillips HM, Bamforth SD. Pax9 and Gbx2 Interact in the Pharyngeal Endoderm to Control Cardiovascular Development. Journal of Cardiovascular Development and Disease. 2020; 7(2):20. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7020020
Chicago/Turabian StyleStothard, Catherine A., Silvia Mazzotta, Arjun Vyas, Jurgen E. Schneider, Timothy J. Mohun, Deborah J. Henderson, Helen M. Phillips, and Simon D. Bamforth. 2020. "Pax9 and Gbx2 Interact in the Pharyngeal Endoderm to Control Cardiovascular Development" Journal of Cardiovascular Development and Disease 7, no. 2: 20. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7020020