Notch-1 Signaling Modulates Macrophage Polarization and Immune Defense against Mycobacterium avium paratuberculosis Infection in Inflammatory Diseases


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Bacterial Culture
2.3. Quantitative Real-Time PCR (RT-PCR)
2.4. Measurement of MAP Viability and Load
2.5. Measurement of Active Cadpase-3 and IL-6 Proteins Levels by Enzyme-linked Immunosorbent Assay (ELISA)
2.6. Statistical Analysis
3. Results
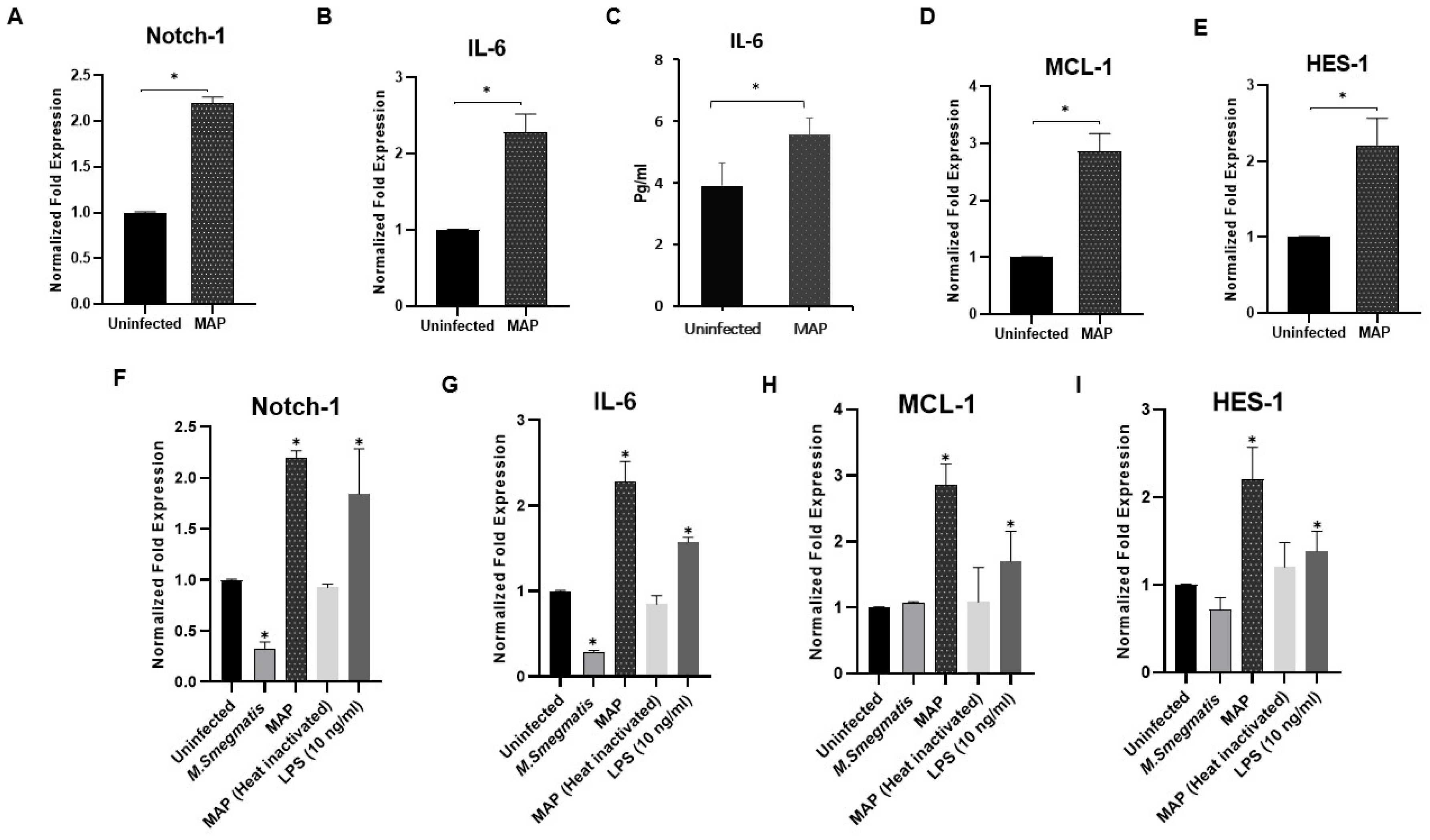
3.1. MAP Infection Induces Notch-1, IL-6 and MCL-1 Expression in Macrophage
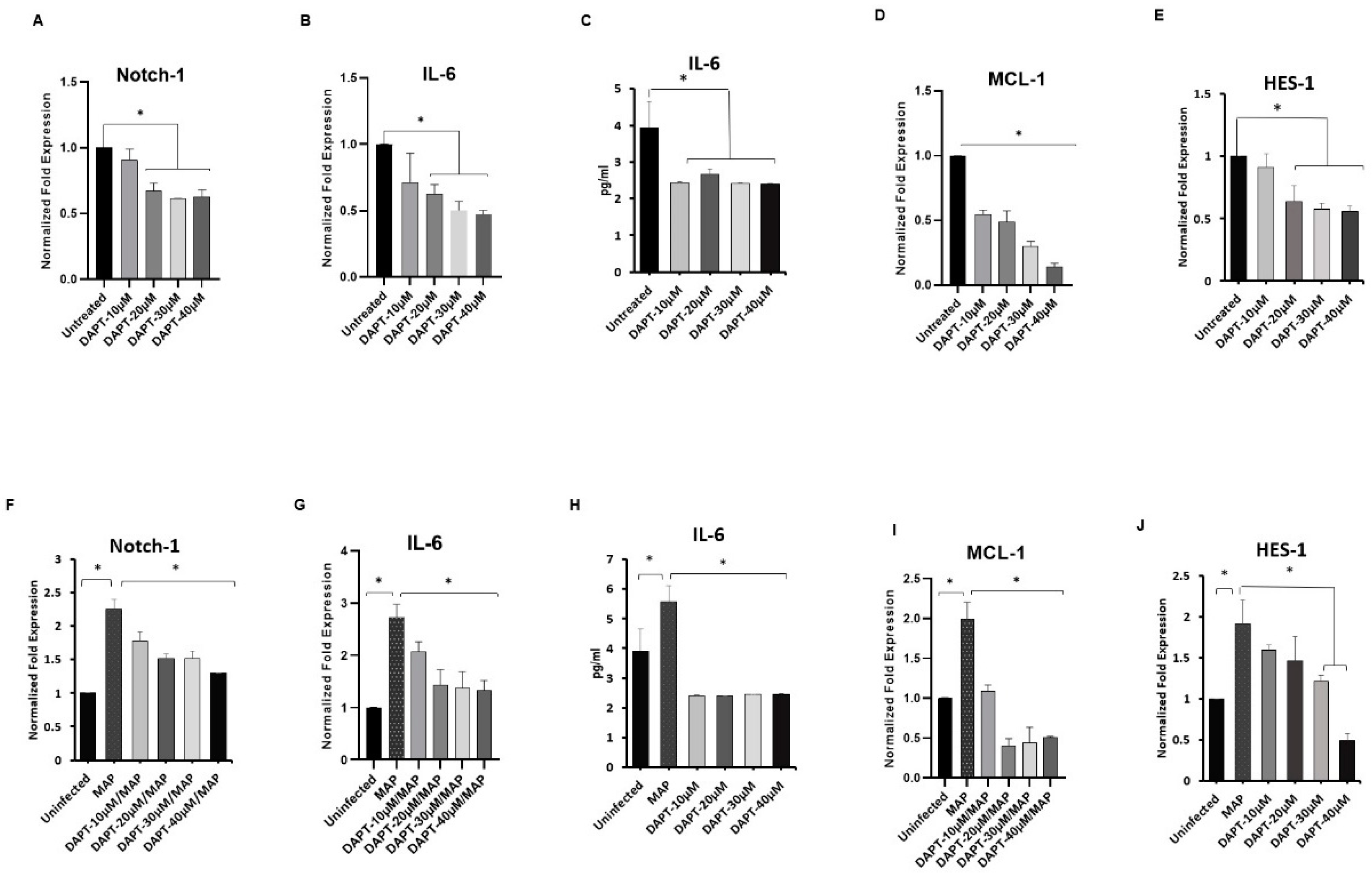
3.2. Effect of DAPT on Expression of Notch-1, IL-6 and, MCL-1 in Macrophages
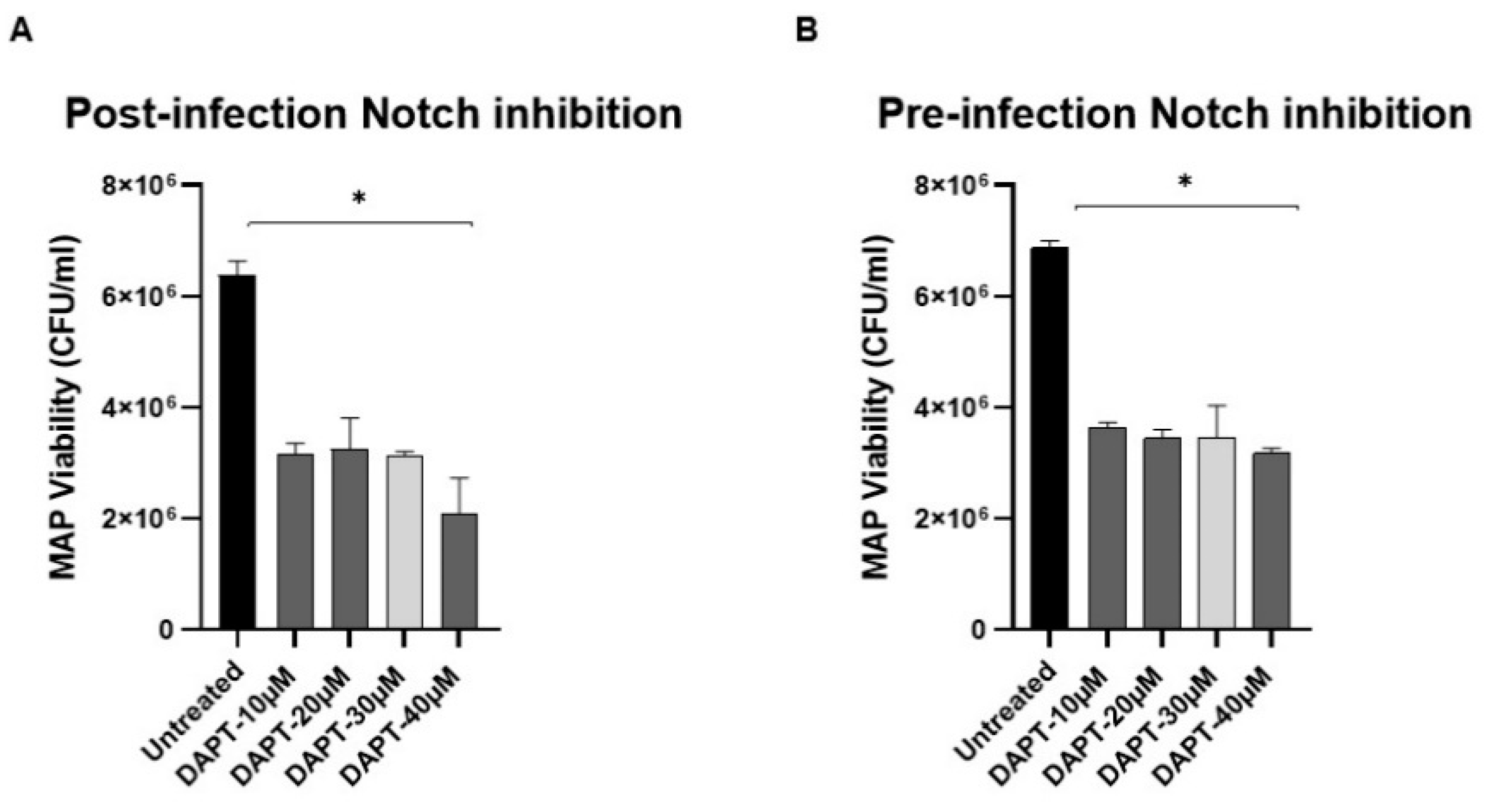
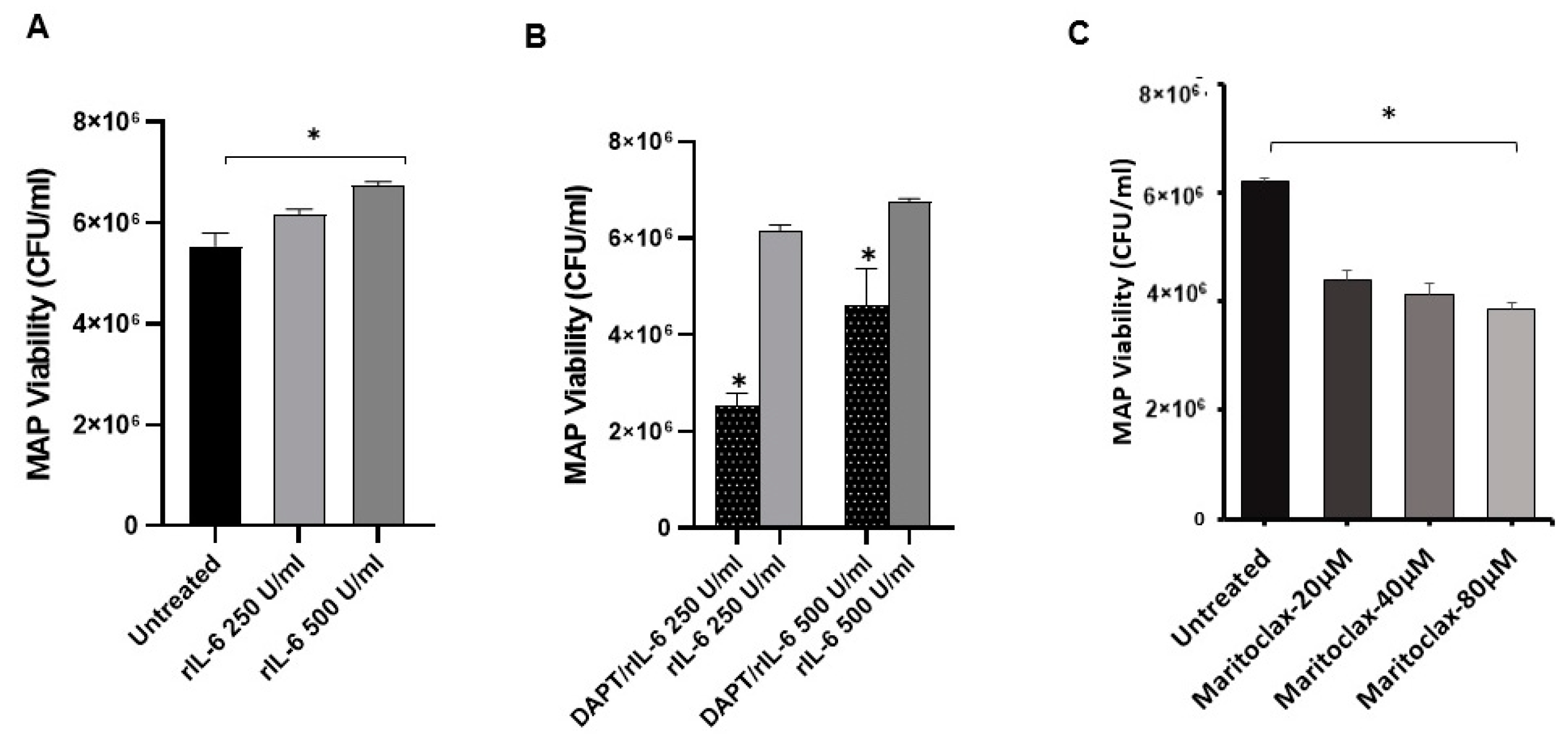
3.3. Effect of DAPT on MAP Survival in Macrophage via Notch-1
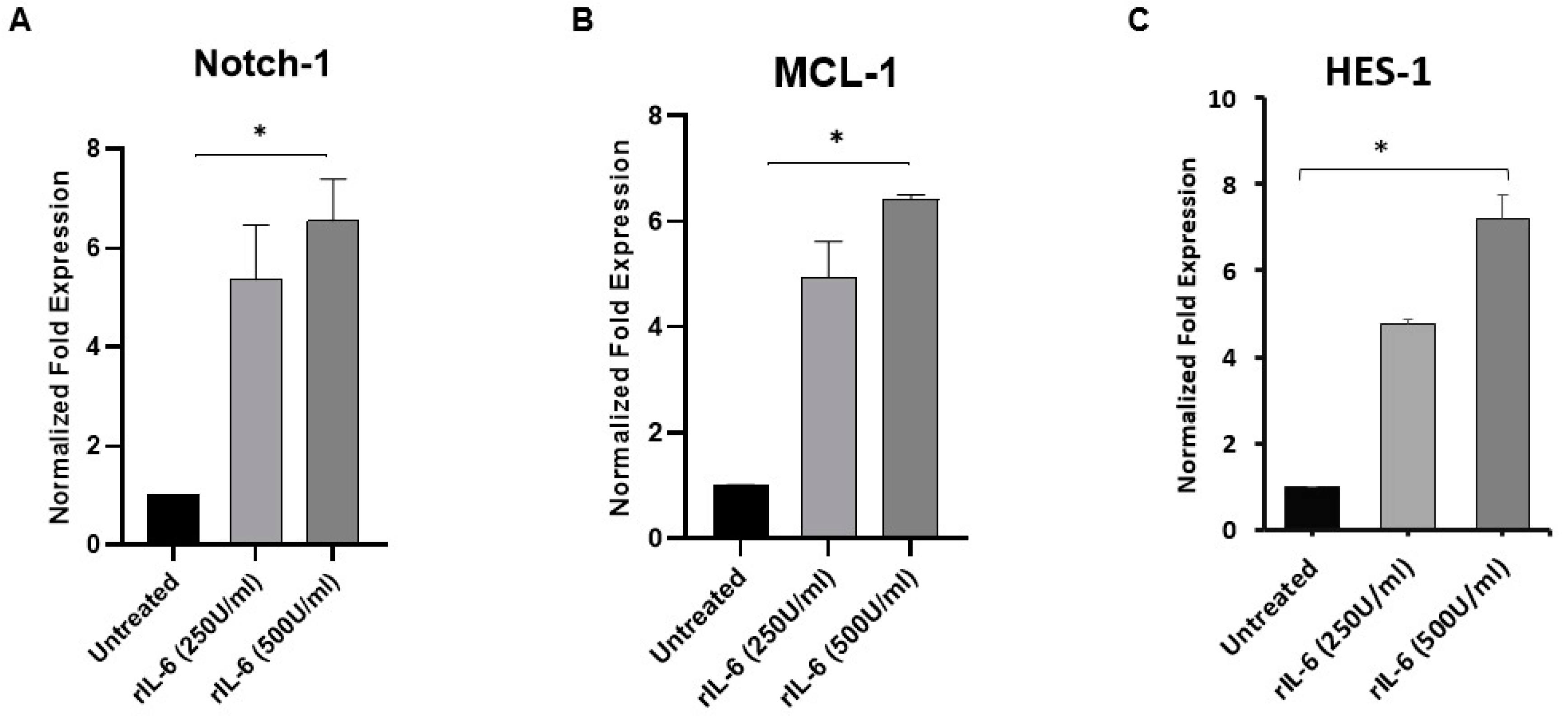
3.4. rIL-6 Induces Notch-1, MCL-1 and HES-1 Expression in THP-1-Derived Macrophage
3.5. Effects of rIL-6 on MAP Viability in Infected Macrophages via Notch Signaling
3.6. Notch Signaling Alters Apoptosis in MAP-Infected Macrophages
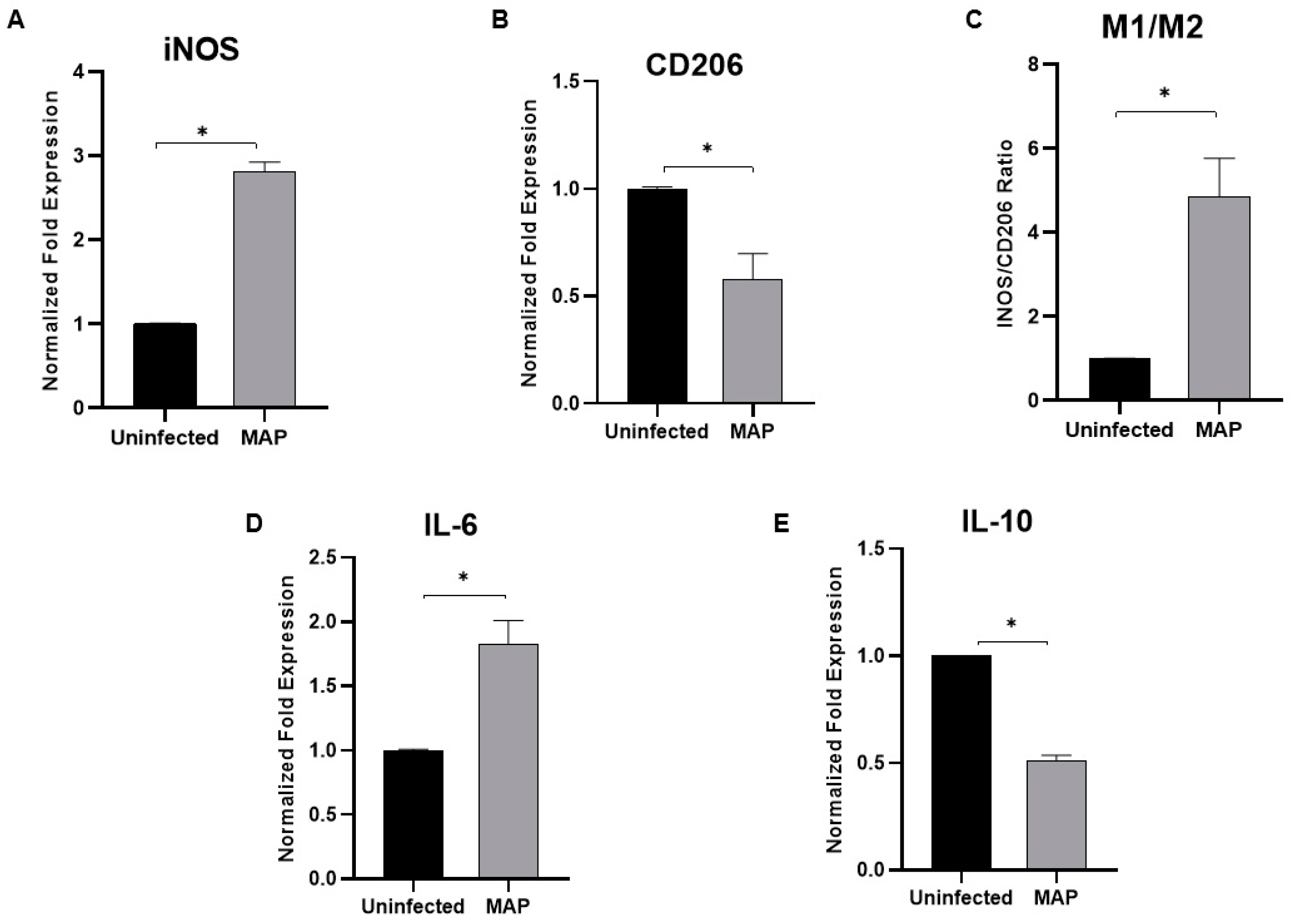
3.7. MAP Infection Modulates Macrophage Polarization
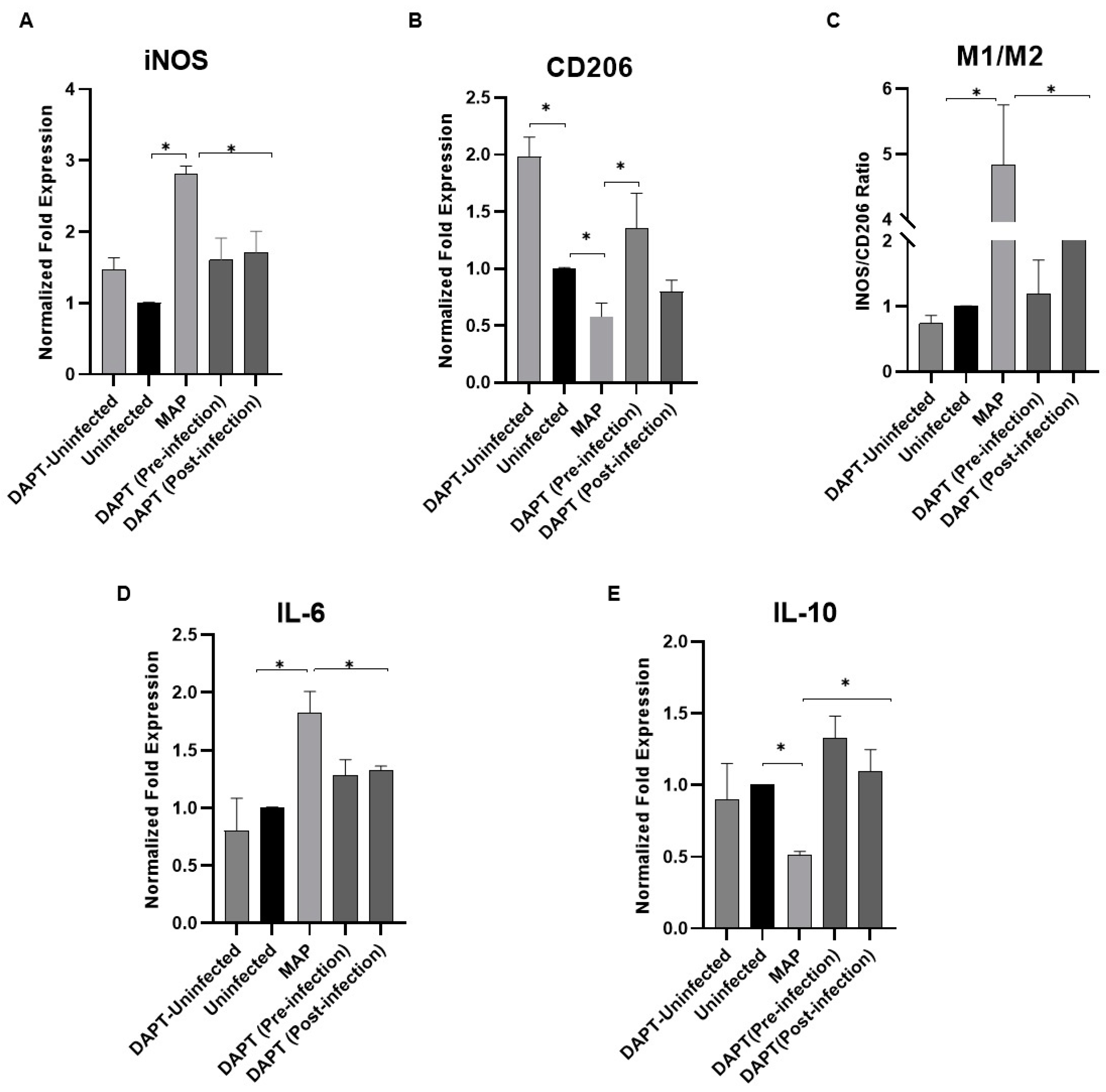
3.8. Determination of Notch-1 Signaling Role in Macrophage Polarization in MAP-Infected Macrophages
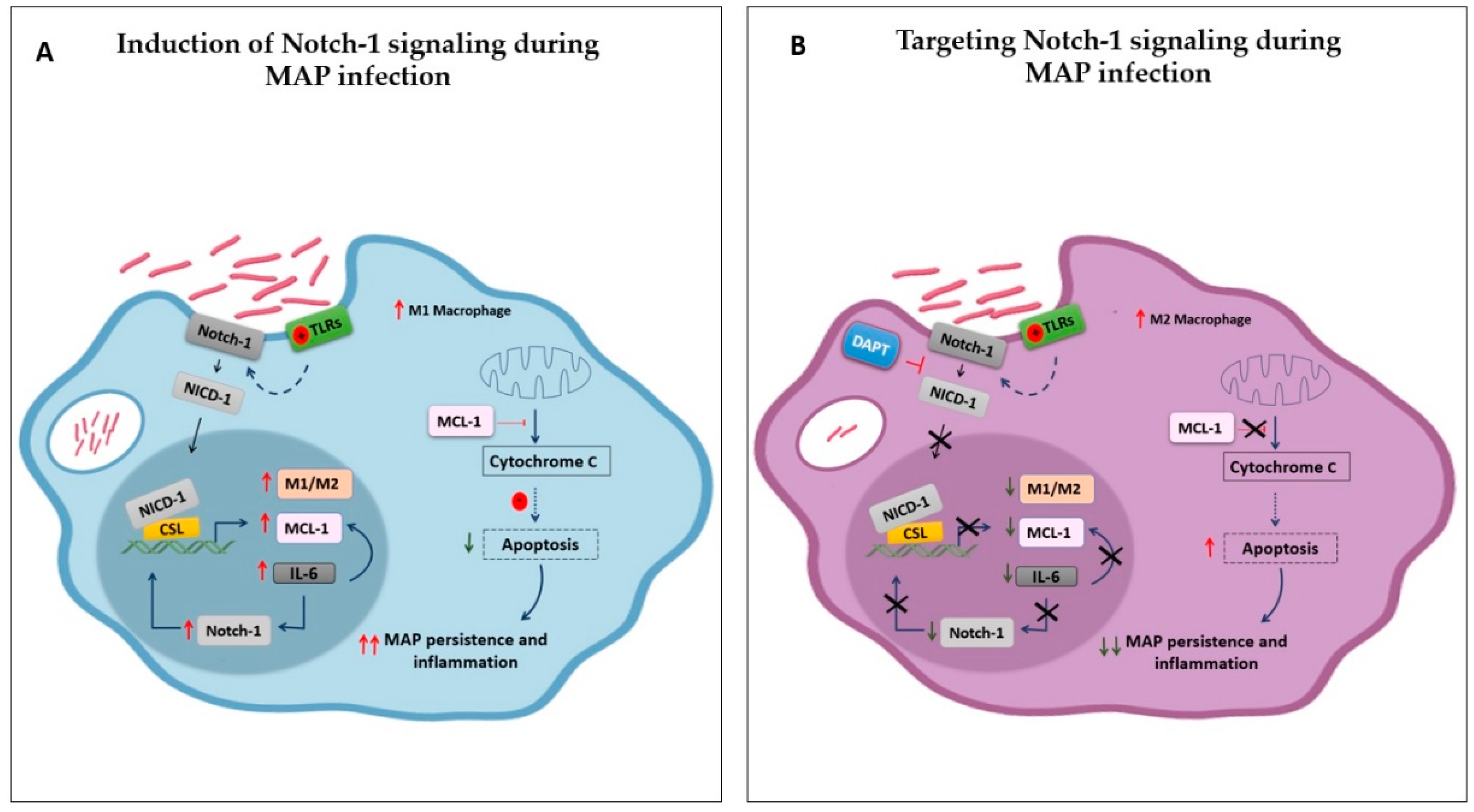
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keewan, E.; Naser, S.A. The role of Notch signaling in macrophages during inflammation and infection: Implication in rheumatoid arthritis? Cells 2020, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Radtke, F.; Fasnacht, N.; MacDonald, H.R. Notch signaling in the immune system. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, F.; MacDonald, H.R. Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat. Reviews Immunol. 2013, 13, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, S.; Tripathi, B.N.; Singh, N. Mechanisms of Mycobacterium avium subsp. paratuberculosis induced apoptosis and necrosis in bovine macrophages. Vet. Microbiology 2013, 165, 392–401. [Google Scholar] [CrossRef]
- Naser, S.A.; Ghobrial, G.; Romero, C.; Valentine, J.F. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet 2004, 364, 1039–1044. [Google Scholar] [CrossRef]
- Naser, S.A.; Thanigachalam, S.; Dow, C.T.; Collins, M.T. Exploring the role of Mycobacterium avium subspecies paratuberculosis in the pathogenesis of type 1 diabetes mellitus: A pilot study. Gut Pathog. 2013, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Sharp, R.C.; Beg, S.A.; Naser, S.A. Polymorphisms in protein tyrosine phosphatase non-receptor type 2 and 22 (PTPN2/22) are linked to hyper-proliferative T-Cells and susceptibility to mycobacteria in rheumatoid arthritis. Front. Cell. Infect. Microbiol. 2018, 8, 11. [Google Scholar] [CrossRef]
- Sly, L.M.; Hingley-Wilson, S.M.; Reiner, N.E.; McMaster, W.R. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J. Immunol. 2003, 170, 170,430–437. [Google Scholar] [CrossRef] [Green Version]
- Winau, F.; Weber, S.; Sad, S.; De Diego, J.; Hoops, S.L.; Breiden, B.; Sandhoff, K.; Brinkmann, V.; Kaufmann, S.H.; Schaible, U.E. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 2006, 24, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Osborne, B. Arbiter of differentiation and death: Notch signaling meets apoptosis. J. Cell. Physiol. 1999, 181, 393–409. [Google Scholar] [CrossRef]
- Palaga, T.; Ratanabunyong, S.; Pattarakankul, T.; Sangphech, N.; Wongchana, W.; Hadae, Y.; Kueanjinda, P. Notch signaling regulates expression of Mcl-1 and apoptosis in PPD-treated macrophages. Cell. Mol. Immunol. 2013, 10, 444–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. Febs Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Ma, Y.; Cole, S.M.; Zander, C.; Chen, K.H.; Karras, J.; Pope, R.M. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood 2003, 102, 344–352. [Google Scholar] [CrossRef]
- Niemand, C.; Nimmesgern, A.; Haan, S.; Fischer, P.; Schaper, F.; Rossaint, R.; Heinrich, P.C.; Müller-Newen, G. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J. Immunol. 2003, 170, 3263–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qasem. A.; Naser, S.A. TNFα inhibitors exacerbate Mycobacterium paratuberculosis infection in tissue culture: A rationale for poor response of patients with Crohn’s disease to current approved therapy. BMJ Open Gastroenterol. 2018, 5, e000216. [Google Scholar] [CrossRef] [Green Version]
- Wongchana, W.; Palaga, T. Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell. Mol. Immunol. 2012, 9, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Mutvei, A.P.; Chivukula, I.V.; Andersson, E.R.; Ramsköld, D.; Sandberg, R.; Lee, K.L.; Kronqvist, P.; Mamaeva, V.; Östling, P.; et al. Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKα/IKKβ. Oncogene 2013, 32, 4892–4902. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Ushio, A.; Arakaki, R.; Yamada, A.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Crucial roles of macrophages in the pathogenesis of autoimmune disease. World J. Immunol. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.; Tang, S.M.; Canner, J.P.; Morishige, K.; Arboleda-Velasquez, J.F.; Cardoso, A.A.; Carlesso, N.; Aster, J.C.; Aikawa, M. Delta-Like 4 Induces Notch Signaling in Macrophages. Circulation 2007, 115, 2948–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Schaller, M.; Hogaboam, C.M.; Standiford, T.J.; Sandor, M.; Lukacs, N.W.; Chensue, S.W.; Kunkel, S.L. TLR9 regulates the mycobacteria-elicited pulmonary granulomatous immune response in mice through DC-derived Notch ligand delta-like 4. J. Clin. Investig. 2009, 119, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Narayana, Y.; Balaji, K.N. NOTCH1 up-regulation and signaling involved in Mycobacterium bovis BCG-induced SOCS3 expression in macrophages. J. Biol. Chem. 2008, 283, 12501–12511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Liu, H.; Zhou, Y.; Yan, D.; Chen, J.; Ma, D.; Feng, Y.; Qin, L.; Liu, F.; Huang, X.; et al. Notch4 negatively regulates the inflammatory response to Mycobacterium tuberculosis infection by inhibiting TAK1 activation. J. Infect. Dis. 2018, 218, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Allen, R.M.; Carson, I.V.W.F.; Schaller, M.; Cavassani, K.A.; Hogaboam, C.M.; Lukacs, N.W.; Matsukawa, A.; Kunkel, S.L. The critical role of Notch ligand Delta-like 1 in the pathogenesis of influenza A virus (H1N1) infection. PLoS Pathog. 2011, 7, e1002341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehaffy, C.; Belisle, J.T.; Dobos, K.M. Mycobacteria and their sweet proteins: An overview of protein glycosylation and lipoglycosylation in M. tuberculosis. Tuberculosis 2019, 115, 1–3. [Google Scholar] [CrossRef]
- Hildebrand, D.; Uhle, F.; Sahin, D.; Krauser, U.; Weigand, M.A.; Heeg, K. The interplay of notch signaling and STAT3 in TLR-activated human primary monocytes. Front. Cell. Infect. Microbiol. 2018, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Riendeau, C.J.; Kornfeld, H. THP-1 cell apoptosis in response to Mycobacterial infection. Infect. Immun. 2003, 71, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.T.; Sommer, S.; Kabara, E.A.; Verman, N.; Kuelbs, M.A.; Saama, P.; Halgren, R.; Coussens, P.M. Gene expression profiling of monocyte-derived macrophages following infection with Mycobacterium avium subspecies avium and Mycobacterium avium subspecies paratuberculosis. Physiol. Genom. 2006, 28, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Kiszewski, A.E.; Becerril, E.; Aguilar, L.D.; Kader, I.T.; Myers, W.; Portaels, F.; Hernandez Pando, R. The local immune response in ulcerative lesions of Buruli disease. Clin. Exp. Immunol. 2006, 143, 445–451. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′-3′) | Amplicon Length (bp) |
|---|---|---|
| β-actin | CTCATCTTGTTTTCTGCGCAAGTT CTTCCCTCCTCAGATCATTGCTC | 226 |
| Notch-1 | TGAAATTCAGGGCCCCTCC GCATCGGGCACCTGAAC | 162 |
| HES-1 | CAGATCAATGCCATGACCTACCC GGAAAGCAAACTGGCCATCG | 250 |
| MCL-1 | TGGTGGTGGTTGGTTAAAAGTCA GTGGAGTTCTTCCATGTAGAGGAC | 152 |
| IL-6 | AGGAGAAGATTCCAAAGATGTAGCC TGCTCTAGAACCCAGCAAAGAC | 228 |
| iNOS | GGAGCAACGTTGAGGAAATAAGACT AAGAGCCAGAAGCGCTATCAC | 252 |
| CD206 | GGAGGATTCCATGTATTTGTGAGC AAATGAGTGAAGTGAAATCAGTTACCT | 510 |
| IL-10 | ATGTCTAGTTCAGGCAGTCCCA GGGCTTGCTCTTGCAAAACC | 272 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keewan, E.; Naser, S.A. Notch-1 Signaling Modulates Macrophage Polarization and Immune Defense against Mycobacterium avium paratuberculosis Infection in Inflammatory Diseases. Microorganisms 2020, 8, 1006. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8071006
Keewan E, Naser SA. Notch-1 Signaling Modulates Macrophage Polarization and Immune Defense against Mycobacterium avium paratuberculosis Infection in Inflammatory Diseases. Microorganisms. 2020; 8(7):1006. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8071006
Chicago/Turabian StyleKeewan, Esra’a, and Saleh A. Naser. 2020. "Notch-1 Signaling Modulates Macrophage Polarization and Immune Defense against Mycobacterium avium paratuberculosis Infection in Inflammatory Diseases" Microorganisms 8, no. 7: 1006. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8071006