The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Founding Elements of the Taxon Hypothesis Paradigm
- The theory of scientific hypotheses and their falsification has greatly influenced the development of the TH paradigm (e.g., [12,13]). However, we acknowledge that the best taxonomic papers published before Popper’s works included taxonomic descriptions amenable to falsification in the sense that high-quality taxon descriptions contain list of studied specimens and their properties. Therefore, other researchers can restudy specimens lodged in public collections and falsify primary taxon descriptions. However, our understanding that taxon descriptions can be viewed as proper scientific hypotheses emerged only when the theory of falsification (and the debate that ensued) became available. The TH paradigm seeks to capture scientific hypotheses and to provide a venue for their falsification through all levels of the tree of life and across time. The aim of this paper is not to discuss the limitations of falsification—we just want to emphasize its influence on the TH paradigm in its early stages of conception in the late 1980s.
- Zavadski’s book [14] features rich information on species criteria (SC), namely, morphological, biochemical, geographical, ecological, genetical, and physiological properties of the species. He had a view that all—or at least most—of these criteria must be used for the discrimination of species. Zavadski also introduced the practical species standard (PSS), which is a set of instructions on how species boundaries and content are defined. His view was that species theory or species concepts must be kept separately from PSS. Our TH paradigm takes into consideration that (1) all species criteria should be considered when delimiting species; and (2) the paradigm can be accompanied with one to many practical species standards. The UNITE identification and communication system is one example of the PSS of the TH paradigm. Therefore, this discussion of the TH paradigm is illustrated by examples from the UNITE system. These examples can also be called UNITE PSS sensu Zavadski. The major difference is that the TH paradigm is widened to include taxa at all levels, not only species.
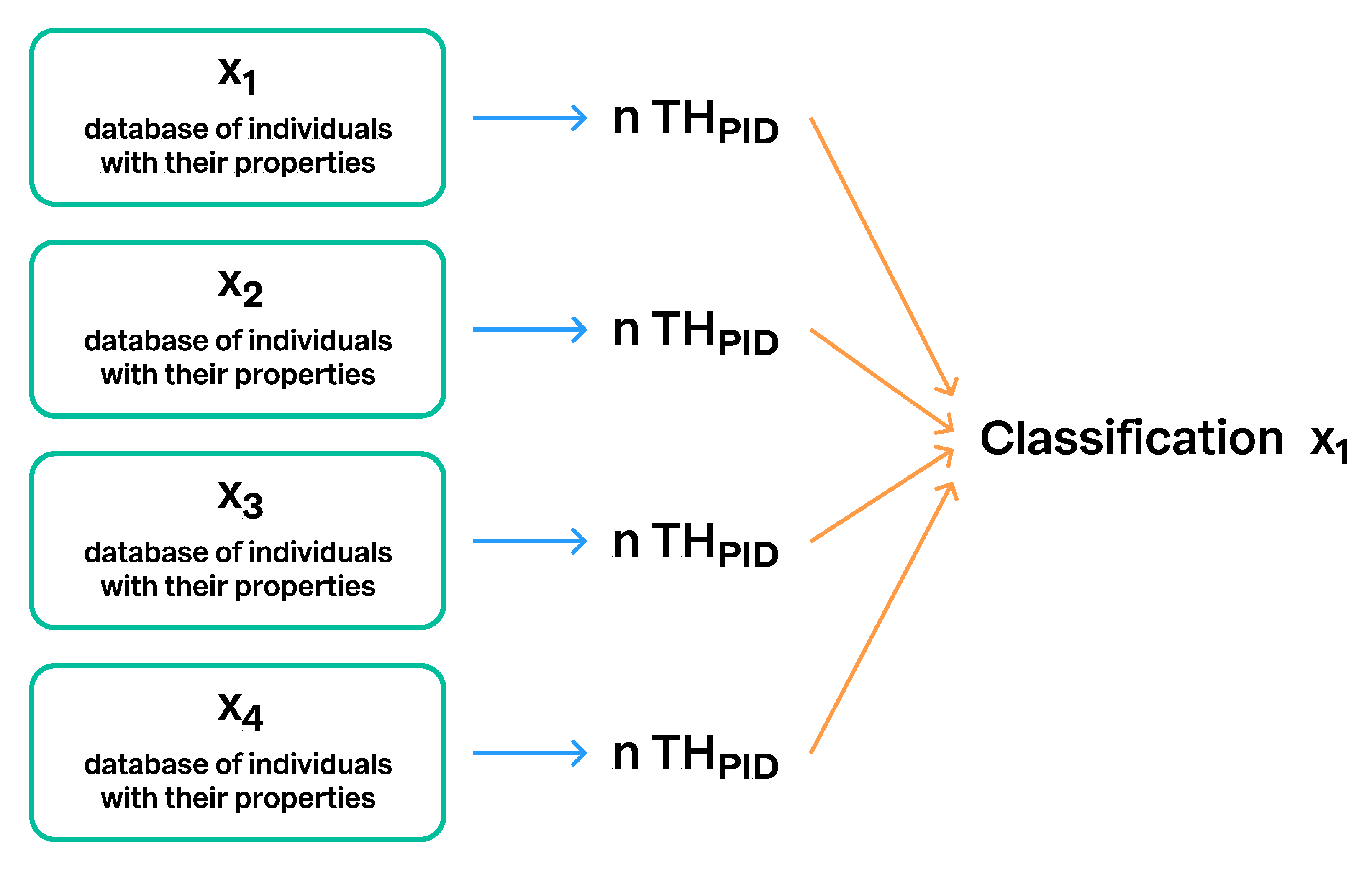
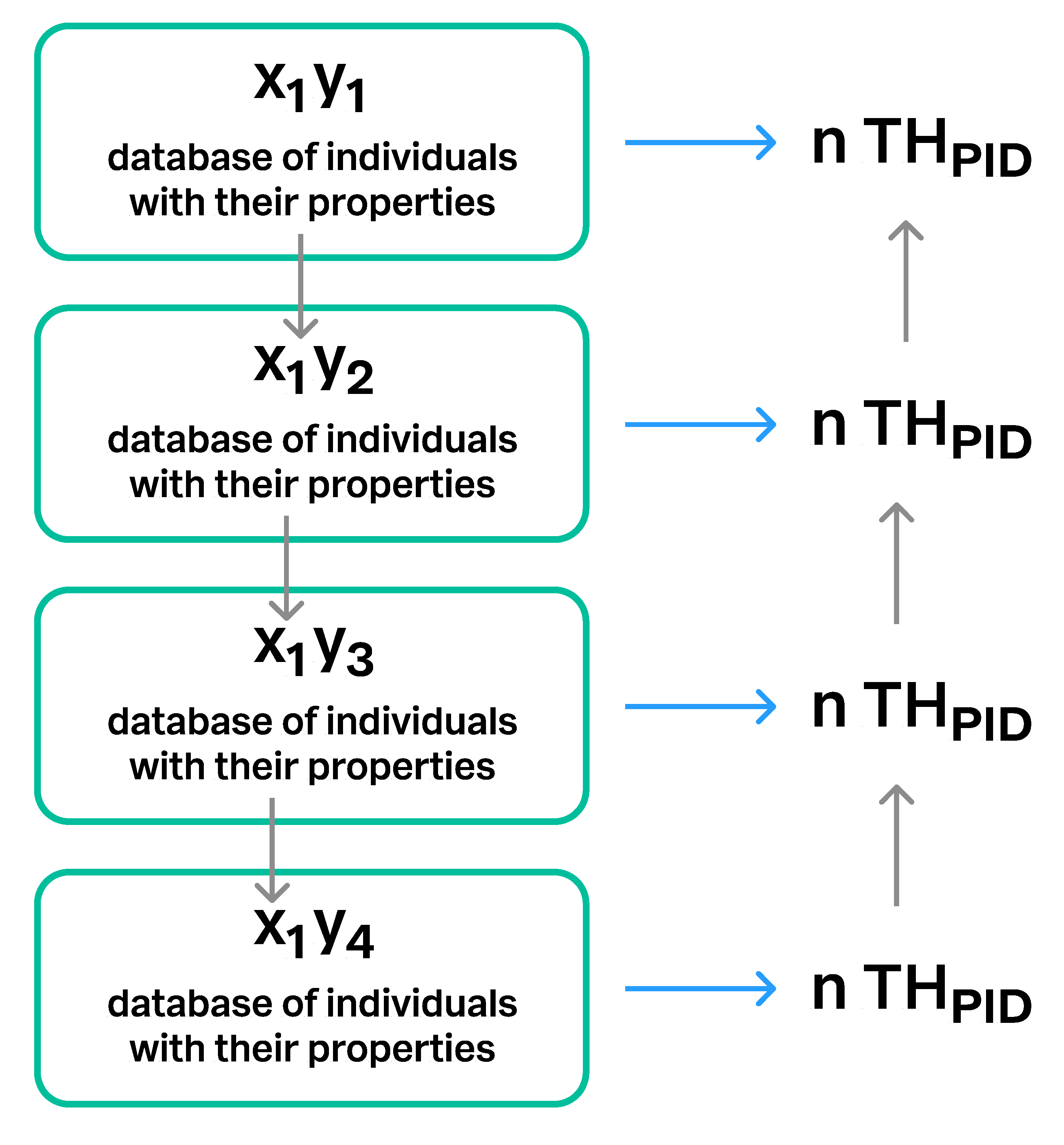
- Dallwitz [15,16] developed the Descriptive Language for Taxonomy (DELTA), which is a computer-based system for encoding and management of taxonomic characters or attributes. The DELTA format allows the user to atomize all properties (characters sensu Dallwitz) of the biological individuals and taxa, and then build datasets of encoded properties called Items. Originally, the Items were conceived for the computational processing of taxon descriptions and for the automated generation of identification keys. THs have some similarities to the DELTA system. They, too, are datasets of taxon properties similar to DELTA Items. The major difference is that the TH datasets include individuals, and that properties are always attached to the individuals. The DELTA Items, however, are taxon descriptions, where the properties of the individuals are summarized such that the property of each individual is lost. An exception would be when Items include properties of a single individual. In the TH paradigm, individuals and their properties are conceptually free to float between datasets. They may appear in another TH when new hypotheses are computed. However, the same individuals in different THs are linked through the unique PIDs. This feature is also implemented in the UNITE system.
2.2. UNITE—A Practical Example of the Taxon Hypothesis Paradigm
2.2.1. UNITE SH Calculations
2.2.2. UNITE TH Construction
2.3. PlutoF—The Data Management Platform for Taxon Hypotheses
2.4. Implementation of the UNITE Species Hypothesis System in GBIF
3. Results
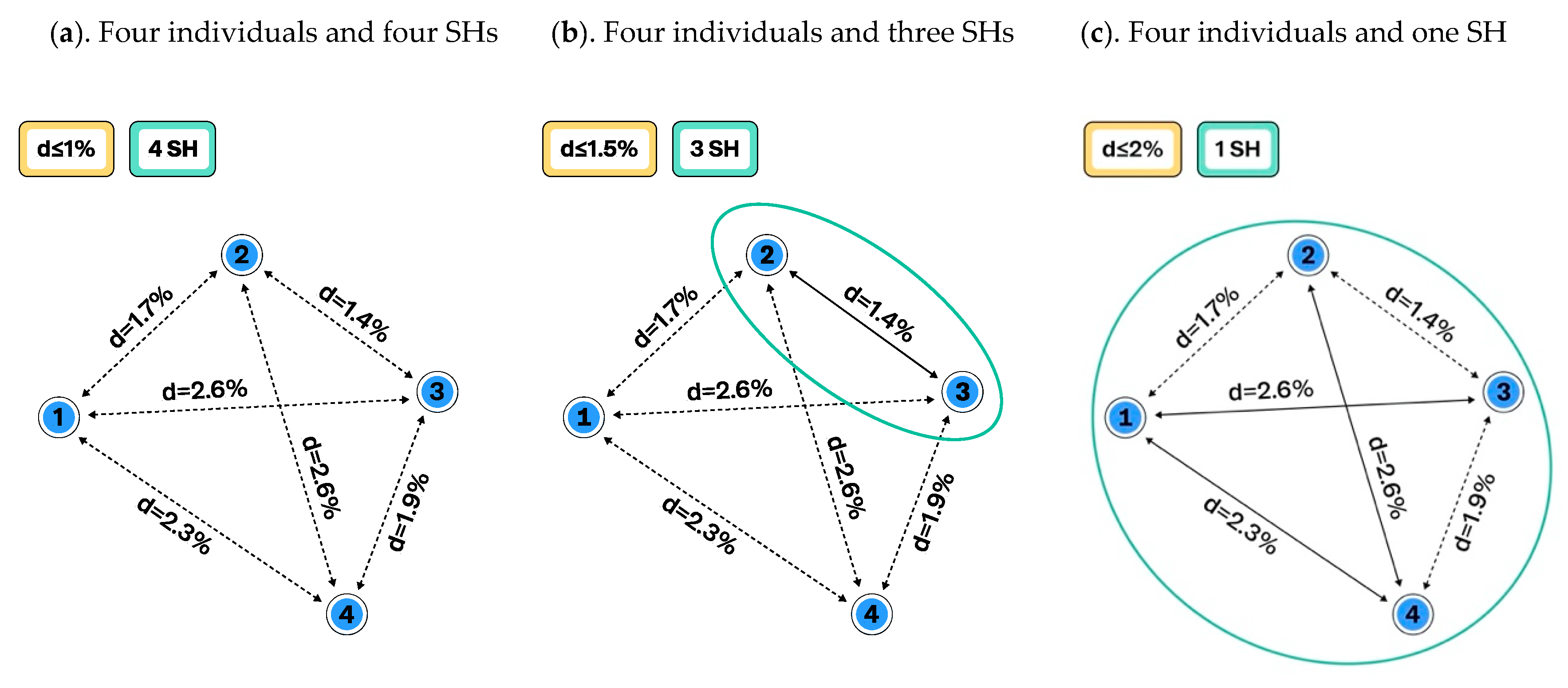
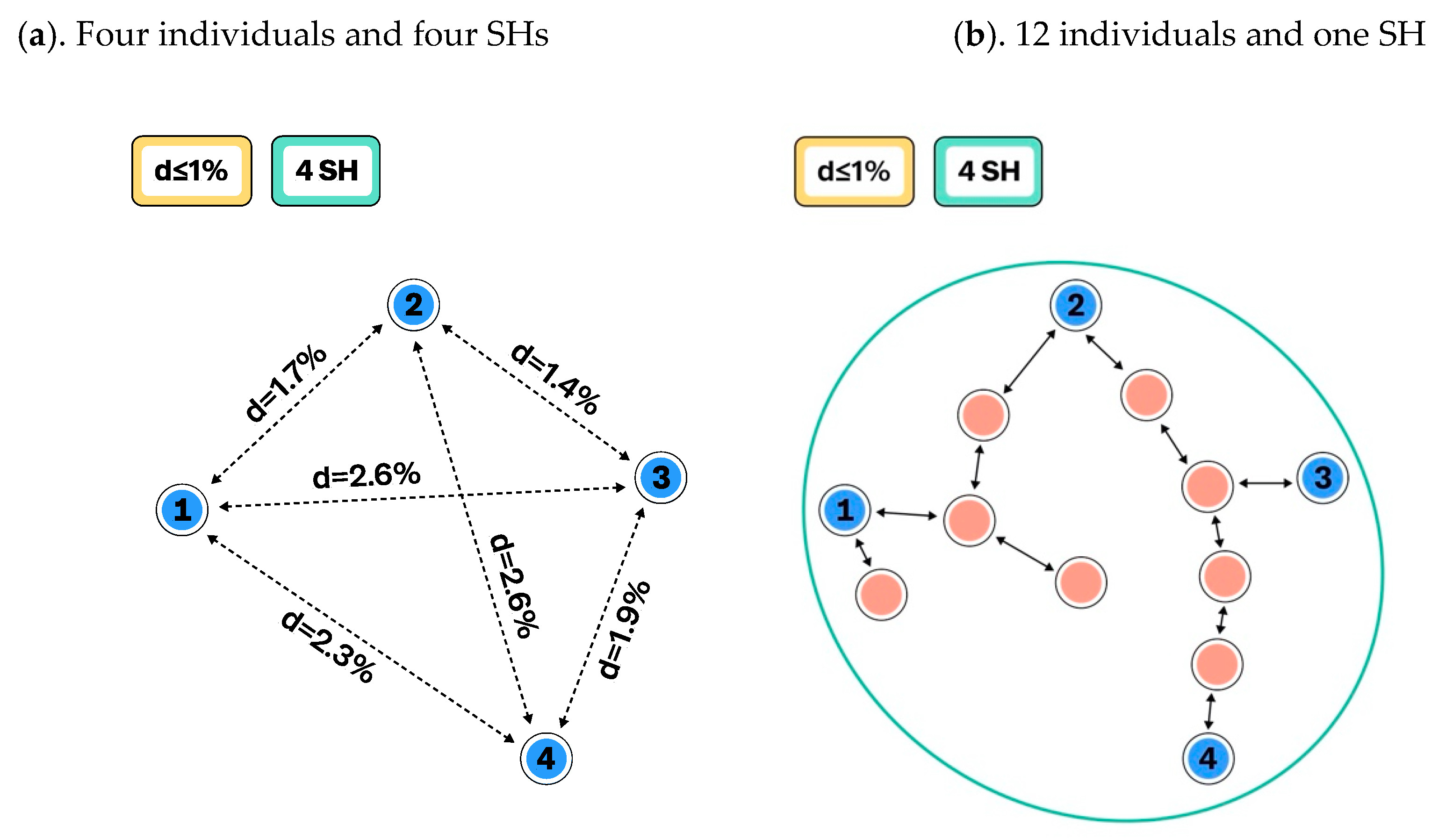
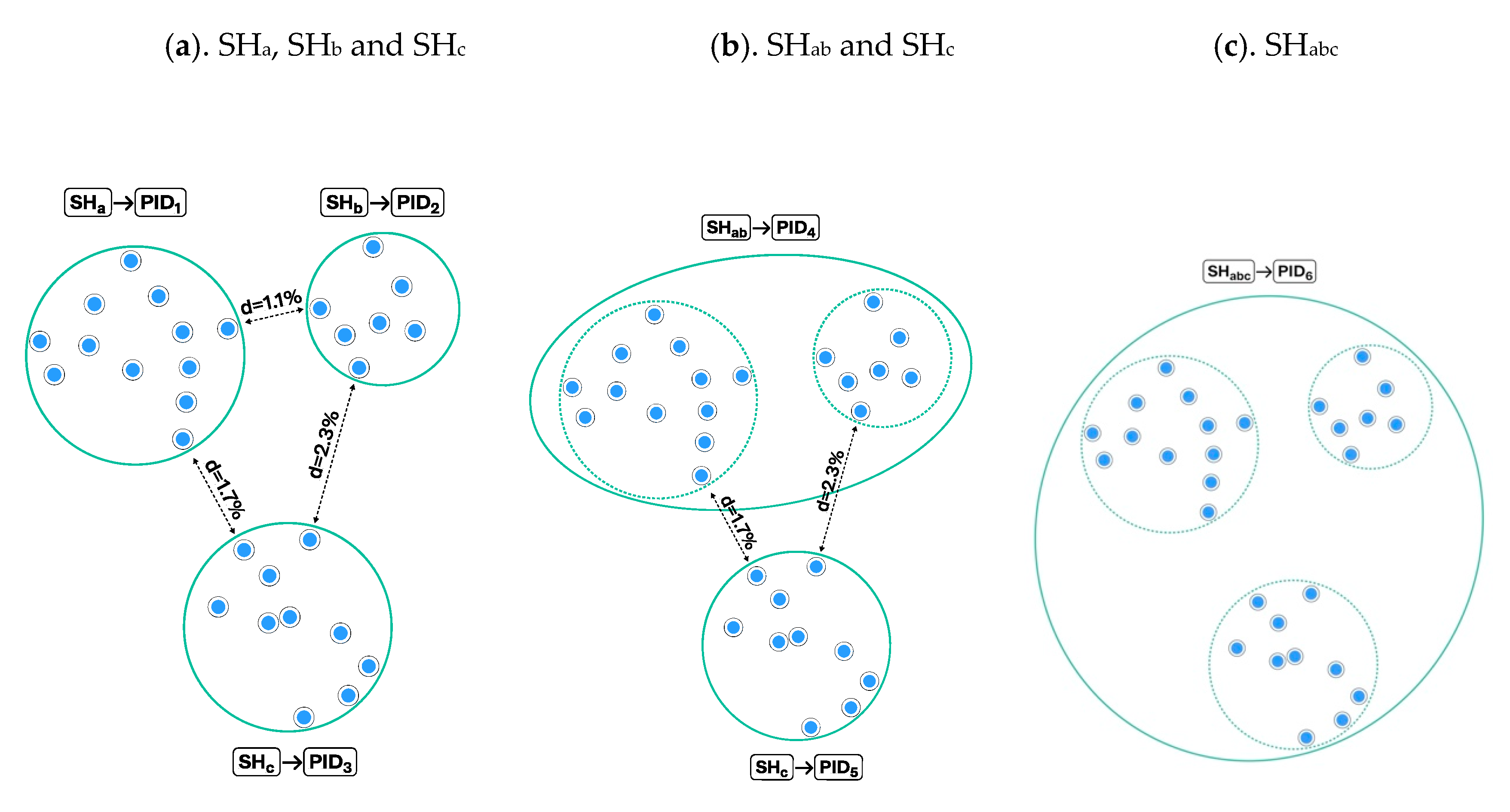
3.1. Construction of Species Hypotheses
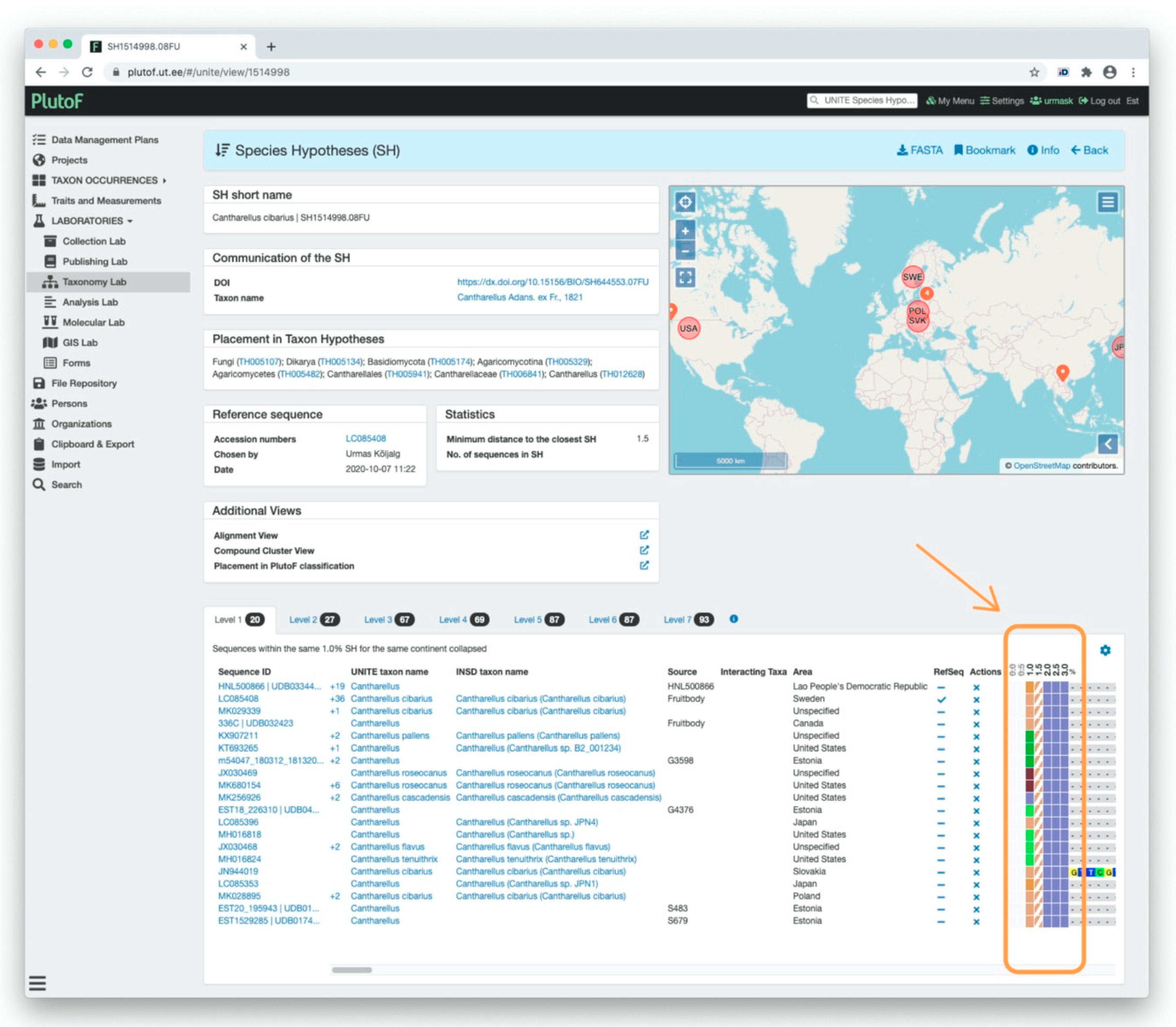
UNITE Example: Computation and Visualization of SHs in UNITE
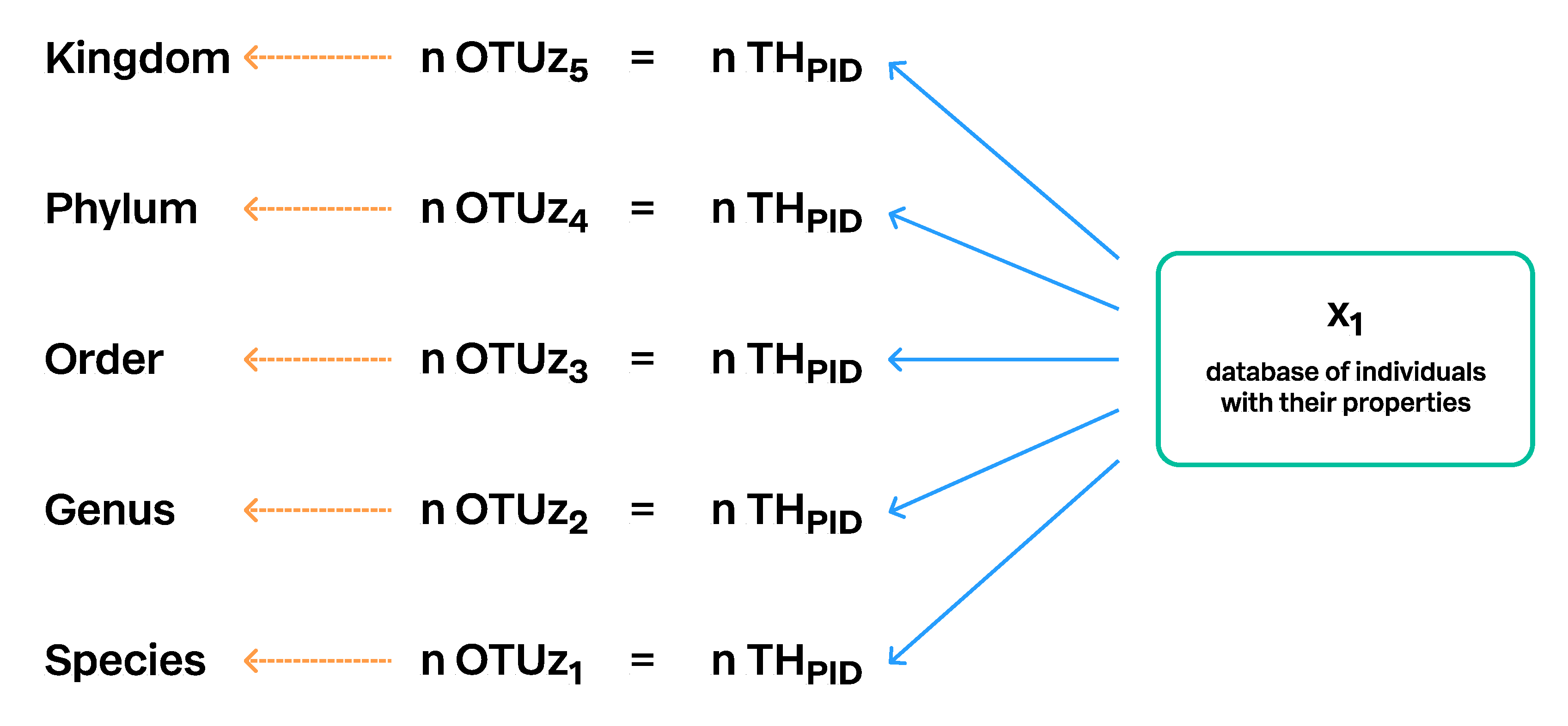
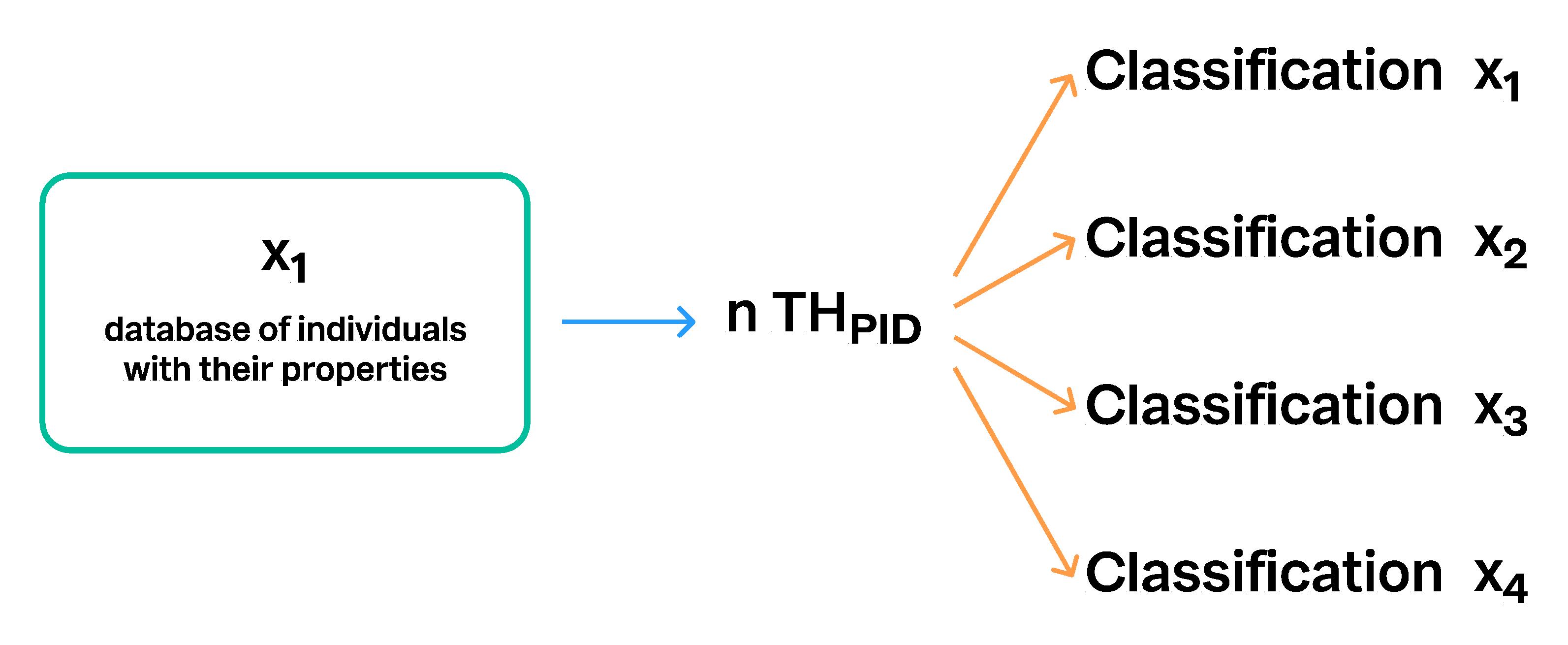
3.2. Communication of SHs via Persistent Identifiers (PID) and Taxon Names
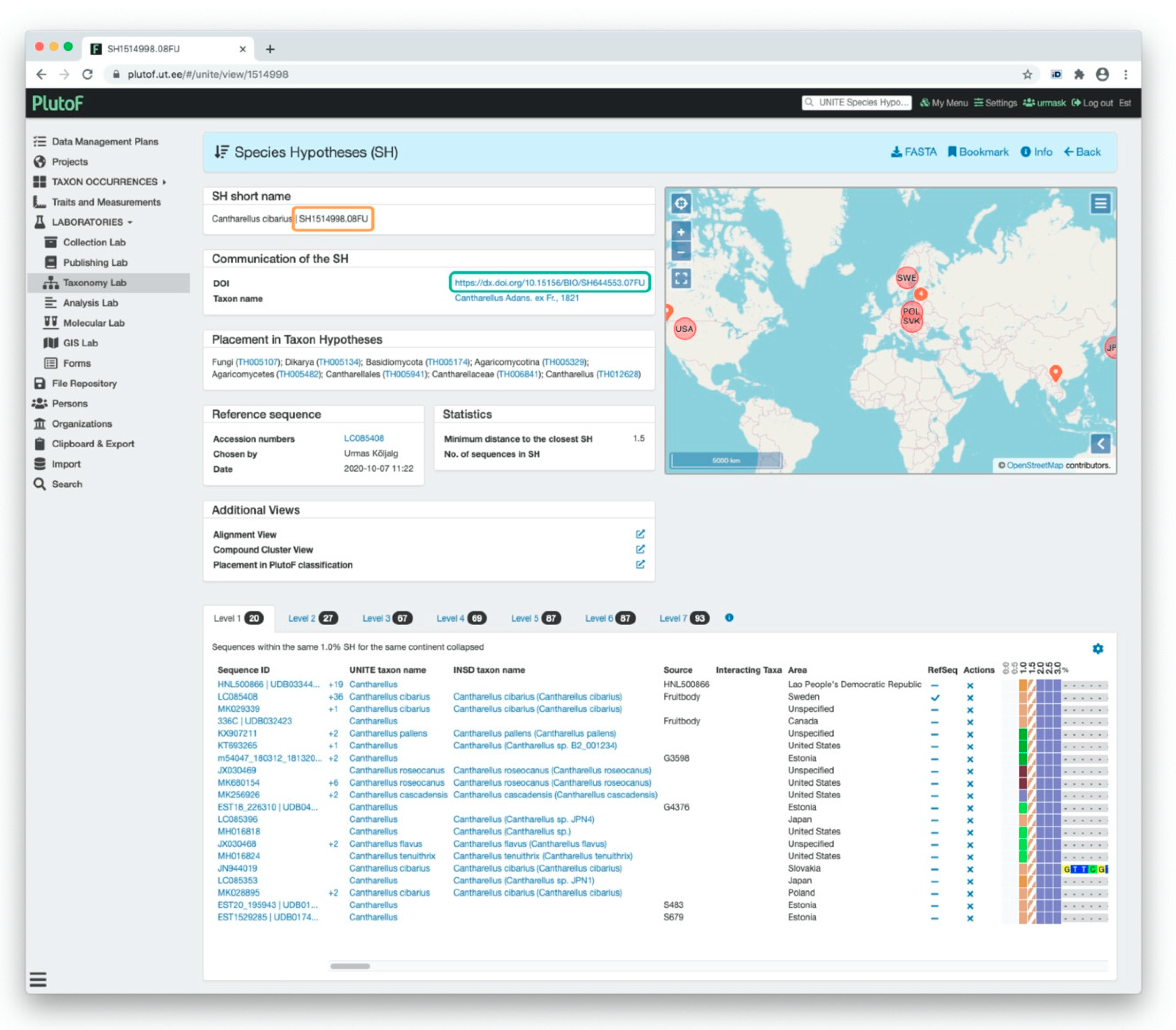
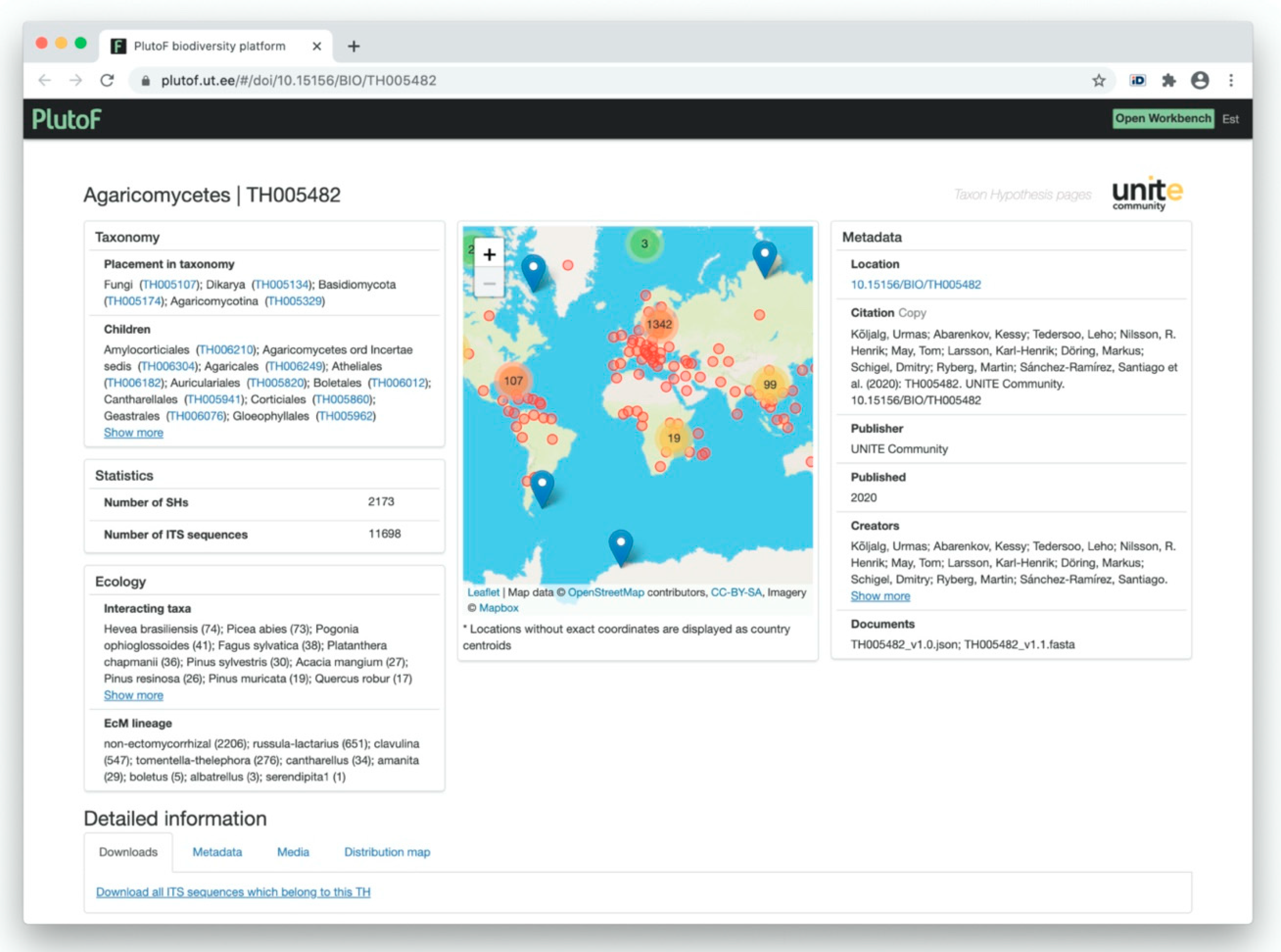
3.2.1. UNITE Example: Communication of SHs via Digital Object Identifiers (DOIs)
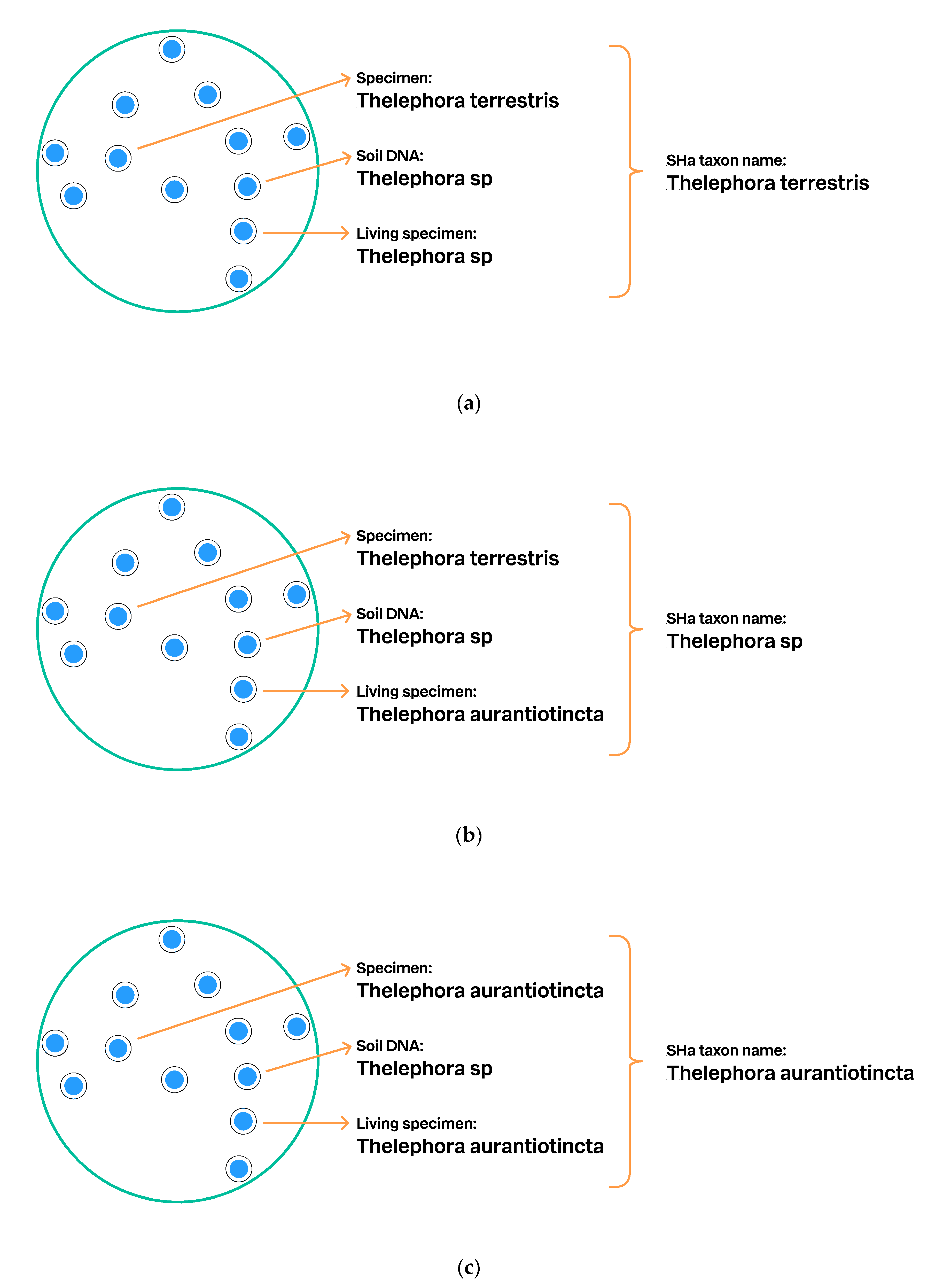
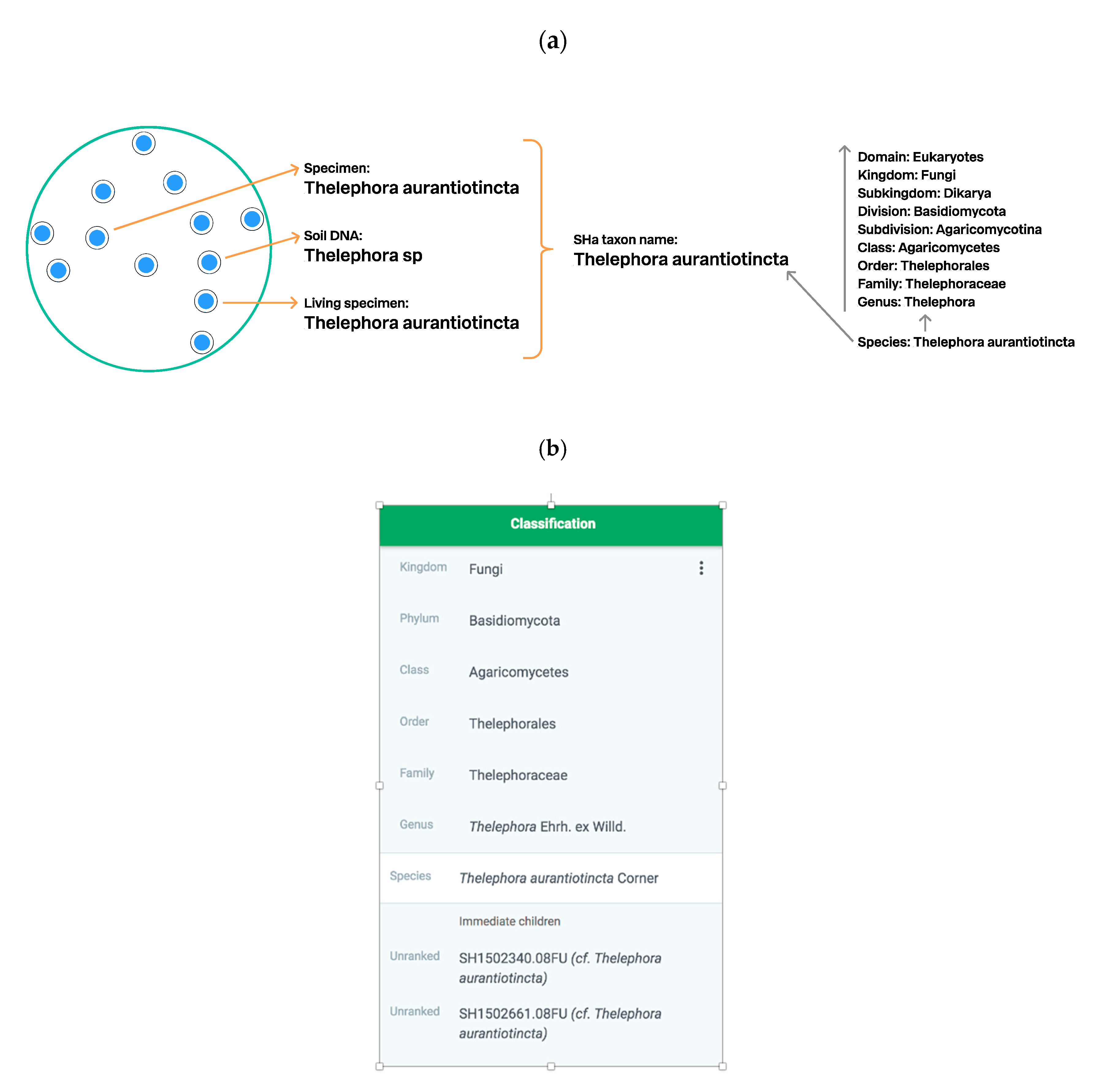
3.2.2. UNITE Example: Communication of SHs via the Taxon Name
3.3. Connecting SHs to the Taxonomic Backbone
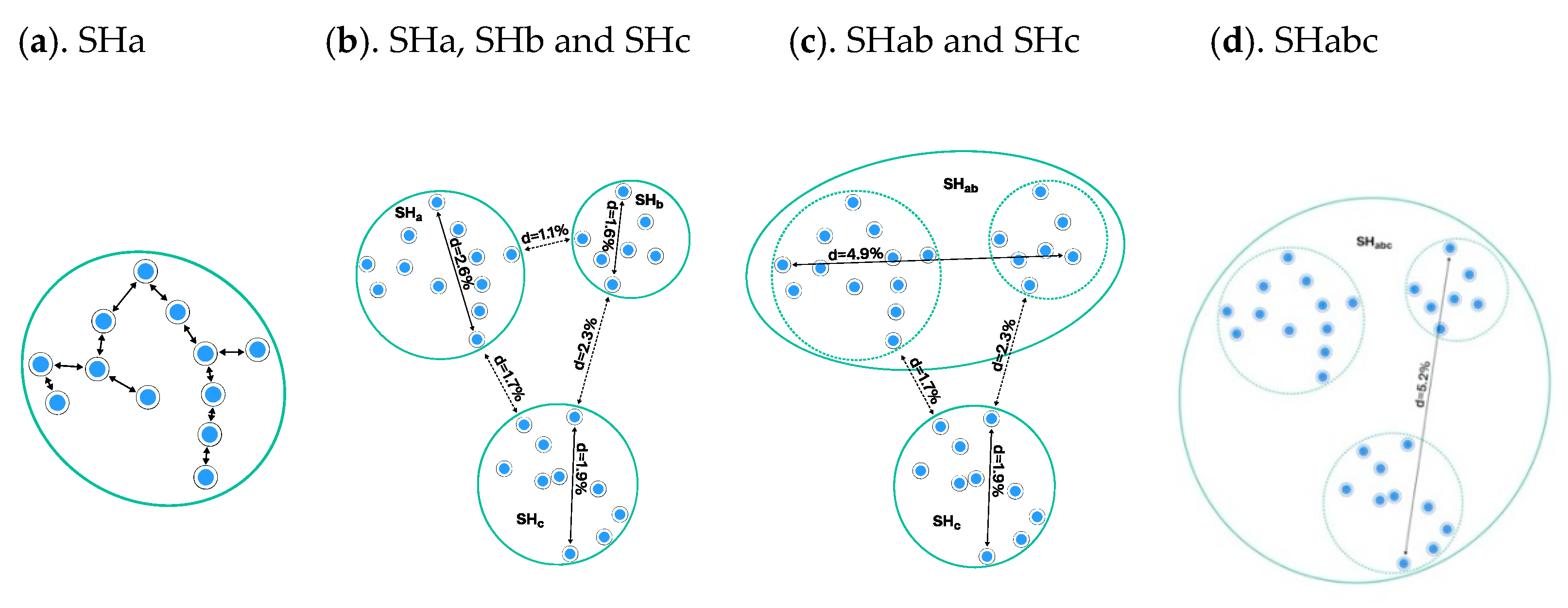
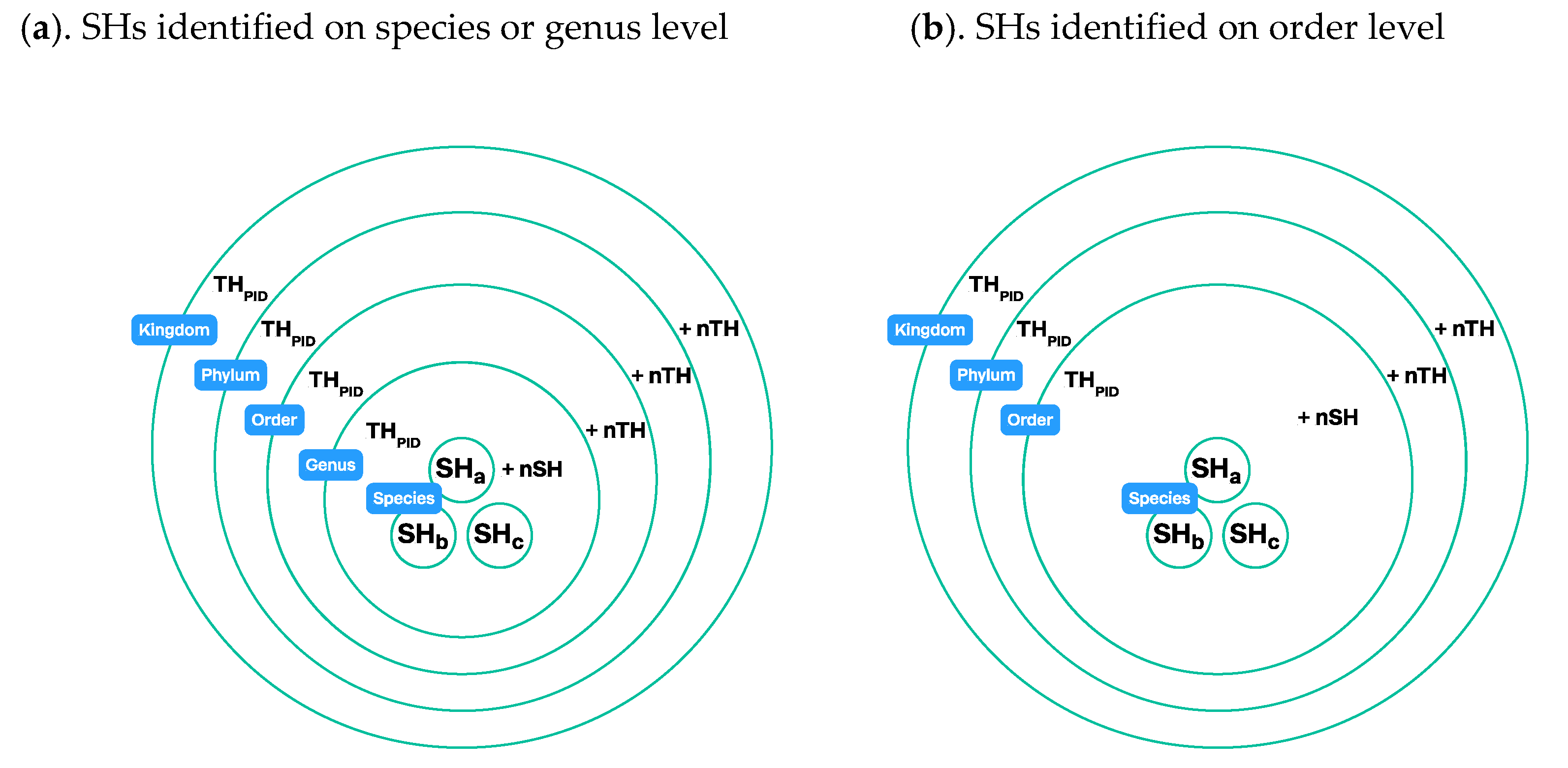
3.4. Construction of Taxon Hypotheses at Higher Levels
3.4.1. Using a Taxonomic Backbone
3.4.2. Using Operational Taxonomic Units
3.4.3. UNITE Example: Taxon Hypotheses
3.5. Communication of Taxon Hypotheses
3.6. Discovery of Formally Undescribed Species
4. Discussion
4.1. Liaison of the TH Paradigm with Taxonomy and Nomenclature
4.2. TH Paradigm and Metagenomics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Taxon Hypothesis
Appendix A.2. Species Hypothesis Is a Distinct Case of the Taxon Hypothesis
Appendix A.3. Individuals
References
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.; Worm, B. How many species are there on Earth and in the ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 1–17. [Google Scholar]
- Hibbett, D.S. After the gold rush, or before the flood? Evolutionary morphology of mushroom-forming fungi (Agaricomycetes) in the early 21st century. Mycol. Res. 2007, 111, 1001–1018. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Sánchez-Ramírez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.; May, T.; Ryberg, M.; Abarenkov, K. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Diversity, taxonomy and phylogeny of the Fungi. Biol. Rev. 2019, 94, 2101–2137. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Robert, V.; Vu, D.; Amor, A.B.H.; Van De Wiele, N.; Brouwer, C.; Jabas, B.; Szoke, S.; Dridi, A.; Triki, M.; Ben Daoud, S.; et al. MycoBank gearing up for new horizons. IMA Fungus 2013, 4, 371–379. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- GBIF Registry. Available online: https://www.gbif.org/grscicoll (accessed on 28 October 2020).
- Ratnasingham, S.; Hebert, P.D. Bold: The Barcode of Life Data System. Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Kõljalg, U.; Nilsson, H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.T.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of Fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- Popper, K.R. The Logic of Scientific Discovery (as Logik der Forschung); Verlag von Julius Springer: Vienna, Austria, 1934; pp. 1–513. [Google Scholar]
- Chalmers, A.F. What is This Thing Called Science? Queensland University Press: St Lucia, QLD, Australia; Open University Press: London, UK, 1976; pp. 1–157. [Google Scholar]
- Zavadski, K.M. Species and Speciation; Nauka: Leningrad, Russia, 1969; pp. 1–396. (In Russian) [Google Scholar]
- Dallwitz, M.J. A flexible computer program for generating identification keys. Syst. Zool. 1974, 23, 50–57. [Google Scholar] [CrossRef]
- Dallwitz, M.J. A general system for coding taxonomic descriptions. Taxon 1980, 29, 41–46. [Google Scholar] [CrossRef]
- International Nucleotide Sequence Database Collaboration (INSDC). Available online: http://www.insdc.org (accessed on 28 October 2020).
- NCBI Data Portal. Available online: https://www.ncbi.nlm.nih.gov (accessed on 28 October 2020).
- UNITE Database. Available online: https://unite.ut.ee (accessed on 28 October 2020).
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.J.G.M.; Sánchez-García, M.; Ebersberger, I.; De Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Nomenclature for incompletely specified bases in nucleic acid sequences: Recommendations. Nucleic Acids Res. 1984, 13, 3021–3030. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaeffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [Green Version]
- Abarenkov, K.; Tedersoo, L.; Nilsson, R.H.; Vellak, K.; Saar, I.; Veldre, V.; Parmasto, E.; Prous, M.; Aan, A.; Ots, M.; et al. PlutoF—A Web Based Workbench for Ecological and Taxonomic Research, with an Online Implementation for Fungal ITS Sequences. Evol. Bioinform. 2010, 6, 189–196. [Google Scholar] [CrossRef]
- Mons, B.; Neylon, C.; Velterop, J.; Dumontier, M.; da Silva Santos, L.O.B.; Wilkinson, M.D. Cloudy, increasingly FAIR; revisiting the FAIR Data guiding principles for the European Open Science Cloud. Inf. Serv. Use 2017, 37, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Wieczorek, J.; Bloom, D.; Guralnick, R.; Blum, S.; Döring, M.; Giovanni, R.; Robertson, T.; Vieglais, D. Darwin Core: An evolving community-developed Biodiversity Data Standard. PLoS ONE 2012, 7, e29715. [Google Scholar] [CrossRef]
- Yilmaz, P.; Kottmann, R.; Field, D.; Knight, R.; Cole, J.R.; Amaralzettler, L.; Gilbert, J.; Karsch-Mizrachi, I.; Johnston, A.; Cochrane, G.; et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 2011, 29, 415–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brase, J. DataCite—A Global Registration Agency for Research Data. In Proceedings of the Fourth International Conference on Cooperation and Promotion of Information Resources in Science and Technology, Beijing, China, 21–23 November 2009; pp. 257–261. [Google Scholar] [CrossRef] [Green Version]
- GBIF Backbone Taxonomy. Available online: https://0-doi-org.brum.beds.ac.uk/10.15468/39omei (accessed on 27 October 2020).
- Blaxter, M.; Mann, J.; Chapman, T.; Whitton, C.; Floyd, R.; Abebe, E. Defining operational taxonomic units using DNA barcode data. Phil. Trans. R. Soc. B 2015, 360, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Adding Sequence-Based Identifiers to Backbone Taxonomy Reveals ‘Dark Taxa’ Fungi. Available online: https://www.gbif.org/news/2LrgV5t3ZuGeU2WIymSEuk/adding-sequence-based-identifiers-to-backbone-taxonomy-reveals-dark-taxa-fungi (accessed on 27 October 2020).
- UNITE Community, Abarenkov, K. UNITE—Unified System for the DNA Based Fungal Species Linked to the Classification. 2019, Version 1.2. PlutoF. Available online: https://0-doi-org.brum.beds.ac.uk/10.15468/mkpcy3 (accessed on 27 October 2020).
- Ratnasingham, S.; Hebert, P.D.N. A DNA-based registry for all animal species: The barcode Index Number (BIN) System. PLoS ONE 2013, 8, e66213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBIF Introduces New Version of the Backbone Taxonomy. Available online: https://www.gbif.org/news/2UfGq1L6iXbSu0ElamvVlH/gbif-introduces-new-version-of-the-backbone-taxonomy (accessed on 27 October 2020).
- The International Barcode of Life Consortium. International Barcode of Life project (iBOL) Barcode Index Numbers (BINs). 2016. Available online: https://0-doi-org.brum.beds.ac.uk/10.15468/wvfqoi (accessed on 27 October 2020).
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; et al. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytologist. 2005, 166, 1062–1068. [Google Scholar] [CrossRef]
- Sokal, R.R.; Sneath, P.H. Principles of Numerical Taxonomy; W.H. Freeman and Co.: San Francisco, CA, USA, 1963; pp. 1–359. [Google Scholar]
- Hibbett, D. The invisible dimension of fungal diversity. Science 2016, 351, 1150–1151. [Google Scholar] [CrossRef]
- Kõljalg, U.; Tedersoo, L.; Nilsson, R.H.; Abarenkov, K. Digital identifiers for fungal species. Science 2016, 352, 1182–1183. [Google Scholar] [CrossRef]
- Frøslev, T.G.; Jeppesen, T.S.; Dima, B. Cortinarius koldingensis—A new species of Cortinarius, subgenus Phlegmacium related to Cortinarius sulfurinus. Mycol. Prog. 2015, 14, 73. [Google Scholar] [CrossRef]
- Torres-Cruz, T.J.; Tobias, T.L.B.; Almatruk, M.; Hesse, C.N.; Kuske, C.R.; Desirò, A.; Benucci, G.M.N.; Bonito, G.; Stajich, J.E.; Dunlap, C.; et al. Bifiguratus adelaidae gen. et sp. nov., a new member of Mucoromycotina in endophytic and soil-dwelling habitats. Mycologia 2017, 109, 363–378. [Google Scholar] [CrossRef] [Green Version]
- Voitk, A.; Saar, I.; Trudell, S.; Spirin, V.; Beug, M.; Kõljalg, U. Polyozellus multiplex (Thelephorales) is a species complex containing four new species. Mycologia 2017, 109, 975–992. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Wurzbacher, C.; Bahram, M.; Coimbra, V.R.; Larsson, E.; Tedersoo, L.; Eriksson, J.; Duarte, C.; Svantesson, S.; Sánchez-García, M.; et al. Top 50 most wanted fungi. MycoKeys 2016, 12, 29–40. [Google Scholar] [CrossRef] [Green Version]
- DarwinCore. Available online: https://dwc.tdwg.org (accessed on 28 October 2020).
- Access to Biological Collection Data (ABCD). Available online: https://www.tdwg.org/standards/abcd/ (accessed on 28 October 2020).
- Genomic Standards consortium (MIxS). Available online: https://gensc.org/mixs/ (accessed on 28 October 2020).
- Biodiversity Information Standards (TDWG). Available online: https://www.tdwg.org/standards/ (accessed on 28 October 2020).
- Penev, L.; Lyal, C.H.C.; Weitzman, A.; Morse, D.R.; King, D.; Sautter, G.; Georgjev, T.; Morris, R.A.; Catapano, T.; Agosti, D. XML schemas and mark-up practices of taxonomic literature. Zookeys 2011, 150, 89–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawksworth, D.L. A new dawn for the naming of fungi: Impacts of decisions made in Melbourne in July 2011 on the future publication and regulation of fungal names. IMA Fungus 2011, 2, 155–162. [Google Scholar] [CrossRef]
- May, T.; Redhead, S.A.; Bensch, K.; Hawksworth, D.L.; Lendemer, J.; Lombard, L.; Turland, N.J. Chapter F of the International Code of Nomenclature for algae, fungi, and plants as approved by the 11th International Mycological Congress, San Juan, Puerto Rico, July 2018. IMA Fungus 2019, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.T.; Tindall, B.J.; Garrity, G.M. International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2019, 69, S1–S111. [Google Scholar]
- Garnett, S.T.; Christidis, L.; Conix, S.; Costello, M.J.; Zachos, F.E.; Bánki, O.S.; Bao, Y.; Barik, S.K.; Buckeridge, J.S.; Hobern, D.; et al. Principles for creating a single authoritative list of the world’s species. PLoS Biol. 2020, 18, e3000736. [Google Scholar] [CrossRef] [PubMed]
- Turland, N.J.; Wiersema, J.H.; Barrie, F.R.; Greuter, W.; Hawksworth, D.L.; Herendeen, P.S.; Knapp, S.; Kusber, W.-H.; Li, D.-Z.; Marhold, K.; et al. International Code of Nomenclature for algae, fungi, and plants (Shenzhen Code) adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017. In Regnum Vegetabile; Koeltz Botanical Books: Glashütten, Germany, 2018; p. 159. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- World Federation for Culture Collections (WFCC). Available online: http://www.wfcc.info/index.php/home/ (accessed on 28 October 2020).
- Thiers, B. Index Herbariorum: A Global Directory of Public Herbaria and Associated Staff. New York Botanical Garden’s Virtual Herbarium. Available online: http://sweetgum.nybg.org/science/ih/ (accessed on 28 October 2020).
- Distributed System of Scientific Collections (DiSSCo). Available online: https://www.dissco.eu (accessed on 28 October 2020).
- Hardisty, A.R.; Ma, K.; Nelson, G.; Fortes, J. ‘openDS’—A new standard for digital specimens and other natural science digital object types. Biodivers. Inf. Sci. Stand. 2019, 3, e37033. [Google Scholar] [CrossRef]
- Vlk, L.; Tedersoo, L.; Antl, T.; Větrovský, T.; Abarenkov, K.; Pergl, J.; Albrechtova, J.; Vosátka, M.; Baldrian, P.; Pyšek, P.; et al. Early successional ectomycorrhizal fungi are more likely to naturalize outside their native range than other ectomycorrhizal fungi. New Phytol. 2020, 227, 1289–1293. [Google Scholar] [CrossRef]
- Porter, T.M.; Hajibabaei, M. Putting COI metabarcoding in context: The utility of exact sequence variants (ESVs) in biodiversity analysis. Front. Ecol. Evol. 2020, 8, 248. [Google Scholar] [CrossRef]
- Põlme, S.; Abarenkov, K.; Nilsson, R.H.; Lindahl, B.D.; Clemmensen, K.; Kauserud, H.; Nguyen, N.; Kjøller, K.; Bates, S.T.; Baldrian, P.; et al. Fungal Traits: A user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Divers. under review.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification Level | Taxon Name; TH DOI Code | Taxon Name; TH DOI Code | Taxon Name; TH DOI Code | Taxon Name; TH DOI Code | Taxon Name; TH DOI Code |
|---|---|---|---|---|---|
| Kingdom | Fungi; TH005107 | Fungi; TH005107 | Fungi; TH005107 | Amoebozoa; TH005117 | Viridiplantae; TH005112 |
| Phylum | Basidiomycota; TH005174 | Ascomycota; TH005190 | Ascomycota; TH005190 | Eumycetozoa; TH005218 | Anthophyta; TH005194 |
| Class | Agaricomycetes; TH005482 | Lecanoromycetes; TH005556 | Eurotiomycetes; TH005377 | Dictyostelea; TH005492 | Eudicotyledonae; TH005330 |
| Order | Agaricales; TH006249 | Lecanorales; TH006023 | Eurotiales; TH005852 | Dictyosteliida; TH005599 | Fagales; TH005787 |
| Family | Schizophyllaceae; TH006801 | Parmeliaceae; TH006508 | Aspergillacea; TH006460 | Dictyosteliidae; TH007268 | Fagaceae; TH008199 |
| Species | Schizophyllum commune; SH1565276.08FU | Vulpicida juniperinus; SH1716443.08FU | Aspergillus fumigatus; SH2189906.08FU | Polysphondylium violaceum; SH1514152.08FU | Quercus suber; SH1599838.08FU |
| Species, Publication Year | UNITE SH; the First Publication Year | Reference |
|---|---|---|
| Bifiguratus adelaidae 2017 | https://unite.ut.ee/bl_forw_sh.php?sh_name=SH022292.06FU#fndtn-panel1; 2013 | [43] |
| Cortinarius koldingensis 2015 | https://unite.ut.ee/bl_forw_sh.php?sh_name=SH201833.06FU#fndtn-panel1; 2013 | [42] |
| Polyozellus atrolazulinus 2018 | https://unite.ut.ee/bl_forw_sh.php?sh_name=SH028342.07FU#fndtn-panel1; 2014 | [44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kõljalg, U.; Nilsson, H.R.; Schigel, D.; Tedersoo, L.; Larsson, K.-H.; May, T.W.; Taylor, A.F.S.; Jeppesen, T.S.; Frøslev, T.G.; Lindahl, B.D.; et al. The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa. Microorganisms 2020, 8, 1910. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121910
Kõljalg U, Nilsson HR, Schigel D, Tedersoo L, Larsson K-H, May TW, Taylor AFS, Jeppesen TS, Frøslev TG, Lindahl BD, et al. The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa. Microorganisms. 2020; 8(12):1910. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121910
Chicago/Turabian StyleKõljalg, Urmas, Henrik R. Nilsson, Dmitry Schigel, Leho Tedersoo, Karl-Henrik Larsson, Tom W. May, Andy F. S. Taylor, Thomas Stjernegaard Jeppesen, Tobias Guldberg Frøslev, Björn D. Lindahl, and et al. 2020. "The Taxon Hypothesis Paradigm—On the Unambiguous Detection and Communication of Taxa" Microorganisms 8, no. 12: 1910. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8121910