Early Embryonic Expression of AP-2α Is Critical for Cardiovascular Development

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Breeding
2.3. Imaging
2.4. Immunohistochemistry
2.5. RT-PCR
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
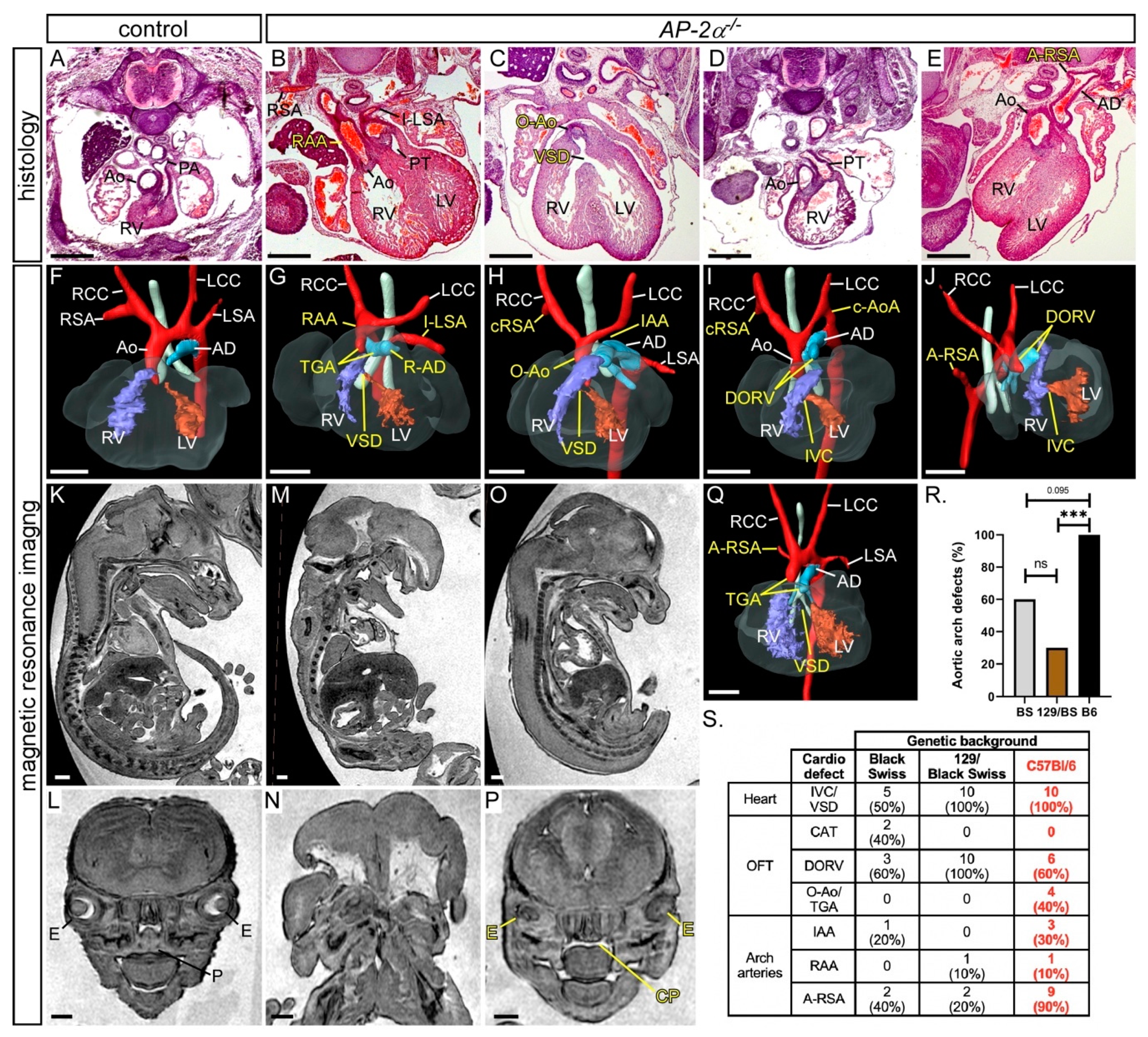
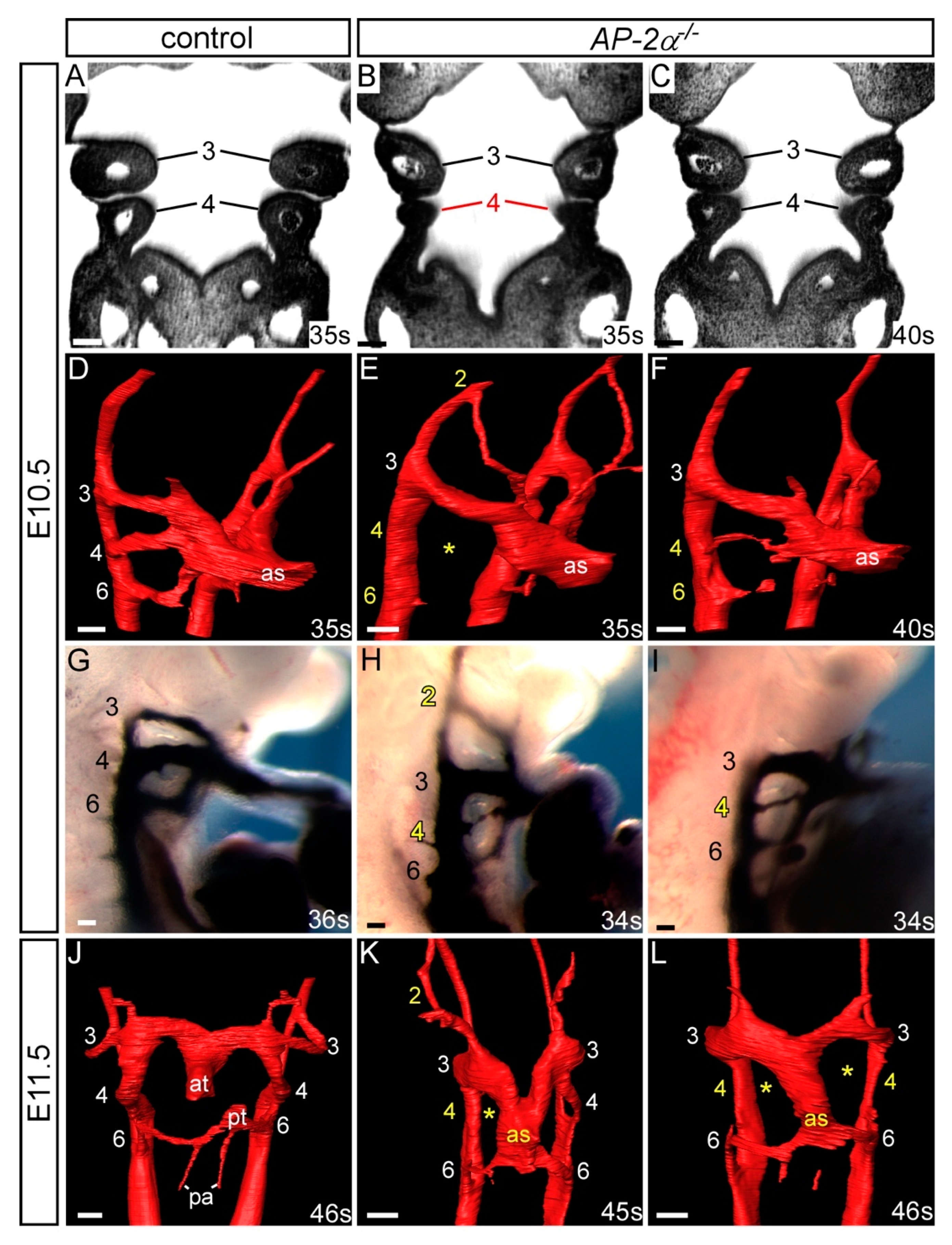
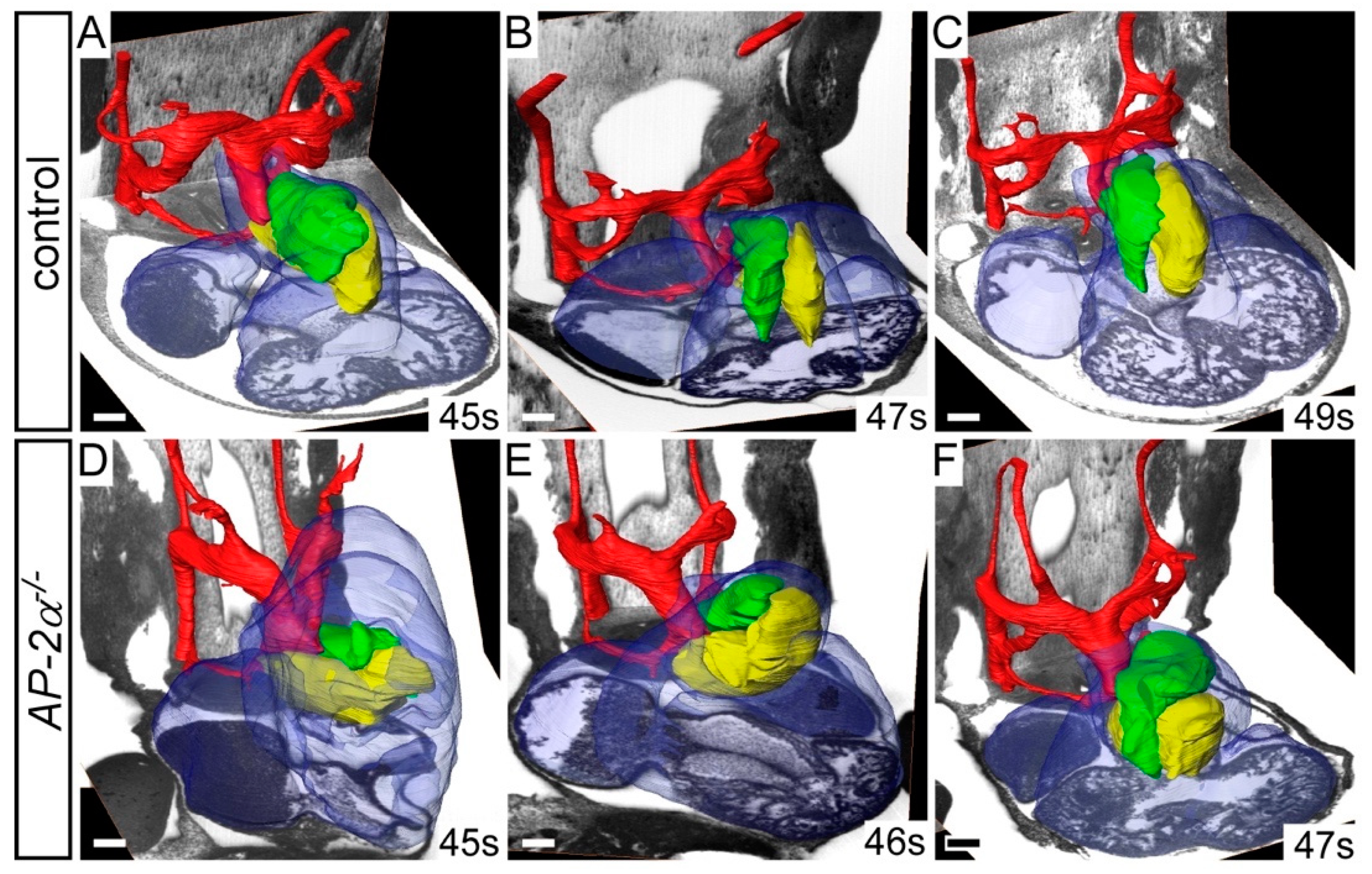
3.1. Cardiovascular Phenotype in C57Bl/6J AP-2α−/− Embryos
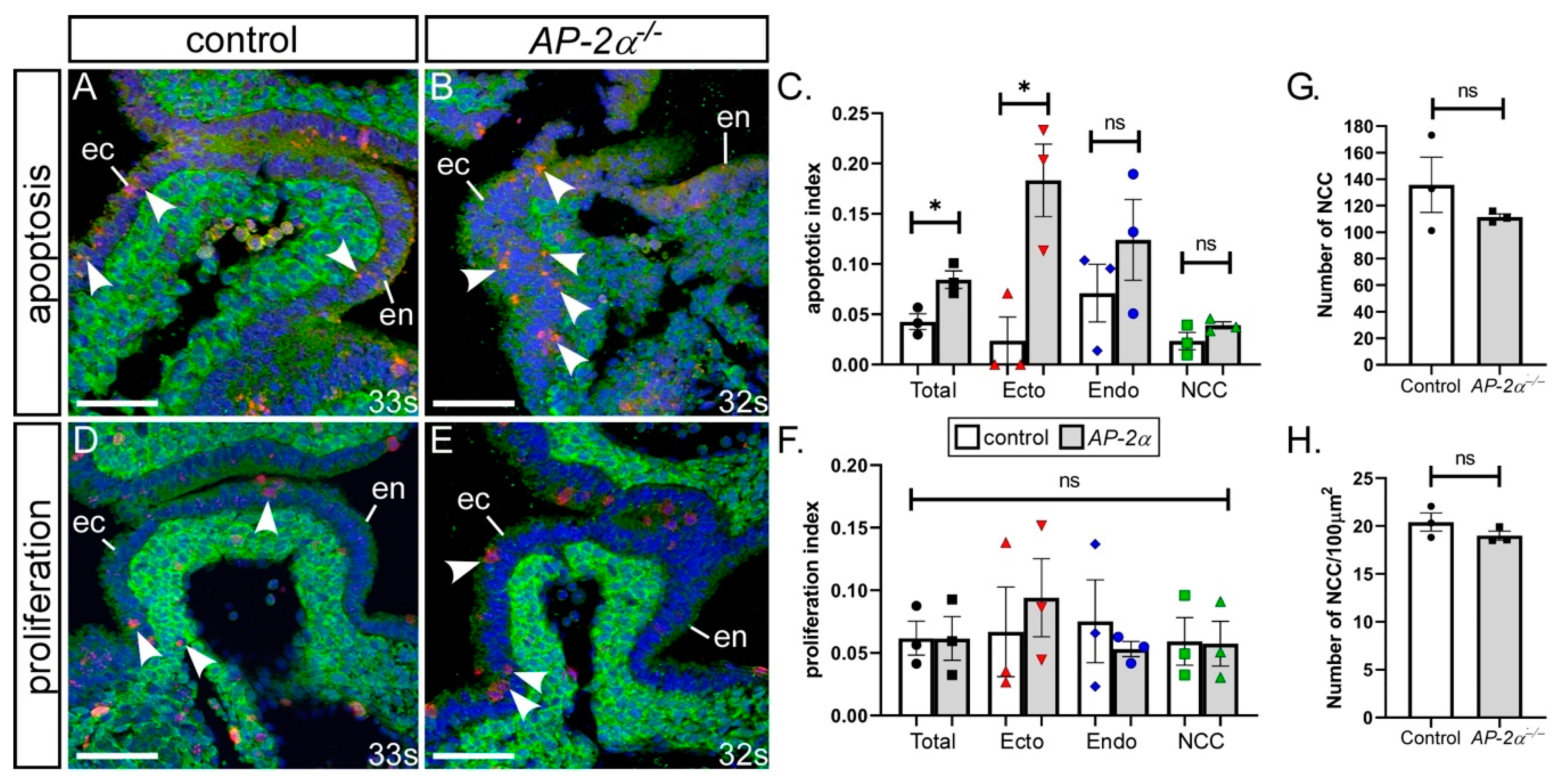
3.2. Cell Fates in AP-2α−/− Embryos
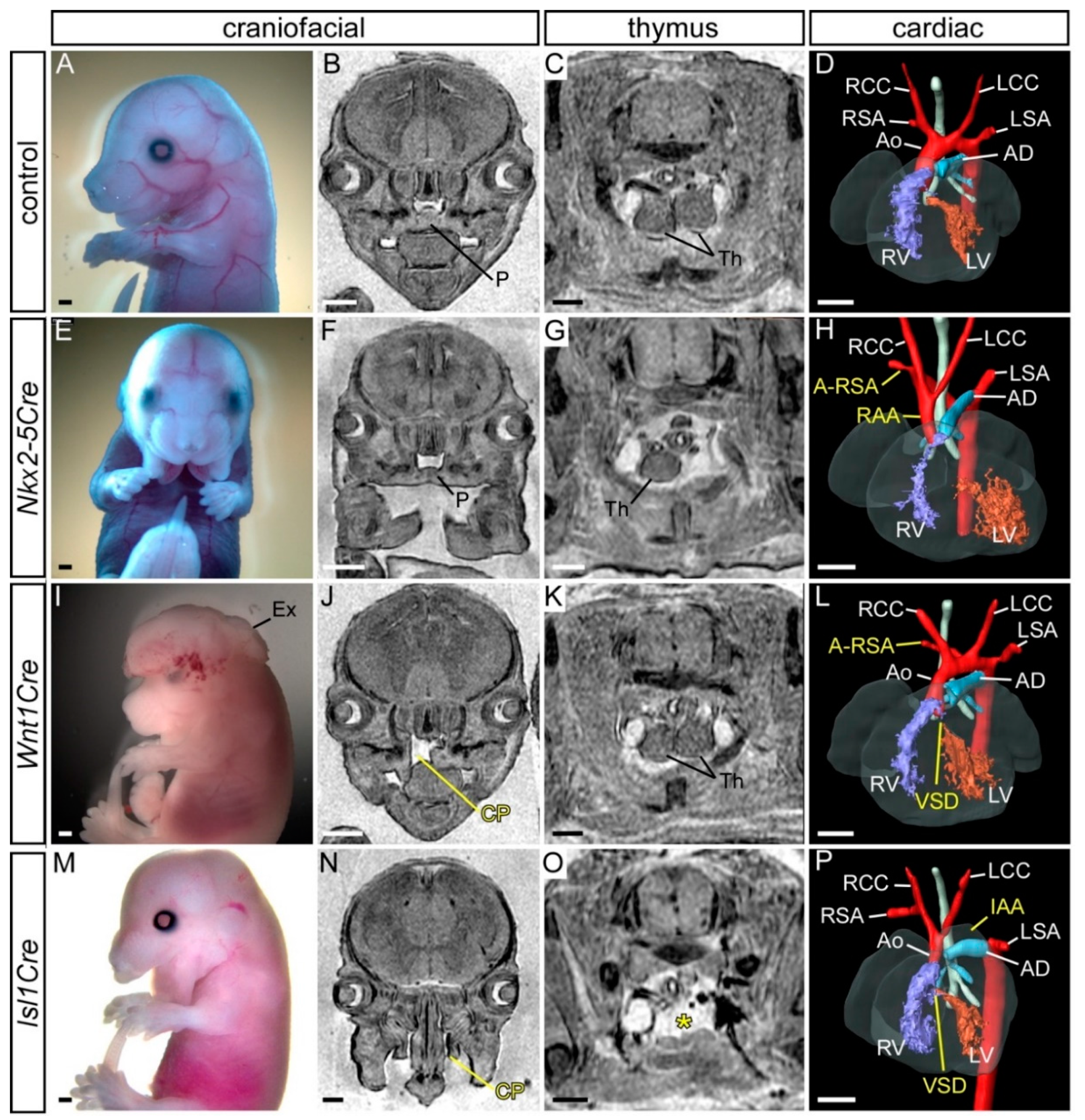
3.3. Conditional Deletion of AP-2α from the Pharyngeal Surface Ectoderm
3.4. Conditional Deletion of AP-2α from the Neural Crest
3.5. Simultaneous Deletion of AP-2α from the Surface Ectoderm and the Neural Crest
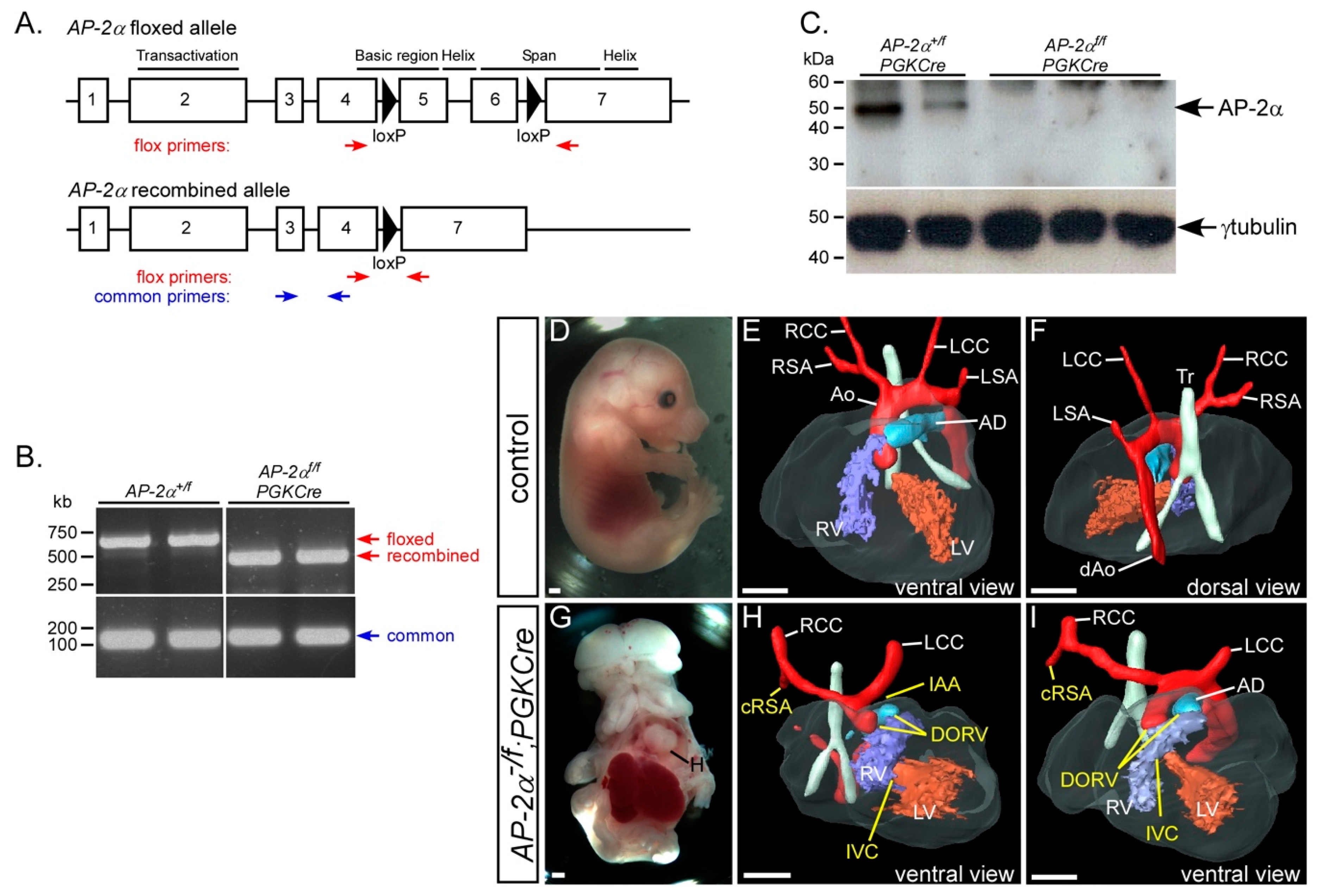
3.6. Early Embryonic Recombination of the AP-2αflox Allele
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoffman, J.I.E. Incidence, mortality and natural history. In Paediatric Cardiology; Anderson, R.H., Baker, E.J., Macartney, F.J., Rigby, M.L., Shinebourne, E.A., Tynan, M., Eds.; Churchill Livingstone: Edinburgh, London, UK, 2002; pp. 111–139. [Google Scholar]
- Burn, J.; Goodship, J. Congenital heart disease. In Principles and Practice of Medical Genetics; Rimoin, D.L., Connor, J.M., Pyeritz, R.E., Korf, B.R., Eds.; Churchill Livingstone: Edinburgh, London, UK, 2002. [Google Scholar]
- Anderson, R.H.; Brown, N.A.; Bamforth, S.D.; Chaudhry, B.; Henderson, D.J.; Mohun, T.J. Development of the Outflow Tract; Oxford University Press (OUP): Oxford, UK, 2018; pp. 226–239. [Google Scholar]
- Anderson, R.H.; Chaudhry, B.; Mohun, T.J.; Bamforth, S.D.; Hoyland, D.; Phillips, H.M.; Webb, S.; Moorman, A.F.; Brown, N.A.; Henderson, D.J. Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc. Res. 2012, 95, 108–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, A. Development of the pharyngeal arches. Am. J. Med Genet. 2003, 119, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Hiruma, T.; Nakajima, Y.; Nakamura, H. Development of pharyngeal arch arteries in early mouse embryo. J. Anat. 2002, 201, 15–29. [Google Scholar] [CrossRef]
- Bamforth, S.D.; Chaudhry, B.; Bennett, M.; Wilson, R.; Mohun, T.J.; Van Mierop, L.H.; Henderson, D.J.; Anderson, R.H. Clarification of the identity of the mammalian fifth pharyngeal arch artery. Clin. Anat. 2012, 26, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W.; Lott, I.T.; et al. Retinoic Acid Embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef]
- Diez-Pardo, J.A.; Tovar, J.A.; Yu, J.; Gonzalez, S.; Martinez, L. Effects of retinoic acid on the neural crest-controlled organs of fetal rats. Pediatr. Surg. Int. 2003, 19, 355–358. [Google Scholar] [CrossRef]
- Hogers, B.; DeRuiter, M.C.; Groot, A.G.-D.; E Poelmann, R. Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc. Res. 1999, 41, 87–99. [Google Scholar] [CrossRef]
- Yashiro, K.; Shiratori, H.; Hamada, H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 2007, 450, 285–288. [Google Scholar] [CrossRef]
- Bentham, J.; Bhattacharya, S. Genetic Mechanisms Controlling Cardiovascular Development. Ann. N. Y. Acad. Sci. 2008, 1123, 10–19. [Google Scholar] [CrossRef]
- Porras, D.; Brown, C.B. Temporal–spatial ablation of neural crest in the mouse results in cardiovascular defects. Dev. Dyn. 2007, 237, 153–162. [Google Scholar] [CrossRef]
- Olaopa, M.; Zhou, H.-M.; Snider, P.; Wang, J.; Schwartz, R.J.; Moon, A.M.; Conway, S.J. Pax3 is essential for normal cardiac neural crest morphogenesis but is not required during migration nor outflow tract septation. Dev. Boil. 2011, 356, 308–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stothard, C.A.; Mazzotta, S.; Vyas, A.; Schneider, J.E.; Mohun, T.J.; Henderson, D.J.; Phillips, H.M.; Bamforth, S.D. Pax9 and Gbx2 Interact in the Pharyngeal Endoderm to Control Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.M.; Stothard, C.A.; Qureshi, W.M.S.; Kousa, A.I.; Briones-Leon, J.A.; Khasawneh, R.R.; O’Loughlin, C.; Sanders, R.; Mazzotta, S.; Dodds, R.; et al. Pax9 is required for cardiovascular development and interacts with Tbx1 in the pharyngeal endoderm to control 4th pharyngeal arch artery morphogenesis. Development 2019, 146, dev177618. [Google Scholar] [CrossRef] [Green Version]
- Calmont, A.; Ivins, S.; Van Bueren, K.L.; Papangeli, I.; Kyriakopoulou, V.; Andrews, W.D.; Martin, J.F.; Moon, A.M.; Illingworth, E.A.; Basson, M.A.; et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 2009, 136, 3173–3183. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Haynie, J.; Yang, X.; Han, B.; Kiatchoosakun, S.; Restivo, J.; Yuan, S.; Prabhakar, N.R.; Herrup, K.; Conlon, R.A.; et al. The essential role of Cited2, a negative regulator for HIF-1, in heart development and neurulation. Proc. Natl. Acad. Sci. USA 2002, 99, 10488–10493. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Barbera, J.P.; Rodríguez, T.A.; Greene, N.D.; Weninger, W.J.; Simeone, A.; Copp, A.J.; Beddington, R.S.P.; Dunwoodie, S.L. Folic acid prevents exencephaly in Cited2 deficient mice. Hum. Mol. Genet. 2002, 11, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamforth, S.D.; Bragança, J.; Eloranta, J.J.; Murdoch, J.N.; Marques, F.I.; Kranc, K.R.; Farza, H.; Henderson, D.J.; Hurst, H.C.; Bhattacharya, S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 2001, 29, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.D.; Bragança, J.; Farthing, C.R.; Schneider, J.E.; Broadbent, C.; Michell, A.C.; Clarke, K.; Neubauer, S.; Norris, D.; A Brown, N.; et al. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 2004, 36, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hagopian-Donaldson, S.; Serbedzija, G.; Elsemore, J.; Plehn-Dujowich, D.; McMahon, A.P.; Flavell, R.A.; Williams, T. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 1996, 381, 238–241. [Google Scholar] [CrossRef]
- Schorle, H.; Meier, P.; Buchert, M.; Jaenisch, R.; Mitchell, P.J. Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 1996, 381, 235–238. [Google Scholar] [CrossRef]
- Brewer, S.; Jiang, X.; Donaldson, S.; Williams, T.; Sucov, H.M. Requirement for AP-2α in cardiac outflow tract morphogenesis. Mech. Dev. 2002, 110, 139–149. [Google Scholar] [CrossRef]
- Weh, E.; Reis, L.M.; Happ, H.C.; Levin, A.V.; Wheeler, P.G.; David, K.L.; Carney, E.; Angle, B.; Hauser, N.; Semina, E.V. Whole exome sequence analysis of Peters anomaly. Qual. Life Res. 2014, 133, 1497–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midha, N.; Sidhu, T.; Chaturvedi, N.; Sinha, R.; Shende, D.R.; Dada, T.; Gupta, V.; Sihota, R. Systemic Associations of Childhood Glaucoma: A Review. J. Pediatr. Ophthalmol. Strabismus 2018, 55, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Traboulsi, E.I.; Maumenee, I.H. Peters’ Anomaly and Associated Congenital Malformations. Arch. Ophthalmol. 1992, 110, 1739–1742. [Google Scholar] [CrossRef]
- Milunsky, J.M.; Maher, T.M.; Zhao, G.; Wang, Z.; Mulliken, J.B.; Chitayat, D.; Clemens, M.; Stalker, H.J.; Bauer, M.; Burch, M.; et al. Genotype-phenotype analysis of the branchio-oculo-facial syndrome. Am. J. Med Genet. Part A 2010, 155, 22–32. [Google Scholar] [CrossRef]
- Reiber, J.; Sznajer, Y.; Posteguillo, E.G.; Müller, D.; Lyonnet, S.; Baumann, C.; Just, W. Additional clinical and molecular analyses ofTFAP2Ain patients with the branchio-oculo-facial syndrome. Am. J. Med Genet. Part A 2010, 152, 994–999. [Google Scholar] [CrossRef]
- Gestri, G.; Osborne, R.J.; Wyatt, A.W.; Gerrelli, D.; Gribble, S.; Stewart, H.; Fryer, A.; Bunyan, D.J.; Prescott, K.; Collin, J.R.O.; et al. Reduced TFAP2A function causes variable optic fissure closure and retinal defects and sensitizes eye development to mutations in other morphogenetic regulators. Qual. Life Res. 2009, 126, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Hammer, S.; Toenjes, M.; Lange, M.; Fischer, J.J.; Dunkel, I.; Mebus, S.; Grimm, C.; Hetzer, R.; Berger, F.; Sperling, S. Characterization of TBX20 in human hearts and its regulation by TFAP2. J. Cell. Biochem. 2008, 104, 1022–1033. [Google Scholar] [CrossRef]
- Satoda, M.; Zhao, F.; Diaz, G.A.; Burn, J.; Goodship, J.; Davidson, H.R.; Pierpont, M.E.M.; Gelb, B.D. Mutations in TFAP2B cause Char syndrome, a familial form of patent ductus arteriosus. Nat. Genet. 2000, 25, 42–46. [Google Scholar] [CrossRef]
- Arkell, R.; Beddington, R.S. BMP-7 influences pattern and growth of the developing hindbrain of mouse embryos. Developement 1997, 124, 1–12. [Google Scholar]
- Mitchell, P.J.; Timmons, P.M.; Hebert, J.M.; Rigby, P.W.; Tjian, R. Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 1991, 5, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, M.R.; Kirby, M.L. Neural crest and cardiovascular development: A 20-year perspective. Birth Defects Res. Part C: Embryo Today: Rev. 2003, 69, 2–13. [Google Scholar] [CrossRef]
- Brewer, S.; Feng, W.; Huang, J.; Sullivan, S.; Williams, T. Wnt1-Cre-mediated deletion of AP-2α causes multiple neural crest-related defects. Dev. Boil. 2004, 267, 135–152. [Google Scholar] [CrossRef] [Green Version]
- Hébert, J.M.; McConnell, S.K. Targeting of cre to the Foxg1 (BF-1) Locus Mediates loxP Recombination in the Telencephalon and Other Developing Head Structures. Dev. Boil. 2000, 222, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, K.A.; DeMayo, F.J.; Braun, R.M.; Reecy, J.M.; Schwartz, R.J. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genes 2001, 31, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Danielian, P.; Muccino, D.; Rowitch, D.H.; Michael, S.K.; McMahon, A.P. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Boil. 1998, 8, 1323–S2. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cai, C.-L.; Lin, L.; Qyang, Y.; Chung, C.; Monteiro, R.; Mummery, C.L.; Fishman, G.I.; Cogen, A.; Evans, S.M. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 2006, 133, 1575–1585. [Google Scholar] [CrossRef] [Green Version]
- Lallemand, Y.; Luria, V.; Haffner-Krausz, R.; Lonai, P. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res. 1998, 7, 105–112. [Google Scholar] [CrossRef]
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef]
- Srinivas, S.; Watanabe, T.; Lin, C.-S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Boil. 2001, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Markel, P.; Shu, P.; Ebeling, C.; Carlson, G.A.; Nagle, D.L.; Smutko, J.S.; Moore, K.J. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat. Genet. 1997, 17, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.D.; Schneider, J.E.; Bhattacharya, S. High-Throughput Analysis of Mouse Embryos by Magnetic Resonance Imaging. Cold Spring Harb. Protoc. 2012, 2012, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, S.; Mohun, T.J.; Weninger, W.J. Visualizing Vertebrate Embryos with Episcopic 3D Imaging Techniques. Sci. World J. 2009, 9, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Weninger, W.J.; Mohun, T.J. Three-dimensional analysis of molecular signals with episcopic imaging techniques. Methods Mol. Biol. 2007, 411, 35–46. [Google Scholar] [PubMed]
- Johnson, A.-L.; Bamforth, S.D. Molecular Pathways and Animal Models of d-Transposition of the Great Arteries. In Congenital Heart Diseases: The Broken Heart; Rickert-Sperling, S., Kelly, R.G., Driscoll, D.J., Eds.; Springer Vienna: Vienna, Austria, 2016; pp. 449–458. [Google Scholar] [CrossRef]
- Unolt, M.; Putotto, C.; Silvestri, L.M.; Marino, D.; Scarabotti, A.; Massaccesi, V.; Caiaro, A.; Versacci, P.; Marino, B. Transposition of great arteries: New insights into the pathogenesis. Front. Pediatr. 2013, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Cerrato, F.; Xu, H.; Vitelli, F.; Morishima, M.; Vincentz, J.; Furuta, Y.; Ma, L.; Martin, J.F.; Baldini, A.; et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 2005, 132, 5307–5315. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Ogden, L.A.; Talbot, A.; Evans, S.; Cai, C.-L.; Black, B.L.; Frank, D.U.; Moon, A.M. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 2006, 133, 2419–2433. [Google Scholar] [CrossRef] [Green Version]
- Engleka, K.A.; Manderfield, L.J.; Brust, R.; Li, L.; Cohen, A.; Dymecki, S.M.; Epstein, J.A. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ. Res. 2012, 110, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, S.T.; Bamforth, S.D.; Chen, C.-M.; Farthing, C.R.; Franklyn, A.; Broadbent, C.; Schneider, J.E.; Saga, Y.; Lewandoski, M.; Bhattacharya, S. Epiblastic Cited2 deficiency results in cardiac phenotypic heterogeneity and provides a mechanism for haploinsufficiency. Cardiovasc. Res. 2008, 79, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Van Otterloo, E.; Li, H.; Jones, K.L.; Williams, T. AP-2α and AP-2β cooperatively orchestrate homeobox gene expression during branchial arch patterning. Development 2018, 145, dev157438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, A.E.; Vasudevan, H.N.; O’Neill, A.K.; Soriano, P.; Bush, J.O. The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Boil. 2013, 379, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Astrof, S. Neural crest cell-autonomous roles of fibronectin in cardiovascular development. Development 2015, 143, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, E.; Arnold, H.-H.; Rigby, P.; Wold, B. Know Your Neighbors: Three Phenotypes in Null Mutants of the Myogenic bHLH Gene MRF4. Cell 1996, 85, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Bergwerff, M.; DeRuiter, M.C.; Hall, S.; E Poelmann, R.; Groot, A.G.-D. Unique vascular morphology of the fourth aortic arches: Possible implications for pathogenesis of type-B aortic arch interruption and anomalous right subclavian artery. Cardiovasc. Res. 1999, 44, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, E.A.; Vitelli, F.; Su, H.; Morishima, M.; Huynh, T.; Pramparo, T.; Jurecic, V.; Ogunrinu, G.; Sutherland, H.F.; Scambler, P.; et al. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 2001, 410, 97–101. [Google Scholar] [CrossRef]
- Randall, V.; McCue, K.; Roberts, C.; Kyriakopoulou, V.; Beddow, S.; Barrett, A.; Vitelli, F.; Prescott, K.; Shaw-Smith, C.; Devriendt, K.; et al. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J. Clin. Investig. 2009, 119, 3301–3310. [Google Scholar] [CrossRef] [Green Version]
- Kunz, L.H.; Gilbert, W.M.; Towner, D. Increased incidence of cardiac anomalies in pregnancies complicated by gastroschisis. Am. J. Obstet. Gynecol. 2005, 193, 1248–1252. [Google Scholar] [CrossRef]
- Gibbin, C.; Touch, S.; Broth, R.E.; Berghella, V. Abdominal wall defects and congenital heart disease. Ultrasound Obstet. Gynecol. 2003, 21, 334–337. [Google Scholar] [CrossRef]
- Koçak, G.; Önal, Ç.; Koçak, A.; Karakurt, C.; Ateş, Ö.; Çayli, S.R.; Yoloğlu, S. Prevalence and Outcome of Congenital Heart Disease in Patients with Neural Tube Defect. J. Child Neurol. 2008, 23, 526–530. [Google Scholar] [CrossRef]
- Xu, P.-X.; Zheng, W.; Laclef, C.; Maire, P.; Maas, R.L.; Peters, H.; Xu, X. Eya1 is required for the morphogenesis of mammalian thymus, parathyroid and thyroid. Development 2002, 129, 3033–3044. [Google Scholar]
- Guo, C.; Sun, Y.; Zhou, B.; Adam, R.M.; Li, X.; Pu, W.T.; Morrow, B.E.; Moon, A.; Li, X. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J. Clin. Investig. 2011, 121, 1585–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molin, D.G.; DeRuiter, M.C.; Wisse, L.J.; Azhar, M.; Doetschman, T.; Poelmann, R.; Groot, A.C.G.-D. Altered apoptosis pattern during pharyngeal arch artery remodelling is associated with aortic arch malformations in Tgfβ2 knock-out mice. Cardiovasc. Res. 2002, 56, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Roest, P.A.; Nordstrand, H.; Wisse, L.J.; Poelmann, R.; Eriksson, U.; Groot, A.C.G.-D. Disturbed morphogenesis of cardiac outflow tract and increased rate of aortic arch anomalies in the offspring of diabetic rats. Birth Defects Res. Part A: Clin. Mol. Teratol. 2004, 70, 927–938. [Google Scholar] [CrossRef]
- Macatee, T.L.; Hammond, B.P.; Arenkiel, B.R.; Francis, L.; Frank, D.U.; Moon, A.M. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development 2003, 130, 6361–6374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, R.; Javidan, Y.; Zhang, T.; Nelson, S.; Schilling, T.F. AP2-dependent signals from the ectoderm regulate craniofacial development in the zebrafish embryo. Development 2005, 132, 3127–3138. [Google Scholar] [CrossRef] [Green Version]
- Brewer, S.; Williams, T. Loss of AP-2α impacts multiple aspects of ventral body wall development and closure. Dev. Boil. 2004, 267, 399–417. [Google Scholar] [CrossRef] [Green Version]
- Trainor, P. Specification of neural crest cell formation and migration in mouse embryos. Semin. Cell Dev. Boil. 2005, 16, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, M.P.; Hutchins, G.M. Arrested rotation of the outflow tract may explain double-outlet right ventricle. Circ. 1988, 77, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- LoMonico, M.P.; Bostrom, M.P.G.; Moore, G.W.; Hutchins, G.M. Arrested Rotation of the Outflow Tract May Explain Tetralogy of Fallot and Transposition of the Great Arteries. Pediatr. Pathol. 1988, 8, 267–281. [Google Scholar] [CrossRef]
- Scherptong, R.W.; Jongbloed, M.R.; Wisse, L.J.; Bartelings, M.M.; Poelmann, R.; Schalij, M.J.; Groot, A.C.G.-D.; Vicente-Steijn, R. Morphogenesis of outflow tract rotation during cardiac development: The pulmonary push concept. Dev. Dyn. 2012, 241, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Baardman, M.E.; Zwier, M.V.; Wisse, L.J.; De Groot, A.C.G.; Kerstjens-Frederikse, W.S.; Hofstra, R.M.W.; Jurdzinski, A.; Hierck, B.P.; Jongbloed, M.R.M.; Berger, R.M.F.; et al. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis. Model. Mech. 2016, 9, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | n | Abnormal | VSD | DORV | O-Ao | TGA | IAA +/−A-RSA | cAoA +/−A-RSA | RAA +/−A-SA | A-RSA |
|---|---|---|---|---|---|---|---|---|---|---|
| AP-2α−/− | 10 | 10 (100%) | 4 | 6 | 2 | 2 a | 3 | 2 | 1 | 4 |
| AP-2α−/f;Foxg1Cre | 9 | 0 | - | - | - | - | - | - | - | - |
| AP-2α−/f;Nkx2-5Cre | 14 | 2 (14%) | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| AP-2α−/f;Wnt1Cre | 25 b | 3 (12%) | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| AP-2α−/f; Foxg1Cre;Wnt1Cre | 6 | 0 | - | - | - | - | - | - | - | - |
| AP-2α−/f; Nkx2-5Cre;Wnt1Cre | 9 | 1 (11%) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| AP-2α−/f;Isl1Cre | 11 | 4 (36%) c | 2 | 1 | 0 | 0 | 2 | 0 | 0 | 1 |
| AP-2α−/f;PGKCre | 10 | 10 (100%) | 1 | 7 d | 0 | 0 | 1 | 4 | 1 | 4 |
| Genotype | n | PAA | Abnormal | Unilateral Defect | Bilateral Defect | Present | Hypo/Int/Absent | Absent |
|---|---|---|---|---|---|---|---|---|
| AP-2α−/− | 19 | 2 | 11 (58%) | 10 | 1 | 1 | 0 | 0 |
| 4 | 16 (84%) | 6 | 10 | - | 8 | 2 | ||
| 6 | 11 (58%) | 5 | 6 | - | 4 | 2 |
| Genotype | Thymus | Craniofacial |
|---|---|---|
| AP-2α−/f;Foxg1Cre | Bilaterally or unilaterally absent from normal position, reduced in size and/or split (7/9) | Upper facial cleft (7/9) |
| AP-2α−/f;Nkx2-5Cre | Bilaterally or unilaterally absent from normal position, reduced in size and/or split (5/14) | Midline cleft mandible (14/14) |
| AP-2α−/f;Wnt1Cre | Not affected | Cleft palate (5/19); exencephaly (1/19) |
| AP-2α−/f; Foxg1Cre;Wnt1Cre | Bilaterally or unilaterally absent from normal position (6/6) | Upper facial cleft (6/6); exencephaly (2/6) |
| AP-2α−/f; Nkx2-5Cre;Wnt1Cre | Small, split (1/9) | Midline cleft mandible (9/9) |
| AP-2α−/f;Isl1Cre | Absent from normal position (9/11) | Midline cleft mandible and upper facial cleft (8/11); cleft palate (6/11); exencephaly (1/11) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, A.-L.; Schneider, J.E.; Mohun, T.J.; Williams, T.; Bhattacharya, S.; Henderson, D.J.; Phillips, H.M.; Bamforth, S.D. Early Embryonic Expression of AP-2α Is Critical for Cardiovascular Development. J. Cardiovasc. Dev. Dis. 2020, 7, 27. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7030027
Johnson A-L, Schneider JE, Mohun TJ, Williams T, Bhattacharya S, Henderson DJ, Phillips HM, Bamforth SD. Early Embryonic Expression of AP-2α Is Critical for Cardiovascular Development. Journal of Cardiovascular Development and Disease. 2020; 7(3):27. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7030027
Chicago/Turabian StyleJohnson, Amy-Leigh, Jürgen E. Schneider, Timothy J. Mohun, Trevor Williams, Shoumo Bhattacharya, Deborah J. Henderson, Helen M. Phillips, and Simon D. Bamforth. 2020. "Early Embryonic Expression of AP-2α Is Critical for Cardiovascular Development" Journal of Cardiovascular Development and Disease 7, no. 3: 27. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd7030027