Comparative Studies on the Polymorphism and Copy Number Variation of mtSSU rDNA in Ciliates (Protista, Ciliophora): Implications for Phylogenetic, Environmental, and Ecological Research

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
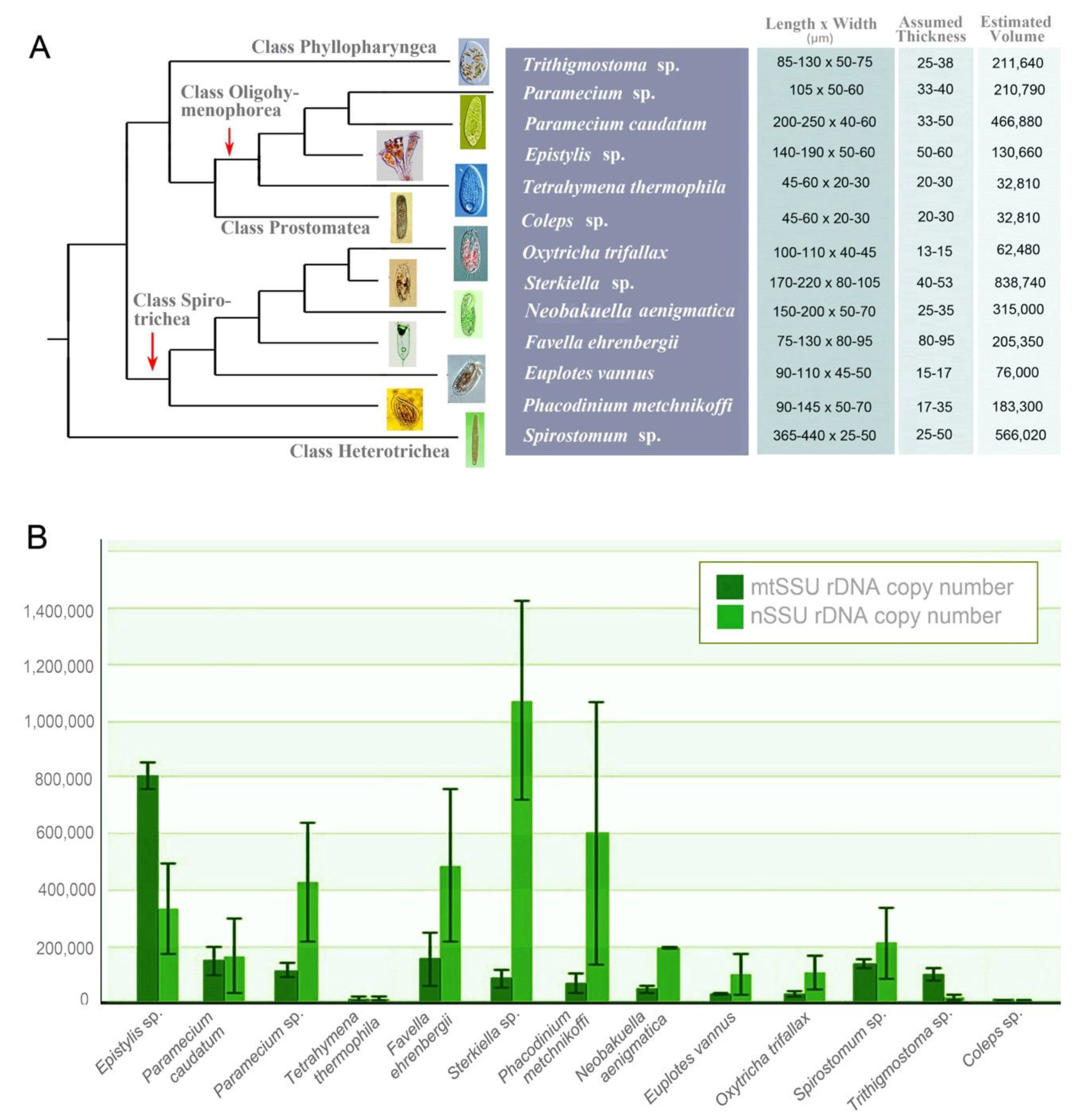
2.1. Taxon Sampling and Identification
2.2. DNA Extraction, PCR Amplification, and Sequencing
2.3. Estimation of mtSSU rDNA Copy Number (Digital PCR) and nSSU rDNA Copy Number (Quantitative PCR)
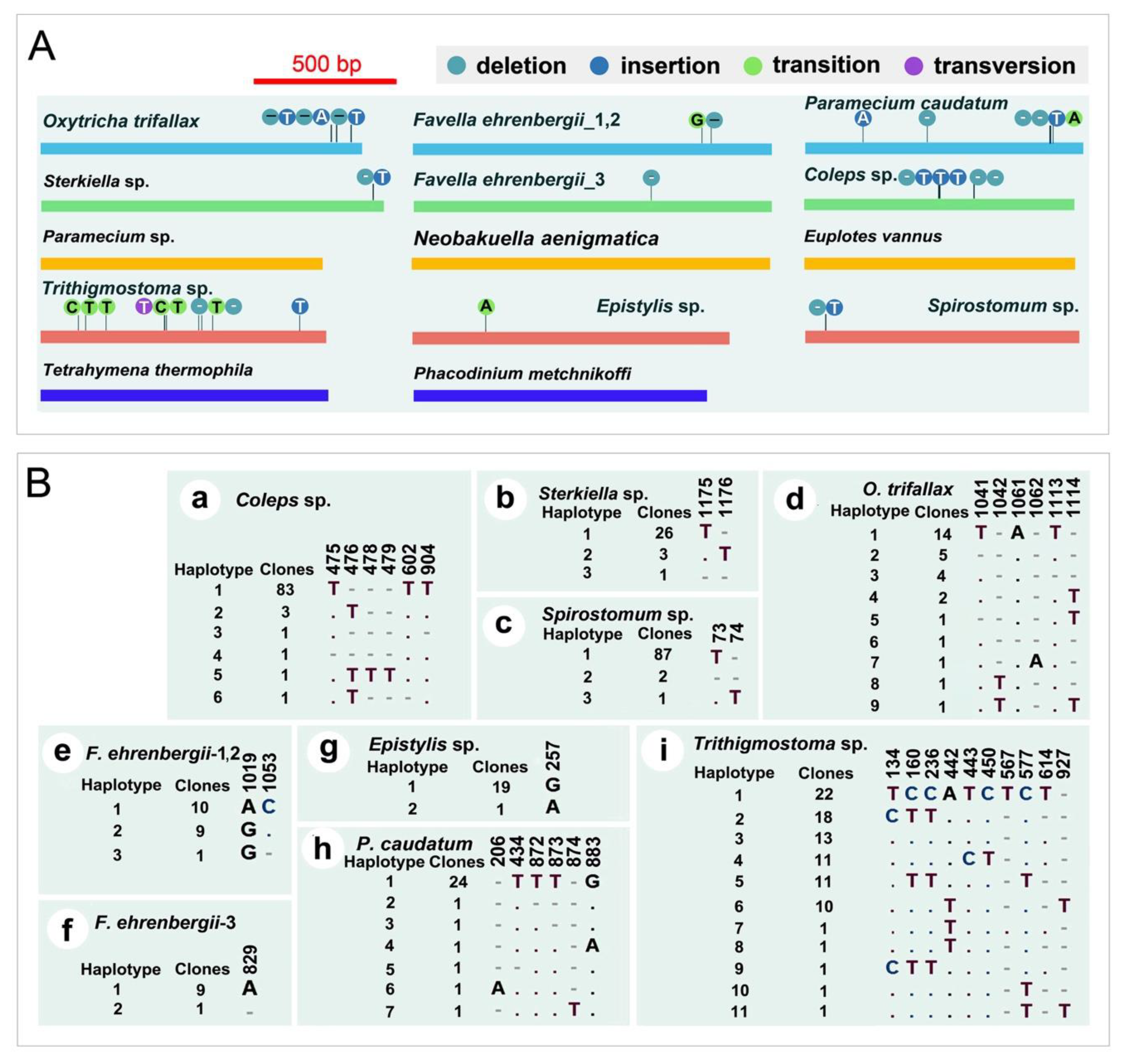
2.4. mtSSU rDNA Polymorphism of Ciliates
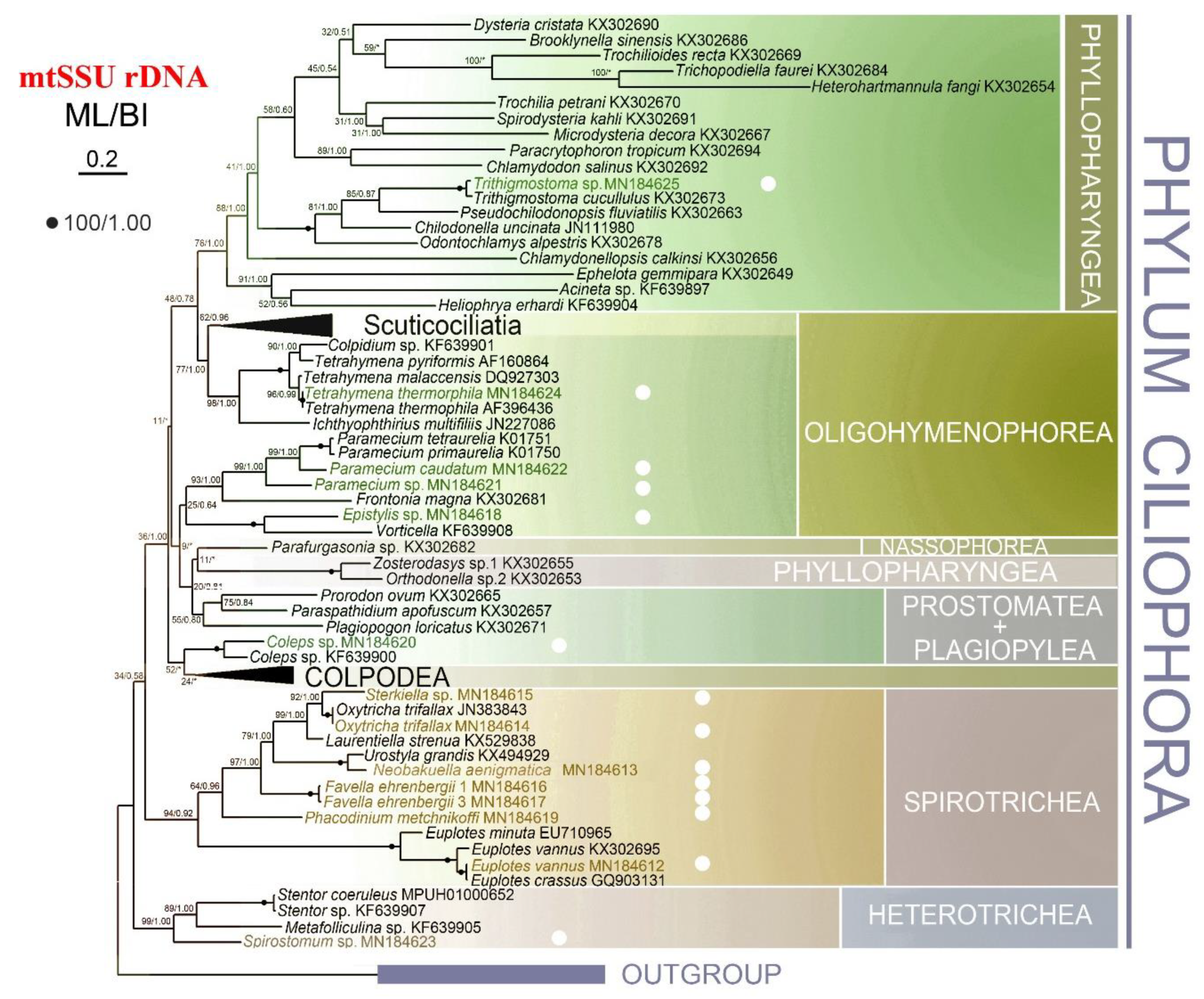
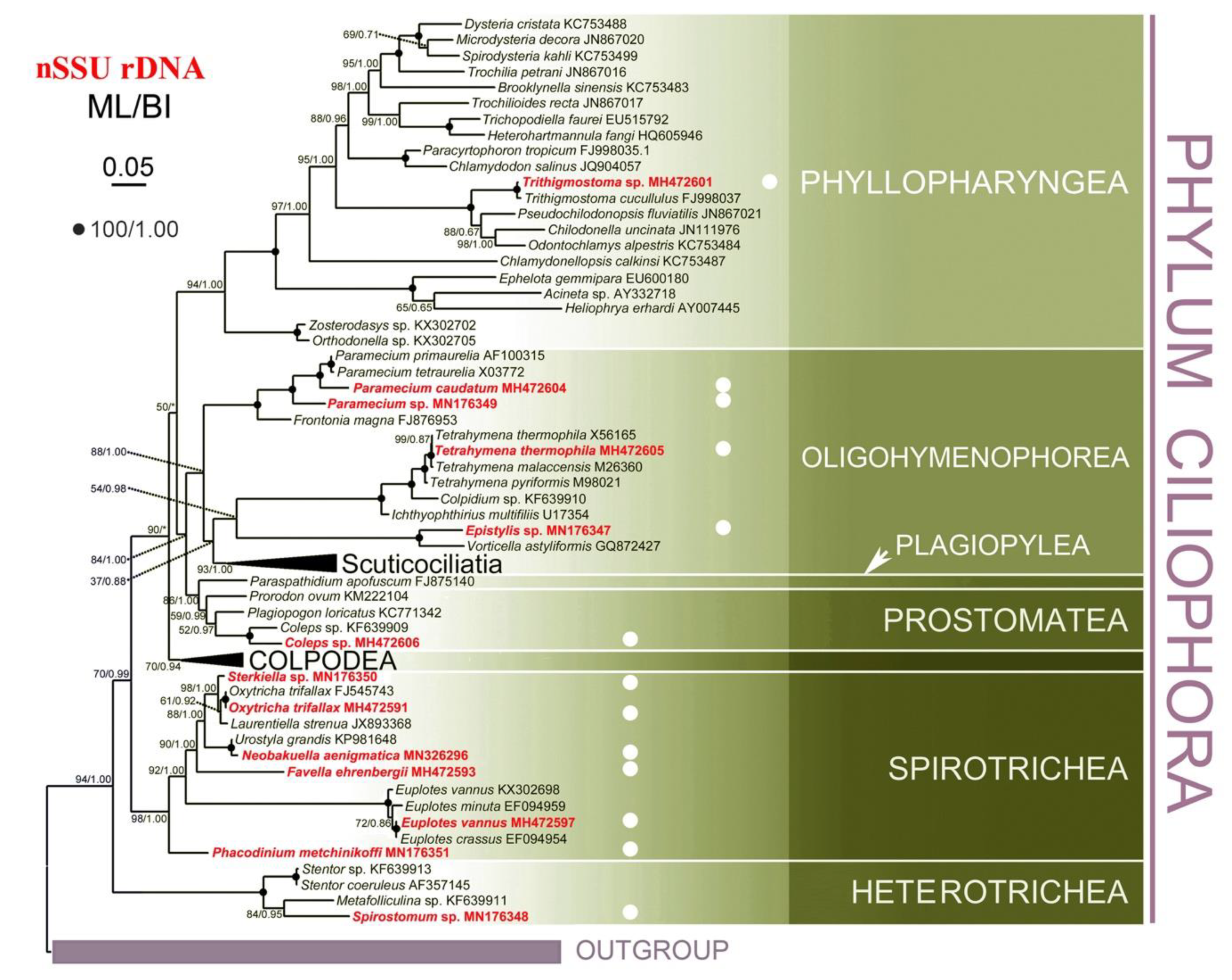
2.5. Phylogenetic Analyses
3. Results
3.1. mtSSU and nSSU rDNA Copy Number in Ciliates
3.2. mtSSU rDNA Polymorphism of Ciliates
3.3. Phylogenetic Analyses
4. Discussion
4.1. mtSSU and nSSU rDNA Copy Number of Ciliates
4.2. mtSSU rDNA Polymorphism of Ciliates
4.3. Phylogenetic Analyses
4.4. Ecological Significance
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Accessibility
References
- Grattepanche, J.D.; Walker, L.; Ott, B.; Pinto, D.P.; Delwiche, C.; Lane, C.; Katz, L. Microbial diversity in the eukaryotic SAR clade. BioEssays 2018, 40, e1700198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adl, S.M.; Simpson, A.G.; Farmer, M.A.; Andersen, R.A.; Anderson, O.R.; Barta, J.R.; Bowser, S.S.; Brugerolle, G.; Fensome, R.A.; Fredericq, S. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J. Eukaryot. Microbiol. 2005, 52, 399–451. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Burger, G.; Durnford, D.G.; Lang, B.F.; Lee, R.W.; Pearlman, R.E.; Roger, A.J.; Gray, M.W. The tree of eukaryotes. Trends Ecol. Evol. 2005, 20, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, L.W.; Grant, J.; Tekle, Y.I.; Lasek-Nesselquist, E.; Morrison, H.G.; Sogin, M.L.; Patterson, D.J.; Katz, L.A. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst. Biol. 2010, 59, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Jiang, Y.H.; Gao, F.; Zheng, W.; Krock, T.J.; Stover, N.A.; Lu, C.; Katz, L.A.; Song, W.B. Genome analyses of the new model protist Euplotes vannus focusing on genome rearrangement and resistance to environmental stressors. Mol. Ecol. Resour. 2019, 19, 1292–1308. [Google Scholar] [CrossRef]
- Pierce, R.W.; Turner, J.T. Ecology of planktonic ciliates in marine food webs. Rev. Aquat. Sci 1992, 6, 139–181. [Google Scholar]
- Zöllner, E.; Hoppe, H.-G.; Sommer, U.; Jürgens, K. Effect of zooplankton-mediated trophic cascades on marine microbial food web components (bacteria, nanoflagellates, ciliates). Limnol. Oceanogr. 2009, 54, 262–275. [Google Scholar] [CrossRef]
- Zhao, X.; Xiong, J.; Mao, F.; Sheng, Y.; Chen, X.; Feng, L.; Dui, W.; Yang, W.; Kapusta, A.; Feschotte, C. RNAi-dependent Polycomb repression controls transposable elements in Tetrahymena. Genes Dev. 2019, 33, 348–364. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, Y.R.; Sheng, Y.; Warren, A.; Gao, S. GPSit: An automated method for evolutionary analysis of nonculturable ciliated microeukaryotes. Mol. Ecol. Resour. 2018, 18, 700–713. [Google Scholar] [CrossRef]
- Sheng, Y.; He, M.; Zhao, F.; Shao, C.; Miao, M. Phylogenetic relationship analyses of complicated class Spirotrichea based on transcriptomes from three diverse microbial eukaryotes: Uroleptopsis citrina, Euplotes vannus and Protocruzia tuzeti. Mol. Phylogenet. Evol. 2018, 129, 338–345. [Google Scholar] [CrossRef]
- Chalker, D.L.; Meyer, E.; Mochizuki, K. Epigenetics of ciliates. Cold Spring Harbor Perspect. Biol. 2013, 5, a017764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turley, C.; Newell, R.; Robins, D. Survival strategies of two small marine ciliates and their role in regulating bacterial community structure under experimental conditions. Mar. Ecol. Prog. Ser. 1986, 33, 59–70. [Google Scholar] [CrossRef]
- Gao, S.; Xiong, J.; Zhang, C.; Berquist, B.R.; Yang, R.; Zhao, M.; Molascon, A.J.; Kwiatkowski, S.Y.; Yuan, D.; Qin, Z. Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes Dev. 2013, 27, 1662–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Li, X.; Song, W.B.; Wang, W.; Gao, S. Cyclin Cyc2p is required for micronuclear bouquet formation in Tetrahymena thermophila. Sci. China Life Sci. 2019, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chen, X.; Sheng, Y.; Liu, Y.; Gao, S. N6-adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res. 2017, 45, 11594–11606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Wang, Y.Y.; Huang, J.B.; Chen, X.; Zhao, X.; Gao, S.; Song, W.B. Our recent progress in epigenetic research using the model ciliate, Tetrahymena thermophila. Mar. Life Sci. Tech. 2019, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yu, Y.; Li, C.; Yan, Q. Comparative study on the gut microbiotas of four economically important Asian carp species. Sci. China Life Sci. 2018, 61, 696–705. [Google Scholar] [CrossRef]
- Zhao, Y.; Yi, Z.; Warren, A.; Song, W.B. Species delimitation for the molecular taxonomy and ecology of the widely distributed microbial eukaryote genus Euplotes (Alveolata, Ciliophora). Proc. R. Soc. B 2018, 285, 20172159. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.K.; Bardgett, R.D.; Smith, P.; Reay, D.S. Microorganisms and climate change: Terrestrial feedbacks and mitigation options. Nat. Rev. Microbiol. 2010, 8, 779–790. [Google Scholar] [CrossRef]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Huang, J.A.; Filker, S.; Stoeck, T.; Bi, Y.; Yu, Y.; Song, W.B. Spatio-temporal patterns of zooplankton in a main-stem dam affected tributary: A case study in the Xiangxi River of the Three Gorges Reservoir, China. Sci. China Life Sci. 2019, 62, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Amaral-Zettler, L.A.; McCliment, E.A.; Ducklow, H.W.; Huse, S.M. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS ONE 2009, 4, e6372. [Google Scholar] [CrossRef]
- Caron, D.A.; Countway, P.D.; Savai, P.; Gast, R.J.; Schnetzer, A.; Moorthi, S.D.; Dennett, M.R.; Moran, D.M.; Jones, A.C. Defining DNA-based operational taxonomic units for microbial-eukaryote ecology. Appl. Environ. Microbiol. 2009, 75, 5797–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Dong, J.; Liu, X.; Massana, R. Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 2013, 164, 369–379. [Google Scholar] [CrossRef]
- Alverson, A.J.; Kolnick, L. Intragenomic nucleotide polymorphism among small subunit (18s) rDNA paralogs in the diatom genus Skeletonema (bacillariophyta) 1. J. Phycol. 2005, 41, 1248–1257. [Google Scholar] [CrossRef]
- Ganley, A.R.D.; Kobayashi, T. Highly efficient concerted evolution in the ribosomal DNA repeats: Total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res. 2007, 17, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, T.T.; Wang, Y.R.; Katz, L.A.; Gao, F.; Song, W.B. Disentangling sources of variation in SSU rDNA sequences from single cell analyses of ciliates: Impact of copy number variation and experimental error. Proc. R. Soc. B 2017, 284, 20170425. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.R.; Wang, C.; Jiang, Y.H.; Katz, L.A.; Gao, F.; Yan, Y. Further analyses of variation of ribosome DNA copy number and polymorphism in ciliates provide insights relevant to studies of both molecular ecology and phylogeny. Sci. China Life Sci. 2019, 62, 203–214. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Y.R.; Wang, C.; Zhang, T.T.; Al-Farraj, S.A.; Gao, F. Further consideration on the phylogeny of the Ciliophora: Analyses using both mitochondrial and nuclear data with focus on the extremely confused class Phyllopharyngea. Mol. Phylogenet. Evol. 2017, 112, 96–106. [Google Scholar] [CrossRef]
- Gao, F.; Li, J.; Song, W.; Xu, D.; Warren, A.; Yi, Z.; Gao, S. Multi-gene-based phylogenetic analysis of oligotrich ciliates with emphasis on two dominant groups: Cyrtostrombidiids and strombidiids (Protozoa, Ciliophora). Mol. Phylogenet. Evol. 2016, 105, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Alan, W.; Zhang, Q.; Gong, J.; Miao, M.; Sun, P.; Xu, D.; Huang, J.; Yi, Z.; Song, W.B. The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the phylum Ciliophora (Eukaryota, Alveolata). Sci. Rep. 2016, 6, 24874. [Google Scholar] [CrossRef] [Green Version]
- Dunthorn, M.; Hall, M.; Foissner, W.; Stoeck, T.; Katz, L.A. Broad taxon sampling of ciliates using mitochondrial small subunit ribosomal DNA. Acta Protozool. 2014, 53, 207–213. [Google Scholar]
- Huang, J.B.; Zhang, T.T.; Zhang, Q.; Li, Y.; Warren, A.; Pan, H.; Yan, Y. Further insights into the highly derived haptorids (Ciliophora, Litostomatea): Phylogeny based on multigene data. Zool. Scr. 2018, 47, 231–242. [Google Scholar] [CrossRef]
- Zhao, Y.; Yi, Z.; Gentekaki, E.; Zhan, A.; Al-Farraj, S.A.; Song, W.B. Utility of combining morphological characters, nuclear and mitochondrial genes: An attempt to resolve the conflicts of species identification for ciliated protists. Mol. Phylogenet. Evol. 2016, 94, 718. [Google Scholar] [CrossRef]
- Gentekaki, E.; Kolisko, M.; Boscaro, V.; Bright, K.J.; Dini, F.; Di Giuseppe, G.; Gong, Y.; Miceli, C.; Modeo, L.; Molestina, R.E.; et al. Large-scale phylogenomic analysis reveals the phylogenetic position of the problematic taxon Protocruzia and unravels the deep phylogenetic affinities of the ciliate lineages. Mol. Phylogenet. Evol. 2014, 78, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Strüder-Kypke, M.C.; Lynn, D.H. Comparative analysis of the mitochondrial cytochrome c oxidase subunit I (COI) gene in ciliates (Alveolata, Ciliophora) and evaluation of its suitability as a biodiversity marker. Syst. Biodivers. 2010, 8, 131–148. [Google Scholar] [CrossRef]
- Zhao, Y.; Gentekaki, E.; Yi, Z.; Lin, X. Genetic differentiation of the mitochondrial cytochrome oxidase c subunit I gene in genus Paramecium (Protista, Ciliophora). PLoS ONE 2013, 8, e77044. [Google Scholar] [CrossRef] [Green Version]
- Katz, L.A.; Kovner, A.M. Alternative processing of scrambled genes generates protein diversity in the ciliate Chilodonella uncinata. J. Exp. Zool. Part B 2010, 314, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Roy, S.W.; Katz, L.A. Analyses of alternatively processed genes in ciliates provide insights into the origins of scrambled genomes and may provide a mechanism for speciation. mBio 2015, 6, e01998. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.T.; Wang, C.; Katz, L.A.; Gao, F. A paradox: Rapid evolution rates of germline-limited sequences are associated with conserved patterns of rearrangements in cryptic species of Chilodonella uncinata (Protista, Ciliophora). Sci. China Life Sci. 2018, 61, 1071–1078. [Google Scholar] [CrossRef]
- van Hoek, A.H.; Akhmanova, A.S.; Huynen, M.A.; Hackstein, J.H. A mitochondrial ancestry of the hydrogenosomes of Nyctotherus ovalis. Mol. Biol. Evol. 2000, 17, 202–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunthorn, M.; Foissner, W.; Katz, L.A. Expanding character sampling for ciliate phylogenetic inference using mitochondrial SSU-rDNA as a molecular marker. Protist 2011, 162, 85–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, L.A.; DeBerardinis, J.; Hall, M.S.; Kovner, A.M.; Dunthorn, M.; Muse, S.V. Heterogeneous rates of molecular evolution among cryptic species of the ciliate morphospecies Chilodonella uncinata. J. Mol. Evol. 2011, 73, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.T.; Fan, X.; Gao, F.; Al-Farraj, S.A.; El-Serehy, H.A.; Song, W.B. Further analyses on the phylogeny of the subclass Scuticociliatia (Protozoa, Ciliophora) based on both nuclear and mitochondrial data. Mol. Phylogenet. Evol. 2019, 139, 106565. [Google Scholar] [CrossRef]
- Wilbert, N. Eine verbesserte Technik der Protargolimprä gnation für Ciliaten. Mikrokosmos 1975, 64, 171–179. [Google Scholar]
- Lynn, D.H. The Ciliated Protozoa: Characterization, Classification and Guide to the Literature, 3rd ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Fokin, S.I.; Przyboś, E.; Chivilev, S.M.; Beier, C.L.; Horn, M.; Skotarczak, B.; Wodecka, B.; Fujishima, M. Morphological and molecular investigations of Paramecium schewiakoffi sp. nov. (Ciliophora, Oligohymenophorea) and current status of distribution and taxonomy of Paramecium spp. Eur. J. Protistol. 2004, 40, 225–243. [Google Scholar] [CrossRef]
- Li, L.; Khan, S.N.; Ji, D.; Shin, M.K.; Berger, H. Morphology and small subunit (SSU) rRNA gene sequence of the new brackish water ciliate Neobakuella flava n.g., n. sp. (Ciliophora, Spirotricha, Bakuellidae) and SSU rRNA gene sequences of six additional hypotrichs from Korea. J. Eukaryot. Microbiol. 2011, 58, 339–351. [Google Scholar] [CrossRef]
- Shao, C.; Lv, Z.; Pan, Y.; Al-Rasheid, K.A.; Yi, Z. Morphology and phylogenetic analysis of two oxytrichid soil ciliates from China, Oxytricha paragranulifera n. sp. and Oxytricha granulifera Foissner and Adam, 1983 (Protista, Ciliophora, Hypotrichia). Int. J. Syst. Evol. Microbiol. 2014, 64, 3016–3027. [Google Scholar] [CrossRef]
- Kumar, S.; Kamra, K.; Bharti, D.; La Terza, A.; Sehgal, N.; Warren, A.; Sapra, G.R. Morphology, morphogenesis, and molecular phylogeny of Sterkiella tetracirrata n. sp. (Ciliophora, Oxytrichidae), from the Silent Valley National Park, India. Eur. J. Protistol. 2015, 51, 86–97. [Google Scholar] [CrossRef]
- Anson, E.L.; Myers, E.W. ReAligner: A program for refining DNA sequence multi-alignments. J. Comput. Biol. 1997, 4, 369–383. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinf. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015, 43, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, J. MrModeltest, a program to evaluate the fit of several models of evolution to a given data and unrooted tree (version 2.2). Program distributed by the author. Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. Available online: http://www. abc. se/~ nylander (accessed on 12 January 2020).
- Suyama, Y.; Miura, K. Size and structural variations of mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1968, 60, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Bisharyan, Y.; Clark, T.G. Calcium-dependent mitochondrial extrusion in ciliated protozoa. Mitochondrion 2011, 11, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Burger, G.; Zhu, Y.; Littlejohn, T.G.; Greenwood, S.J.; Schnare, M.N.; Lang, B.F.; Gray, M.W. Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA. J. Mol. Biol. 2000, 297, 365–380. [Google Scholar] [CrossRef] [Green Version]
- Barth, D.; Berendonk, T.U. The mitochondrial genome sequence of the ciliate Paramecium caudatum reveals a shift in nucleotide composition and codon usage within the genus Paramecium. BMC Genomics 2011, 12, 272. [Google Scholar] [CrossRef] [Green Version]
- de Graaf, R.M.; van Alen, T.A.; Dutilh, B.E.; Kuiper, J.W.; van Zoggel, H.J.; Huynh, M.B.; Görtz, H.-D.; Huynen, M.A.; Hackstein, J.H. The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genomics 2009, 10, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swart, E.C.; Nowacki, M.; Shum, J.; Stiles, H.; Higgins, B.P.; Doak, T.G.; Schotanus, K.; Magrini, V.J.; Minx, P.; Mardis, E.R.; et al. The Oxytricha trifallax mitochondrial genome. Genome Biol. Evol. 2011, 4, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-M.; Min, G.-S.; Kim, S. The mitochondrial genome of the ciliate Pseudourostyla cristata (Ciliophora, Urostylida). Mitochondrial DNA Part B 2019, 4, 66–67. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Gao, Y.; Hou, Y.; Ye, S.; Wang, L.; Sun, J.; Li, Q. Mitochondrial genome sequencing and analysis of scuticociliates (Uronema marinum) isolated from Takifugu rubripes. Mitochondrial DNA Part B 2018, 3, 736–737. [Google Scholar] [CrossRef] [Green Version]
- Hales, K.G. The machinery of mitochondrial fusion, division, and distribution, and emerging connections to apoptosis. Mitochondrion 2004, 4, 285–308. [Google Scholar] [CrossRef]
- Parsons, J.A.; Rustad, R.C. The distribution of DNA among dividing mitochondria of Tetrahymena pyriformis. J. Cell Biol. 1968, 37, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perasso, R.; Beisson, J. Temporal pattern of mitochondrial multiplication during cell-cycle of Paramecium. Biol. Cell. 1978, 32, 275–290. [Google Scholar]
- Kolb-Bachofen, V.; Vogell, W. Mitochondrial proliferation in synchronized cells of Tetrahymena pyriformis: A morphometric study by electron microscopy on the biogenesis of mitochondria during the cell cycle. Exp. Cell Res. 1975, 94, 95–105. [Google Scholar] [CrossRef]
- Włoga, D.; Strzyżewska-Jówko, I.; Gaertig, J.; Jerka-Dziadosz, M. Septins stabilize mitochondria in Tetrahymena thermophila. Eukaryot. Cell 2008, 7, 1373–1386. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Karrer, K.M. Genomic reorganization in ciliated protozoans. Annu. Rev. Genet. 1986, 20, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Prescott, D.M. The DNA of ciliated protozoa. Microbiol. Rev. 1994, 58, 233–267. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.S. The Tetrahymena conjugation junction. In Cell-Cell Channels; Springer: New York, NY, USA, 2006; pp. 39–62. [Google Scholar]
- Dunthorn, M.; Katz, L.A.; Stoeck, T.; Foissner, W. Congruence and indifference between two molecular markers for understanding oral evolution in the Marynidae sensu lato (Ciliophora, Colpodea). Eur. J. Protistol. 2012, 48, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Systematic Classification | mtSSU rDNA Copy Number | nSSU rDNA Copy Number |
|---|---|---|---|
| Epistylis sp. | Oligohymenophorea | 8.1 × 105 ± 4.7 × 104 | 3.3 × 105 ± 1.6 × 105 |
| Paramecium caudatum | Oligohymenophorea | 1.5 × 105 ± 4.9 × 104 | 1.7 × 105 ± 1.3 × 105 |
| Paramecium sp. | Oligohymenophorea | 1.1 × 105 ± 2.4 × 104 | 4.3 × 105 ± 2.1 × 105 |
| Tetrahymena thermophila | Oligohymenophorea | 1.1 × 104 ± 9.6 × 103 | 1.5 × 104 ± 7.1 × 103 |
| Favella ehrenbergii | Spirotrichea | 1.5 × 105 ± 9.7 × 104 | 4.9 × 105 ± 2.7 × 105 |
| Sterkiella sp. | Spirotrichea | 8.4 × 104 ± 3.0 × 104 | 1.1 × 106 ± 3.5 × 105 |
| Phacodinium metchnikoffi | Spirotrichea | 7.1 × 104 ± 3.4 × 104 | 6.0 × 105 ± 4.7 × 105 |
| Neobakuella aenigmatica | Spirotrichea | 4.7 × 104 ± 1.3 × 104 | 1.9 × 105 ± 1.2 × 103 |
| Euplotes vannus | Spirotrichea | 3.1 × 104 ± 3.8 × 103 | 1.0 × 105 ± 7.2 × 104 |
| Oxytricha trifallax | Spirotrichea | 2.9 × 104 ± 9.3 × 103 | 1.1 × 105 ± 5.7 × 104 |
| Spirostomum sp. | Heterotrichea | 1.4 × 105 ± 1.9 × 104 | 2.1 × 105 ± 1.3 × 105 |
| Trithigmostoma sp. | Phyllopharyngea | 1.0 × 105 ± 2.2 × 104 | 1.7 × 104 ± 8.3 × 103 |
| Coleps sp. | Prostomatea | 1.0 × 104 ± 9.5 × 102 | 5.8 × 103 ± 2.1 × 103 |
| Species | Polymorphism Style | # of Total Polymorphism Sites | Number of Clones | Length |
|---|---|---|---|---|
| Epistylis sp. | Transition | 1 | 20 | 1204 |
| Paramecium caudatum | Transition, deletion, insertion | 6 | 30 | 990 |
| Paramecium sp. | No | 0 | 90 | 1016 |
| Tetrahymena thermophila | No | 0 | 30 | 1037 |
| Favella ehrenbergii | Transition, deletion | 2/1 | 30 | 1604/1608 |
| Sterkiella sp. | Deletion, insertion | 2 | 30 | 1213 |
| Phacodinium metchnikoffi | No | 0 | 30 | 1047 |
| Neobakuella aenigmatica | No | 0 | 90 | 1600 |
| Euplotes vannus | No | 0 | 30 | 968 |
| Oxytricha trifallax | Deletion, insertion | 6 | 30 | 1151 |
| Spirostomum sp. | Deletion, insertion | 2 | 90 | 977 |
| Trithigmostoma sp. | Transversion, transition, deletion, insertion | 10 | 90 | 1024 |
| Coleps sp. | Deletion, insertion | 6 | 90 | 959 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Jiang, Y.; Liu, Y.; Li, Y.; Katz, L.A.; Gao, F.; Yan, Y. Comparative Studies on the Polymorphism and Copy Number Variation of mtSSU rDNA in Ciliates (Protista, Ciliophora): Implications for Phylogenetic, Environmental, and Ecological Research. Microorganisms 2020, 8, 316. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030316
Wang Y, Jiang Y, Liu Y, Li Y, Katz LA, Gao F, Yan Y. Comparative Studies on the Polymorphism and Copy Number Variation of mtSSU rDNA in Ciliates (Protista, Ciliophora): Implications for Phylogenetic, Environmental, and Ecological Research. Microorganisms. 2020; 8(3):316. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030316
Chicago/Turabian StyleWang, Yurui, Yaohan Jiang, Yongqiang Liu, Yuan Li, Laura A. Katz, Feng Gao, and Ying Yan. 2020. "Comparative Studies on the Polymorphism and Copy Number Variation of mtSSU rDNA in Ciliates (Protista, Ciliophora): Implications for Phylogenetic, Environmental, and Ecological Research" Microorganisms 8, no. 3: 316. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8030316