
The Complex Role of C-Reactive Protein in Systemic Lupus Erythematosus

 , , and
, , and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CRP as a Biomarker in Rheumatologic Diseases
3. CRP as a Biomarker Indicating Increased Risks of Cerebrovascular and Cardiovascular Diseases
4. Immunoregulatory Functions of CRP and other Pentraxins in SLE
5. Regulation of CRP Synthesis in SLE
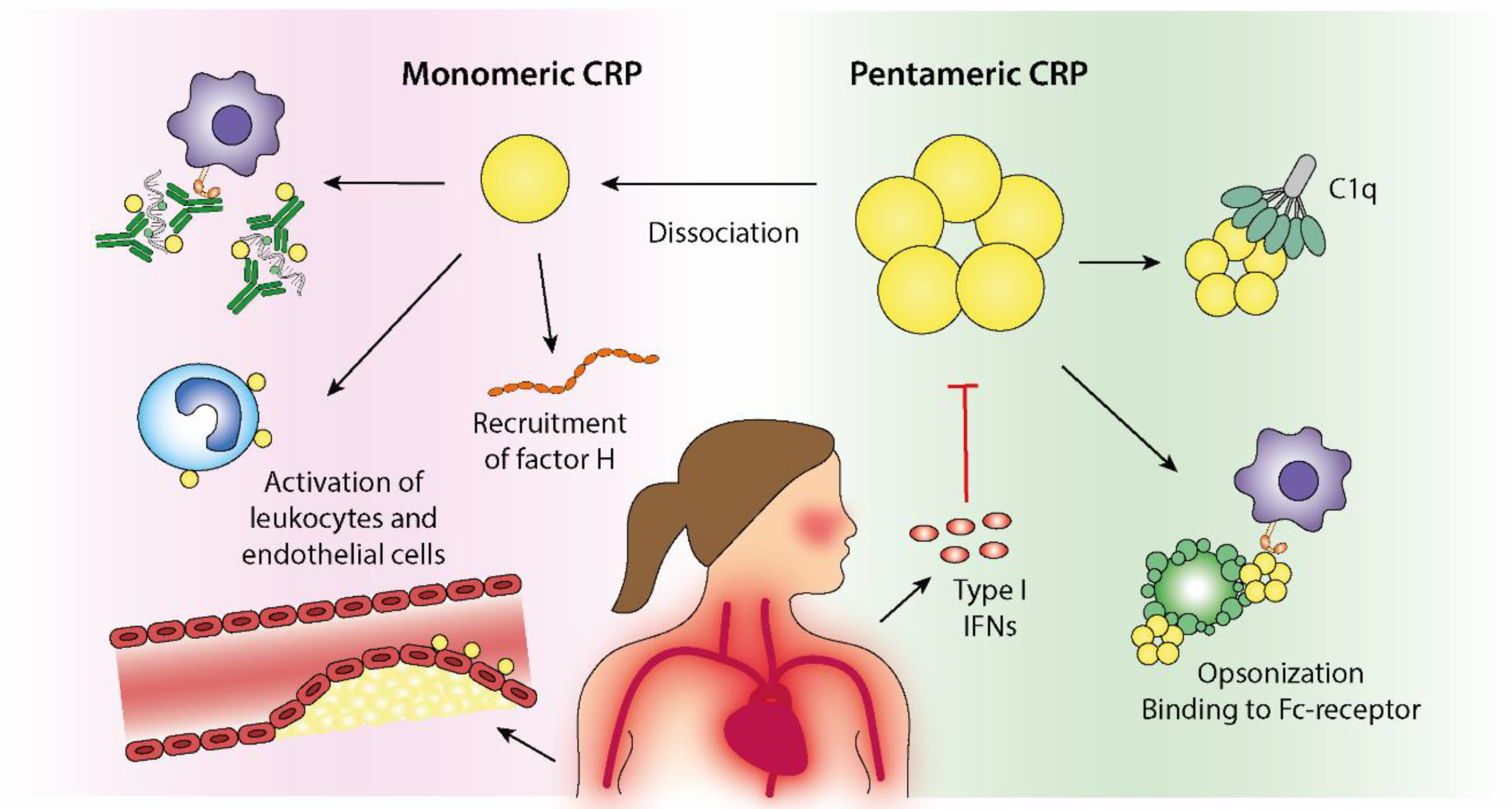
6. Structural Isoforms of CRP with Distinctive Biologic Effects
7. Autoantibodies Directed against CRP in SLE and Related Conditions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tillett, W.S.; Francis, T. Serological Reactions in Pneumonia with a Non-Protein Somatic Fraction of Pneumococcus. J. Exp. Med. 1930, 52, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Kushner, I.; Samols, D. Oswald Avery and the pneumococcus. Pharos Alpha Omega Alpha Honor Med. Soc. 2011, 74, 14–18. [Google Scholar]
- Volanakis, J.E.; Kaplan, M.H. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc. Soc. Exp. Biol. Med. 1971, 136, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Invest. 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, X.; Zou, H.; Dai, Z.; Feng, S.; Zhang, M.; Xiao, G.; Liu, Z.; Cheng, Q. The Basic Characteristics of the Pentraxin Family and Their Functions in Tumor Progression. Front Immunol. 2020, 11, 1757. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Brilland, B.; Vinatier, E.; Subra, J.F.; Jeannin, P.; Augusto, J.F.; Delneste, Y. Anti-Pentraxin Antibodies in Autoimmune Diseases: Bystanders or Pathophysiological Actors? Front Immunol. 2021, 11, 626343. [Google Scholar] [CrossRef]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Roux-Lombard, P.; de Moerloose, P.; Dayer, J.M.; Vischer, T.; Guerne, P.A. Absence of correlation between interleukin 6 and C-reactive protein blood levels in systemic lupus erythematosus compared with rheumatoid arthritis. J. Rheumatol. 1993, 20, 815–821. [Google Scholar]
- Gaitonde, S.; Samols, D.; Kushner, I. C-reactive protein and systemic lupus erythematosus. Arthritis. Rheum. 2008, 59, 1814–1820. [Google Scholar] [CrossRef]
- Kuhlenbaeumer, G.; Huge, A.; Berger, K.; Kessler, C.; Voelzke, H.; Funke, H.; Stoegbauer, F.; Stoll, M.; Ringelstein, E.B. Genetic variants in the C-reactive protein gene are associated with microangiopathic ischemic stroke. Cerebrovasc Dis. 2010, 30, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.I.; Cunninghame Graham, D.S.; Shepherd, C.; Roberton, C.A.; Whittaker, J.; Meeks, J.; Powell, R.J.; Isenberg, D.A.; Walport, M.J.; Vyse, T.J. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum. Mol. Genet. 2004, 13, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Kushner, I. It’s time to redefine inflammation. FASEB J. 2017, 31, 1787–1791. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, B.; Toss, H.; Siegbahn, A.; Venge, P.; Wallentin, L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. N. Engl. J. Med. 2000, 343, 1139–1147. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Wirestam, L.; Pihl, S.; Saleh, M.; Wetterö, J.; Sjöwall, C. Plasma C-Reactive Protein and Pentraxin-3 Reference Intervals During Normal Pregnancy. Front. Immunol. 2021, 12, 722118. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.; Gladman, D.; Helliwell, P.; Marchesoni, A.; Mease, P.; Mielants, H.; Group, C.S. Classification criteria for psoriatic arthritis: Development of new criteria from a large international study. Arthritis. Rheum. 2006, 54, 2665–2673. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; de Boysson, H.; Brouwer, E.; Cassie, R.; Cid, M.C.; Dasgupta, B.; Dejaco, C.; et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, B.; Cimmino, M.A.; Kremers, H.M.; Schmidt, W.A.; Schirmer, M.; Salvarani, C.; Bachta, A.; Dejaco, C.; Duftner, C.; Jensen, H.S.; et al. 2012 Provisional classification criteria for polymyalgia rheumatica: A European League Against Rheumatism/American College of Rheumatology collaborative initiative. Arthritis. Rheum. 2012, 64, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Shin, J.I.; Lee, K.H.; Nakagomi, D.; Quintana, L.F.; Busch, M.; Craven, A.; Luqmani, R.A.; Merkel, P.A.; Mayer, G.; et al. Clinical associations of renal involvement in ANCA-associated vasculitis. Autoimmun. Rev. 2020, 19, 102495. [Google Scholar] [CrossRef] [PubMed]
- Morrow, W.J.; Isenberg, D.A.; Parry, H.F.; Snaith, M.L. C-reactive protein in sera from patients with systemic lupus erythematosus. J. Rheumatol. 1981, 8, 599–604. [Google Scholar] [PubMed]
- Bianchi, M.; Kozyrev, S.V.; Notarnicola, A.; Hultin Rosenberg, L.; Karlsson, A.; Pucholt, P.; Rothwell, S.; Alexsson, A.; Sandling, J.K.; Andersson, H.; et al. Contribution of rare genetic variation to disease susceptibility in a large Scandinavian myositis cohort. Arthritis. Rheumatol. 2021. Epub Ahead of Print. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Almlof, J.C.; Leonard, D.; Sjöwall, C.; Syvanen, A.C.; Rönnblom, L.; Sandling, J.K.; Nordmark, G. Shared and Unique Patterns of DNA Methylation in Systemic Lupus Erythematosus and Primary Sjogren’s Syndrome. Front Immunol. 2019, 10, 1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strangfeld, A.; Eveslage, M.; Schneider, M.; Bergerhausen, H.J.; Klopsch, T.; Zink, A.; Listing, J. Treatment benefit or survival of the fittest: What drives the time-dependent decrease in serious infection rates under TNF inhibition and what does this imply for the individual patient? Ann. Rheum. Dis. 2011, 70, 1914–1920. [Google Scholar] [CrossRef] [Green Version]
- Askling, J. Malignancy and rheumatoid arthritis. Curr. Rheumatol. Rep. 2007, 9, 421–426. [Google Scholar] [CrossRef]
- Manger, B.; Schett, G. Paraneoplastic syndromes in rheumatology. Nat. Rev. Rheumatol. 2014, 10, 662–670. [Google Scholar] [CrossRef]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- de Ferranti, S.D.; Rifai, N. C-reactive protein: A nontraditional serum marker of cardiovascular risk. Cardiovasc. Pathol. 2007, 16, 14–21. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis. Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; Chang, S.C.; Liao, K.P.; Lu, B.; Fine, A.R.; Solomon, D.H.; Costenbader, K.H.; Karlson, E.W. Rheumatoid Arthritis and Mortality Among Women During 36 Years of Prospective Follow-Up: Results From the Nurses’ Health Study. Arthritis. Care Res. 2016, 68, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Avina-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis. Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef]
- Björnadal, L.; Yin, L.; Granath, F.; Klareskog, L.; Ekbom, A. Cardiovascular disease a hazard despite improved prognosis in patients with systemic lupus erythematosus: Results from a Swedish population based study 1964-95. J. Rheumatol. 2004, 31, 713–719. [Google Scholar] [PubMed]
- Arkema, E.V.; Svenungsson, E.; Von Euler, M.; Sjöwall, C.; Simard, J.F. Stroke in systemic lupus erythematosus: A Swedish population-based cohort study. Ann. Rheum. Dis. 2017, 76, 1544–1549. [Google Scholar] [CrossRef]
- Avina-Zubieta, J.A.; To, F.; Vostretsova, K.; De Vera, M.; Sayre, E.C.; Esdaile, J.M. Risk of Myocardial Infarction and Stroke in Newly Diagnosed Systemic Lupus Erythematosus: A General Population-Based Study. Arthritis. Care Res. 2017, 69, 849–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, I.; Parodis, I.; Gunnarsson, I.; Zickert, A.; Hofman-Bang, C.; Wallen, H.; Svenungsson, E. Myocardial infarctions, subtypes and coronary atherosclerosis in SLE: A case-control study. Lupus. Sci. Med. 2021, 8, e000515. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, C.; Ohman, M.L.; Nived, O.; Rantapaa Dahlqvist, S. Cardiovascular event in systemic lupus erythematosus in northern Sweden: Ancidence and predictors in a 7-year follow-up study. Lupus 2012, 21, 452–459. [Google Scholar] [CrossRef]
- Hermansen, M.L.; Lindhardsen, J.; Torp-Pedersen, C.; Faurschou, M.; Jacobsen, S. The risk of cardiovascular morbidity and cardiovascular mortality in systemic lupus erythematosus and lupus nephritis: A Danish nationwide population-based cohort study. Rheumatology 2017, 56, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A., Jr.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the Framingham Study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; du Berger, R.; Cote, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis. Rheum. 2001, 44, 2331–2337. [Google Scholar] [CrossRef]
- Fischer, L.M.; Schlienger, R.G.; Matter, C.; Jick, H.; Meier, C.R. Effect of rheumatoid arthritis or systemic lupus erythematosus on the risk of first-time acute myocardial infarction. Am. J. Cardiol. 2004, 93, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.R.; Liu, C.W.; Wu, Y.W.; Lai, K.R.; Wu, C.Y.; Lin, J.W.; Chan, C.L.; Pan, R.H. Systemic lupus erythematosus is associated with poor outcome after acute myocardial infarction. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1400–1407. [Google Scholar] [CrossRef]
- Rossides, M.; Simard, J.F.; Svenungsson, E.; von Euler, M.; Arkema, E.V. Mortality and Functionality after Stroke in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2017, 44, 1590–1596. [Google Scholar] [CrossRef]
- Svenungsson, E.; Antovic, A. The antiphospholipid syndrome - often overlooked cause of vascular occlusions? J. Intern. Med. 2020, 287, 349–372. [Google Scholar] [CrossRef] [Green Version]
- Goodson, N.J.; Symmons, D.P.; Scott, D.G.; Bunn, D.; Lunt, M.; Silman, A.J. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: A ten-year followup study of a primary care-based inception cohort. Arthritis. Rheum. 2005, 52, 2293–2299. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Möller, B.; Ljung, L.; Magnusson, S.; Smedby, T.; Södergren, A.; Ohman, M.L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: A five year prospective study. Arthritis Res. Ther. 2011, 13, R131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maradit-Kremers, H.; Nicola, P.J.; Crowson, C.S.; Ballman, K.V.; Gabriel, S.E. Cardiovascular death in rheumatoid arthritis: A population-based study. Arthritis. Rheum. 2005, 52, 722–732. [Google Scholar] [CrossRef]
- Kao, A.H.; Wasko, M.C.; Krishnaswami, S.; Wagner, J.; Edmundowicz, D.; Shaw, P.; Cunningham, A.L.; Danchenko, N.; Sutton-Tyrrell, K.; Tracy, R.P.; et al. C-reactive protein and coronary artery calcium in asymptomatic women with systemic lupus erythematosus or rheumatoid arthritis. Am. J. Cardiol. 2008, 102, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Rathouska, J.; Derosa, G.; Maffioli, P.; Nachtigal, P. Statin impact on disease activity and C-reactive protein concentrations in systemic lupus erythematosus patients: A systematic review and meta-analysis of controlled trials. Autoimmun. Rev. 2016, 15, 344–353. [Google Scholar] [CrossRef]
- Gladman, D.; Ginzler, E.; Goldsmith, C.; Fortin, P.; Liang, M.; Urowitz, M.; Bacon, P.; Bombardieri, S.; Hanly, J.; Hay, E.; et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis. Rheum. 1996, 39, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Singh, S.; Link, K.; Petri, M. High-sensitivity C-reactive protein as an associate of clinical subsets and organ damage in systemic lupus erythematosus. Semin. Arthritis. Rheum. 2008, 38, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Enocsson, H.; Wetterö, J.; Skogh, T.; Sjöwall, C. Soluble urokinase plasminogen activator receptor levels reflect organ damage in systemic lupus erythematosus. Transl. Res. 2013, 162, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enocsson, H.; Wirestam, L.; Dahle, C.; Padyukov, L.; Jonsen, A.; Urowitz, M.B.; Gladman, D.D.; Romero-Diaz, J.; Bae, S.C.; Fortin, P.R.; et al. Soluble urokinase plasminogen activator receptor (suPAR) levels predict damage accrual in patients with recent-onset systemic lupus erythematosus. J. Autoimmun. 2020, 106, 102340. [Google Scholar] [CrossRef]
- Dieker, J.; Berden, J.H.; Bakker, M.; Briand, J.P.; Muller, S.; Voll, R.; Sjöwall, C.; Herrmann, M.; Hilbrands, L.B.; van der Vlag, J. Autoantibodies against Modified Histone Peptides in SLE Patients Are Associated with Disease Activity and Lupus Nephritis. PLoS ONE 2016, 11, e0165373. [Google Scholar] [CrossRef]
- Lu, J.; Marnell, L.L.; Marjon, K.D.; Mold, C.; Du Clos, T.W.; Sun, P.D. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 2008, 456, 989–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganrot, P.O.; Kindmark, C.O. C-reactive protein--a phagocytosis-promoting factor. Scand J. Clin. Lab. Investig. 1969, 24, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, R.F.; Osmand, A.P.; Lint, T.F.; Gewurz, H. Interaction of C-reactive protein with lymphocytes and monocytes: Complement-dependent adherence and phagocytosis. J. Immunol. 1976, 117, 774–781. [Google Scholar]
- Manderson, A.P.; Botto, M.; Walport, M.J. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 2004, 22, 431–456. [Google Scholar] [CrossRef]
- Truedsson, L.; Bengtsson, A.A.; Sturfelt, G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity 2007, 40, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.H.; Volanakis, J.E. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J. Immunol. 1974, 112, 2135–2147. [Google Scholar]
- Siegel, J.; Rent, R.; Gewurz, H. Interactions of C-reactive protein with the complement system. I. Protamine-induced consumption of complement in acute phase sera. J. Exp. Med. 1974, 140, 631–647. [Google Scholar] [CrossRef]
- Ji, S.R.; Wu, Y.; Potempa, L.A.; Liang, Y.H.; Zhao, J. Effect of modified C-reactive protein on complement activation: A possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler Thromb. Vasc. Biol. 2006, 26, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Zeller, J.; Bogner, B.; Kiefer, J.; Braig, D.; Winninger, O.; Fricke, M.; Karasu, E.; Peter, K.; Huber-Lang, M.; Eisenhardt, S.U. CRP Enhances the Innate Killing Mechanisms Phagocytosis and ROS Formation in a Conformation and Complement-Dependent Manner. Front. Immunol. 2021, 12, 721887. [Google Scholar] [CrossRef]
- Berman, S.; Gewurz, H.; Mold, C. Binding of C-reactive protein to nucleated cells leads to complement activation without cytolysis. J. Immunol. 1986, 136, 1354–1359. [Google Scholar]
- Jarva, H.; Jokiranta, T.S.; Hellwage, J.; Zipfel, P.F.; Meri, S. Regulation of complement activation by C-reactive protein: Targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8–11. J. Immunol. 1999, 163, 3957–3962. [Google Scholar]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar] [CrossRef]
- Weiler, J.M.; Daha, M.R.; Austen, K.F.; Fearon, D.T. Control of the amplification convertase of complement by the plasma protein beta1H. Proc. Natl. Acad. Sci USA 1976, 73, 3268–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whaley, K.; Ruddy, S. Modulation of the alternative complement pathways by beta 1 H globulin. J. Exp. Med. 1976, 144, 1147–1163. [Google Scholar] [CrossRef]
- Harrison, R.A.; Lachmann, P.J. The physiological breakdown of the third component of human complement. Mol. Immunol. 1980, 17, 9–20. [Google Scholar] [CrossRef]
- Pangburn, M.K.; Schreiber, R.D.; Muller-Eberhard, H.J. Human complement C3b inactivator: Isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J. Exp. Med. 1977, 146, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Mihlan, M.; Stippa, S.; Jozsi, M.; Zipfel, P.F. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ. 2009, 16, 1630–1640. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, X.W.; Song, Y.; Yu, F.; Zhao, M.H. Anti-C1q autoantibodies from active lupus nephritis patients could inhibit the clearance of apoptotic cells and complement classical pathway activation mediated by C1q in vitro. Immunobiology 2014, 219, 980–989. [Google Scholar] [CrossRef]
- Trendelenburg, M.; Marfurt, J.; Gerber, I.; Tyndall, A.; Schifferli, J.A. Lack of occurrence of severe lupus nephritis among anti-C1q autoantibody-negative patients. Arthritis. Rheum. 1999, 42, 187–188. [Google Scholar] [CrossRef]
- Janko, C.; Franz, S.; Munoz, L.E.; Siebig, S.; Winkler, S.; Schett, G.; Lauber, K.; Sheriff, A.; van der Vlag, J.; Herrmann, M. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Front. Immunol. 2011, 2, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöwall, C.; Wetterö, J. Pathogenic implications for autoantibodies against C-reactive protein and other acute phase proteins. Clin. Chim. Acta. 2007, 378, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Gatto, M.; Radu, C.M.; Luisetto, R.; Ghirardello, A.; Bonsembiante, F.; Trez, D.; Valentino, S.; Bottazzi, B.; Simioni, P.; Cavicchioli, L.; et al. Immunization with Pentraxin3 prevents transition from subclinical to clinical lupus nephritis in lupus-prone mice: Insights from renal ultrastructural findings. J. Autoimmun. 2020, 111, 102443. [Google Scholar] [CrossRef]
- Sjöwall, C.; Olin, A.I.; Skogh, T.; Wetterö, J.; Morgelin, M.; Nived, O.; Sturfelt, G.; Bengtsson, A.A. C-reactive protein, immunoglobulin G and complement co-localize in renal immune deposits of proliferative lupus nephritis. Autoimmunity 2013, 46, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.Y.; Li, H.Y.; Fu, G.; Yu, F.; Wu, Y.; Zhao, M.H. Autoantibodies against C-Reactive Protein Influence Complement Activation and Clinical Course in Lupus Nephritis. J. Am. Soc. Nephrol. 2017, 28, 3044–3054. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Hao, J.B.; Chu, H.; Wang, F.M.; Song, D.; Zhu, L.; Yu, F.; Li, Y.Z.; Song, Y.; Zhao, M.H. Genetic variants in FH are associated with renal histopathologic subtypes of lupus nephritis: A large cohort study from China. Lupus 2017, 26, 1309–1317. [Google Scholar] [CrossRef]
- Wang, F.M.; Song, D.; Pang, Y.; Song, Y.; Yu, F.; Zhao, M.H. The dysfunctions of complement factor H in lupus nephritis. Lupus 2016, 25, 1328–1340. [Google Scholar] [CrossRef] [PubMed]
- Karpati, E.; Papp, A.; Schneider, A.E.; Hajnal, D.; Cserhalmi, M.; Csincsi, A.I.; Uzonyi, B.; Jozsi, M. Interaction of the Factor H Family Proteins FHR-1 and FHR-5 With DNA and Dead Cells: Implications for the Regulation of Complement Activation and Opsonization. Front Immunol. 2020, 11, 1297. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, M.; Okemefuna, A.I.; Perkins, S.J.; Mihlan, M.; Huber-Lang, M.; Jozsi, M. Molecular basis of C-reactive protein binding and modulation of complement activation by factor H-related protein 4. Mol. Immunol. 2010, 47, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Mihlan, M.; Hebecker, M.; Dahse, H.M.; Halbich, S.; Huber-Lang, M.; Dahse, R.; Zipfel, P.F.; Jozsi, M. Human complement factor H-related protein 4 binds and recruits native pentameric C-reactive protein to necrotic cells. Mol. Immunol. 2009, 46, 335–344. [Google Scholar] [CrossRef]
- Mihlan, M.; Blom, A.M.; Kupreishvili, K.; Lauer, N.; Stelzner, K.; Bergström, F.; Niessen, H.W.; Zipfel, P.F. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J. 2011, 25, 4198–4210. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, A.P.; Trouw, L.A.; McGrath, F.D.; Hack, C.E.; Blom, A.M. Regulation of complement activation by C-reactive protein: Targeting of the inhibitory activity of C4b-binding protein. J. Immunol. 2006, 176, 7612–7620. [Google Scholar] [CrossRef] [Green Version]
- Sjöwall, C.; Wetterö, J.; Bengtsson, T.; Askendal, A.; Almroth, G.; Skogh, T.; Tengvall, P. Solid-phase classical complement activation by C-reactive protein (CRP) is inhibited by fluid-phase CRP-C1q interaction. Biochem. Biophys. Res. Commun. 2007, 352, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Ohtani, K.; Matsuda, Y.; Mori, K.; Hwang, I.; Suzuki, Y.; Inoue, N.; Wakamiya, N. Collectin CL-P1 utilizes C-reactive protein for complement activation. Biochim. Biophys Acta. 2016, 1860, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Mold, C.; Du Clos, T.W.; Sun, P.D. Pentraxins and Fc Receptor-Mediated Immune Responses. Front Immunol. 2018, 9, 2607. [Google Scholar] [CrossRef]
- Bharadwaj, D.; Stein, M.P.; Volzer, M.; Mold, C.; Du Clos, T.W. The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J. Exp. Med. 1999, 190, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Manolov, D.E.; Rocker, C.; Hombach, V.; Nienhaus, G.U.; Torzewski, J. Ultrasensitive confocal fluorescence microscopy of C-reactive protein interacting with FcgammaRIIa. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2372–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mold, C.; Du Clos, T.W. C-reactive protein increases cytokine responses to Streptococcus pneumoniae through interactions with Fc gamma receptors. J. Immunol. 2006, 176, 7598–7604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Båve, U.; Magnusson, M.; Eloranta, M.L.; Perers, A.; Alm, G.V.; Rönnblom, L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J. Immunol. 2003, 171, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Mold, C.; Clos, T.W. C-reactive protein inhibits plasmacytoid dendritic cell interferon responses to autoantibody immune complexes. Arthritis. Rheum. 2013, 65, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Marjon, K.D.; Marnell, L.L.; Wang, R.; Mold, C.; Du Clos, T.W.; Sun, P. Recognition and functional activation of the human IgA receptor (FcalphaRI) by C-reactive protein. Proc. Natl. Acad. Sci. USA 2011, 108, 4974–4979. [Google Scholar] [CrossRef] [Green Version]
- Calabro, P.; Willerson, J.T.; Yeh, E.T. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation 2003, 108, 1930–1932. [Google Scholar] [CrossRef]
- Gould, J.M.; Weiser, J.N. Expression of C-reactive protein in the human respiratory tract. Infect. Immun. 2001, 69, 1747–1754. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Caldera, A.; Vernal, R.; Paredes, R.; Veloso-Matta, P.; Astorga, J.; Hernandez, M. Human periodontal ligament fibroblasts synthesize C-reactive protein and Th-related cytokines in response to interleukin (IL)-6 trans-signalling. Int. Endod. J. 2018, 51, 632–640. [Google Scholar] [CrossRef]
- Jabs, W.J.; Logering, B.A.; Gerke, P.; Kreft, B.; Wolber, E.M.; Klinger, M.H.; Fricke, L.; Steinhoff, J. The kidney as a second site of human C-reactive protein formation in vivo. Eur. J. Immunol. 2003, 33, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Han, C.; Wang, B.; Pang, X.; Mao, J. Angiotensin II induces the expression of c-reactive protein via MAPK-dependent signal pathway in U937 macrophages. Cell Physiol. Biochem. 2011, 27, 63–70. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, S.L.; Rzewnicki, D.; Samols, D.; Kushner, I. The effect of interleukin-1 on C-reactive protein expression in Hep3B cells is exerted at the transcriptional level. Biochem. J. 1995, 310, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J. Biol. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Cha-Molstad, H.; Samols, D.; Kushner, I. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein beta (C/EBP beta) and Rel p50. J. Immunol. 2001, 166, 2378–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enocsson, H.; Sjöwall, C.; Skogh, T.; Eloranta, M.L.; Rönnblom, L.; Wetterö, J. Interferon-alpha mediates suppression of C-reactive protein: Explanation for muted C-reactive protein response in lupus flares? Arthritis. Rheum. 2009, 60, 3755–3760. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Genin, B.; Mentha, G.; Iynedjian, P.B.; Roux-Lombard, P.; Guerne, P.A. IL-1 receptor antagonist (IL-1Ra) does not inhibit the production of C-reactive protein or serum amyloid A protein by human primary hepatocytes. Differential regulation in normal and tumour cells. Clin. Exp. Immunol. 1995, 100, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Lanham, J.G.; De Beer, F.C. C-reactive protein in SLE. Clin Rheum Dis 1982, 8, 91–103. [Google Scholar] [CrossRef]
- Rönnblom, L.; Eloranta, M.L. The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 2013, 25, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Sonoda, S.; Urano, T.; Yamada, T.; Okada, M. Monitoring both serum amyloid protein A and C-reactive protein as inflammatory markers in infectious diseases. Clin. Chem. 1993, 39, 293–297. [Google Scholar] [CrossRef]
- Eloranta, M.L.; Rönnblom, L. Cause and consequences of the activated type I interferon system in SLE. J. Mol. Med. 2016, 94, 1103–1110. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
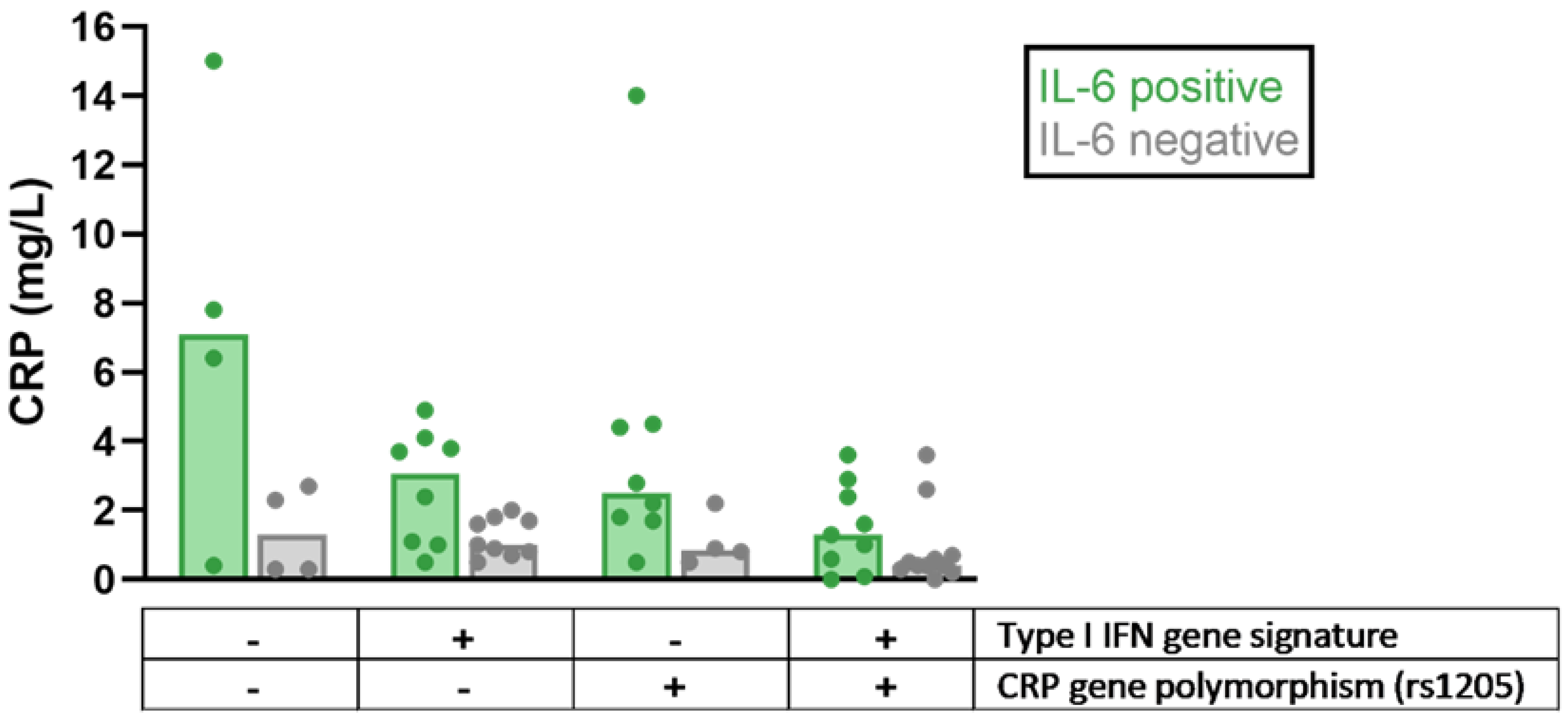
- Enocsson, H.; Sjöwall, C.; Kastbom, A.; Skogh, T.; Eloranta, M.L.; Rönnblom, L.; Wetterö, J. Association of serum C-reactive protein levels with lupus disease activity in the absence of measurable interferon-alpha and a C-reactive protein gene variant. Arthritis. Rheumatol. 2014, 66, 1568–1573. [Google Scholar] [CrossRef] [Green Version]
- Hage, F.G.; Szalai, A.J. C-reactive protein gene polymorphisms, C-reactive protein blood levels, and cardiovascular disease risk. J. Am. Coll. Cardiol. 2007, 50, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalai, A.J.; Alarcon, G.S.; Calvo-Alen, J.; Toloza, S.M.; McCrory, M.A.; Edberg, J.C.; McGwin, G., Jr.; Bastian, H.M.; Fessler, B.J.; Vila, L.M.; et al. Systemic lupus erythematosus in a multiethnic US Cohort (LUMINA). XXX: Association between C-reactive protein (CRP) gene polymorphisms and vascular events. Rheumatology 2005, 44, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Szalai, A.J.; Wu, J.; Lange, E.M.; McCrory, M.A.; Langefeld, C.D.; Williams, A.; Zakharkin, S.O.; George, V.; Allison, D.B.; Cooper, G.S.; et al. Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level. J. Mol. Med. 2005, 83, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Enocsson, H.; Gullstrand, B.; Eloranta, M.L.; Wetterö, J.; Leonard, D.; Rönnblom, L.; Bengtsson, A.A.; Sjöwall, C. C-Reactive Protein Levels in Systemic Lupus Erythematosus Are Modulated by the Interferon Gene Signature and CRP Gene Polymorphism rs1205. Front. Immunol. 2021, 11, 622326. [Google Scholar] [CrossRef]
- Potempa, L.A.; Maldonado, B.A.; Laurent, P.; Zemel, E.S.; Gewurz, H. Antigenic, electrophoretic and binding alterations of human C-reactive protein modified selectively in the absence of calcium. Mol. Immunol. 1983, 20, 1165–1175. [Google Scholar] [CrossRef]
- Verma, S.; Szmitko, P.E.; Yeh, E.T. C-reactive protein: Structure affects function. Circulation 2004, 109, 1914–1917. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.R.; Wu, Y.; Zhu, L.; Potempa, L.A.; Sheng, F.L.; Lu, W.; Zhao, J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J. 2007, 21, 284–294. [Google Scholar] [CrossRef]
- Potempa, L.A.; Siegel, J.N.; Fiedel, B.A.; Potempa, R.T.; Gewurz, H. Expression, detection and assay of a neoantigen (Neo-CRP) associated with a free, human C-reactive protein subunit. Mol. Immunol. 1987, 24, 531–541. [Google Scholar] [CrossRef]
- Schwedler, S.B.; Filep, J.G.; Galle, J.; Wanner, C.; Potempa, L.A. C-reactive protein: A family of proteins to regulate cardiovascular function. Am. J. Kidney. Dis. 2006, 47, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Skogh, T.; Stendahl, O. Complement-mediated delay in immune complex clearance from the blood owing to reduced deposition outside the reticuloendothelial system. Immunology 1983, 49, 53–59. [Google Scholar]
- Motie, M.; Brockmeier, S.; Potempa, L.A. Binding of model soluble immune complexes to modified C-reactive protein. J. Immunol. 1996, 156, 4435–4441. [Google Scholar] [PubMed]
- Eisenhardt, S.U.; Habersberger, J.; Murphy, A.; Chen, Y.C.; Woollard, K.J.; Bassler, N.; Qian, H.; von Zur Muhlen, C.; Hagemeyer, C.E.; Ahrens, I.; et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ. Res. 2009, 105, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.Y.; Liu, X.L.; Liu, Y.T.; Jia, Z.K.; Filep, J.G.; Potempa, L.A.; Ji, S.R.; Wu, Y. Matrix sieving-enforced retrograde transcytosis regulates tissue accumulation of C-reactive protein. Cardiovasc Res. 2019, 115, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Iso, H.; Cui, R.; Date, C.; Kikuchi, S.; Tamakoshi, A.; Group, J.S. C-reactive protein levels and risk of mortality from cardiovascular disease in Japanese: The JACC Study. Atherosclerosis 2009, 207, 291–297. [Google Scholar] [CrossRef]
- Ji, S.R.; Wu, Y.; Potempa, L.A.; Qiu, Q.; Zhao, J. Interactions of C-reactive protein with low-density lipoproteins: Implications for an active role of modified C-reactive protein in atherosclerosis. Int. J. Biochem. Cell Biol. 2006, 38, 648–661. [Google Scholar] [CrossRef]
- Jiang, S.; Bao, Y.; Hou, X.; Fang, Q.; Wang, C.; Pan, J.; Zuo, Y.; Zhong, W.; Xiang, K.; Jia, W. Serum C-reactive protein and risk of cardiovascular events in middle-aged and older chinese population. Am. J. Cardiol. 2009, 103, 1727–1731. [Google Scholar] [CrossRef]
- Molins, B.; Pena, E.; Vilahur, G.; Mendieta, C.; Slevin, M.; Badimon, L. C-reactive protein isoforms differ in their effects on thrombus growth. Arterioscler. Thromb Vasc. Biol. 2008, 28, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Paynter, N.P.; Rifai, N.; Gaziano, J.M.; Cook, N.R. C-reactive protein and parental history improve global cardiovascular risk prediction: The Reynolds Risk Score for men. Circulation 2008, 118, 2243–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res. 2012, 96, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Strang, F.; Scheichl, A.; Chen, Y.C.; Wang, X.; Htun, N.M.; Bassler, N.; Eisenhardt, S.U.; Habersberger, J.; Peter, K. Amyloid plaques dissociate pentameric to monomeric C-reactive protein: A novel pathomechanism driving cortical inflammation in Alzheimer’s disease? Brain Pathol. 2012, 22, 337–346. [Google Scholar] [CrossRef]
- Eisenhardt, S.U.; Thiele, J.R.; Bannasch, H.; Stark, G.B.; Peter, K. C-reactive protein: How conformational changes influence inflammatory properties. Cell Cycle. 2009, 8, 3885–3892. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.R.; Ma, L.; Bai, C.J.; Shi, J.M.; Li, H.Y.; Potempa, L.A.; Filep, J.G.; Zhao, J.; Wu, Y. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J. 2009, 23, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Khreiss, T.; Jozsef, L.; Hossain, S.; Chan, J.S.; Potempa, L.A.; Filep, J.G. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J. Biol. Chem. 2002, 277, 40775–40781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khreiss, T.; Jozsef, L.; Potempa, L.A.; Filep, J.G. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation 2004, 109, 2016–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khreiss, T.; Jozsef, L.; Potempa, L.A.; Filep, J.G. Opposing effects of C-reactive protein isoforms on shear-induced neutrophil-platelet adhesion and neutrophil aggregation in whole blood. Circulation 2004, 110, 2713–2720. [Google Scholar] [CrossRef]
- Khreiss, T.; Jozsef, L.; Potempa, L.A.; Filep, J.G. Loss of pentameric symmetry in C-reactive protein induces interleukin-8 secretion through peroxynitrite signaling in human neutrophils. Circ. Res. 2005, 97, 690–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer, N.; Mihlan, M.; Hartmann, A.; Schlotzer-Schrehardt, U.; Keilhauer, C.; Scholl, H.P.; Charbel Issa, P.; Holz, F.; Weber, B.H.; Skerka, C.; et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J. Immunol. 2011, 187, 4374–4383. [Google Scholar] [CrossRef] [Green Version]
- Molins, B.; Pena, E.; de la Torre, R.; Badimon, L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc. Res. 2011, 92, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.Y.; Ji, S.R.; Bai, C.J.; El Kebir, D.; Li, H.Y.; Shi, J.M.; Zhu, W.; Costantino, S.; Zhou, H.H.; Potempa, L.A.; et al. A redox switch in C-reactive protein modulates activation of endothelial cells. FASEB J. 2011, 25, 3186–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouki, C.; Haas, B.; Chan, J.S.; Potempa, L.A.; Filep, J.G. Loss of pentameric symmetry of C-reactive protein is associated with promotion of neutrophil-endothelial cell adhesion. J. Immunol. 2001, 167, 5355–5361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boncler, M.; Kehrel, B.; Szewczyk, R.; Stec-Martyna, E.; Bednarek, R.; Brodde, M.; Watala, C. Oxidation of C-reactive protein by hypochlorous acid leads to the formation of potent platelet activator. Int. J. Biol. Macromol. 2018, 107, 2701–2714. [Google Scholar] [CrossRef]
- Schwedler, S.B.; Guderian, F.; Dammrich, J.; Potempa, L.A.; Wanner, C. Tubular staining of modified C-reactive protein in diabetic chronic kidney disease. Nephrol. Dial. Transplant 2003, 18, 2300–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slevin, M.; Matou-Nasri, S.; Turu, M.; Luque, A.; Rovira, N.; Badimon, L.; Boluda, S.; Potempa, L.; Sanfeliu, C.; de Vera, N.; et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol. 2010, 20, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Robey, F.A.; Jones, K.D.; Steinberg, A.D. C-reactive protein mediates the solubilization of nuclear DNA by complement in vitro. J. Exp. Med. 1985, 161, 1344–1356. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.A.; Du Clos, T.W.; Khursigara, G.; Picazo, J.J.; Rubin, R.L. Autoantibodies to cryptic epitopes of C-reactive protein and other acute phase proteins in the toxic oil syndrome. J. Autoimmun. 1995, 8, 293–303. [Google Scholar] [CrossRef]
- Bell, S.A.; Faust, H.; Schmid, A.; Meurer, M. Autoantibodies to C-reactive protein (CRP) and other acute-phase proteins in systemic autoimmune diseases. Clin. Exp. Immunol. 1998, 113, 327–332. [Google Scholar] [CrossRef]
- Sjöwall, C.; Eriksson, P.; Almer, S.; Skogh, T. Autoantibodies to C-reactive protein is a common finding in SLE, but not in primary Sjogren’s syndrome, rheumatoid arthritis or inflammatory bowel disease. J. Autoimmun. 2002, 19, 155–160. [Google Scholar] [CrossRef]
- Sjöwall, C.; Bengtsson, A.A.; Sturfelt, G.; Skogh, T. Serum levels of autoantibodies against monomeric C-reactive protein are correlated with disease activity in systemic lupus erythematosus. Arthritis. Res. Ther. 2004, 6, R87–R94. [Google Scholar] [CrossRef] [Green Version]
- Rosenau, B.J.; Schur, P.H. Antibodies to C reactive protein. Ann. Rheum. Dis. 2006, 65, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Sjöwall, C.; Cardell, K.; Boström, E.A.; Bokarewa, M.I.; Enocsson, H.; Ekstedt, M.; Lindvall, L.; Fryden, A.; Almer, S. High prevalence of autoantibodies to C-reactive protein in patients with chronic hepatitis C infection: Association with liver fibrosis and portal inflammation. Hum. Immunol. 2012, 73, 382–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakuszko, K.; Krajewska, M.; Halon, A.; Koscielska-Kasprzak, K.; Myszka, M.; Zabinska, M.; Augustyniak-Bartosik, H.; Rukasz, D.; Weyde, W.; Klinger, M. Pathogenic role of antibodies against monomeric C-reactive protein in tubulointerstitial nephritis and uveitis syndrome. Intern. Med. J. 2014, 44, 809–812. [Google Scholar] [CrossRef]
- Figueredo, M.A.; Rodriguez, A.; Ruiz-Yague, M.; Romero, M.; Fernandez-Cruz, A.; Gomez-de la Concha, E.; Patino, R. Autoantibodies against C-reactive protein: Clinical associations in systemic lupus erythematosus and primary antiphospholipid syndrome. J. Rheumatol. 2006, 33, 1980–1986. [Google Scholar] [PubMed]
- Pesickova, S.S.; Rysava, R.; Lenicek, M.; Vitek, L.; Potlukova, E.; Hruskova, Z.; Jancova, E.; Honsova, E.; Zavada, J.; Trendelenburg, M.; et al. Prognostic value of anti-CRP antibodies in lupus nephritis in long-term follow-up. Arthritis Res. Ther. 2015, 17, 371. [Google Scholar] [CrossRef] [Green Version]
- Mathsson, L.; Ahlin, E.; Sjöwall, C.; Skogh, T.; Rönnelid, J. Cytokine induction by circulating immune complexes and signs of in-vivo complement activation in systemic lupus erythematosus are associated with the occurrence of anti-Sjogren’s syndrome A antibodies. Clin. Exp. Immunol. 2007, 147, 513–520. [Google Scholar] [CrossRef]
- O’Neill, S.G.; Giles, I.; Lambrianides, A.; Manson, J.; D’Cruz, D.; Schrieber, L.; March, L.M.; Latchman, D.S.; Isenberg, D.A.; Rahman, A. Antibodies to apolipoprotein A-I, high-density lipoprotein, and C-reactive protein are associated with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Wener, M.H.; Uwatoko, S.; Mannik, M. Antibodies to the collagen-like region of C1q in sera of patients with autoimmune rheumatic diseases. Arthritis Rheum. 1989, 32, 544–551. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enocsson, H.; Karlsson, J.; Li, H.-Y.; Wu, Y.; Kushner, I.; Wetterö, J.; Sjöwall, C. The Complex Role of C-Reactive Protein in Systemic Lupus Erythematosus. J. Clin. Med. 2021, 10, 5837. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10245837
Enocsson H, Karlsson J, Li H-Y, Wu Y, Kushner I, Wetterö J, Sjöwall C. The Complex Role of C-Reactive Protein in Systemic Lupus Erythematosus. Journal of Clinical Medicine. 2021; 10(24):5837. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10245837
Chicago/Turabian StyleEnocsson, Helena, Jesper Karlsson, Hai-Yun Li, Yi Wu, Irving Kushner, Jonas Wetterö, and Christopher Sjöwall. 2021. "The Complex Role of C-Reactive Protein in Systemic Lupus Erythematosus" Journal of Clinical Medicine 10, no. 24: 5837. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10245837