Detection of Interactions between Proteins by Using Legendre Moments Descriptor to Extract Discriminatory Information Embedded in PSSM

Abstract
:1. Introduction
2. Materials and Methodology
2.1. Godden Standard Datasets
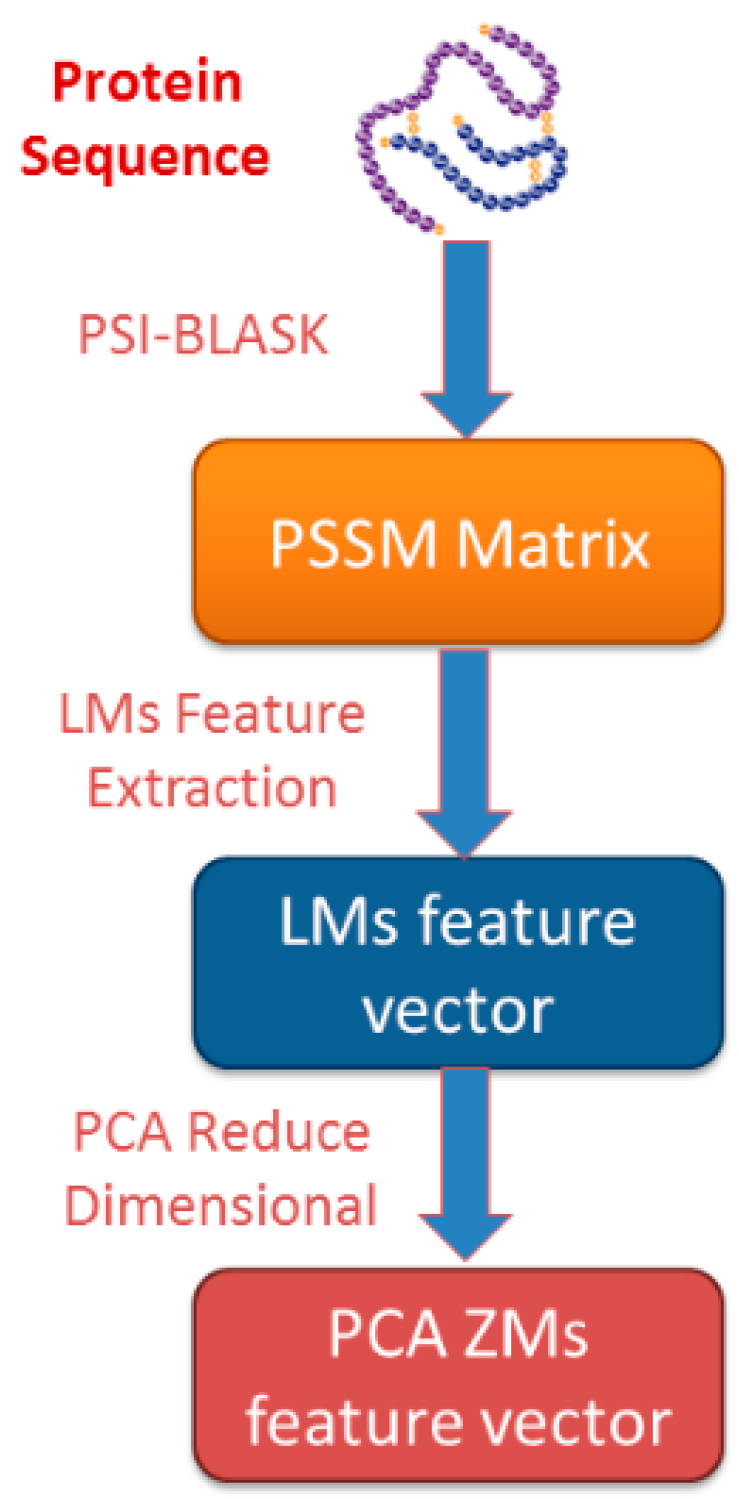
2.2. Position-Specific Scoring Matrix
2.3. Legendre Moments
2.4. Related Machine Learning Models
2.5. PCVM Algorithm
2.6. Initial Parameter Selection and Training
3. Results and Discussion
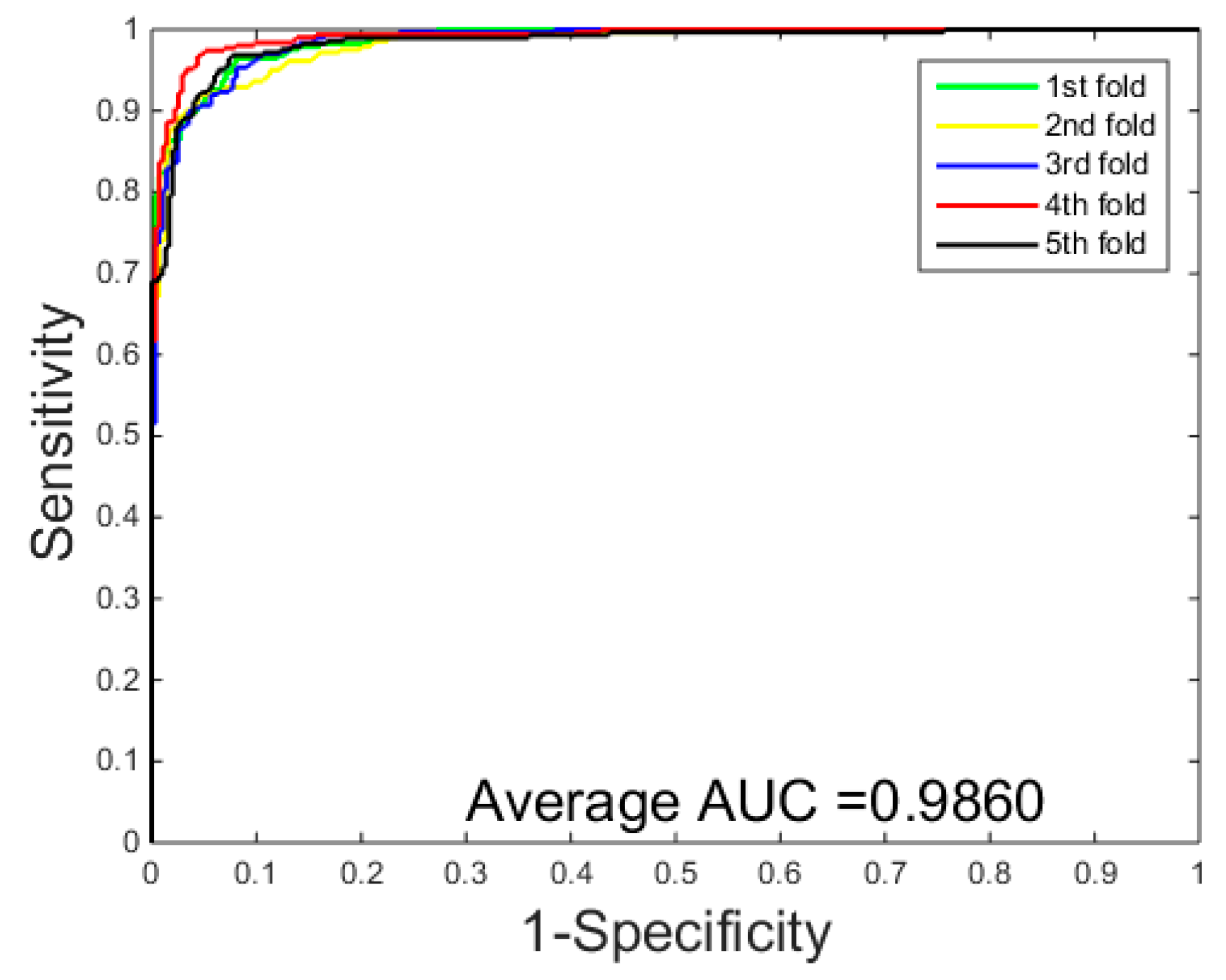
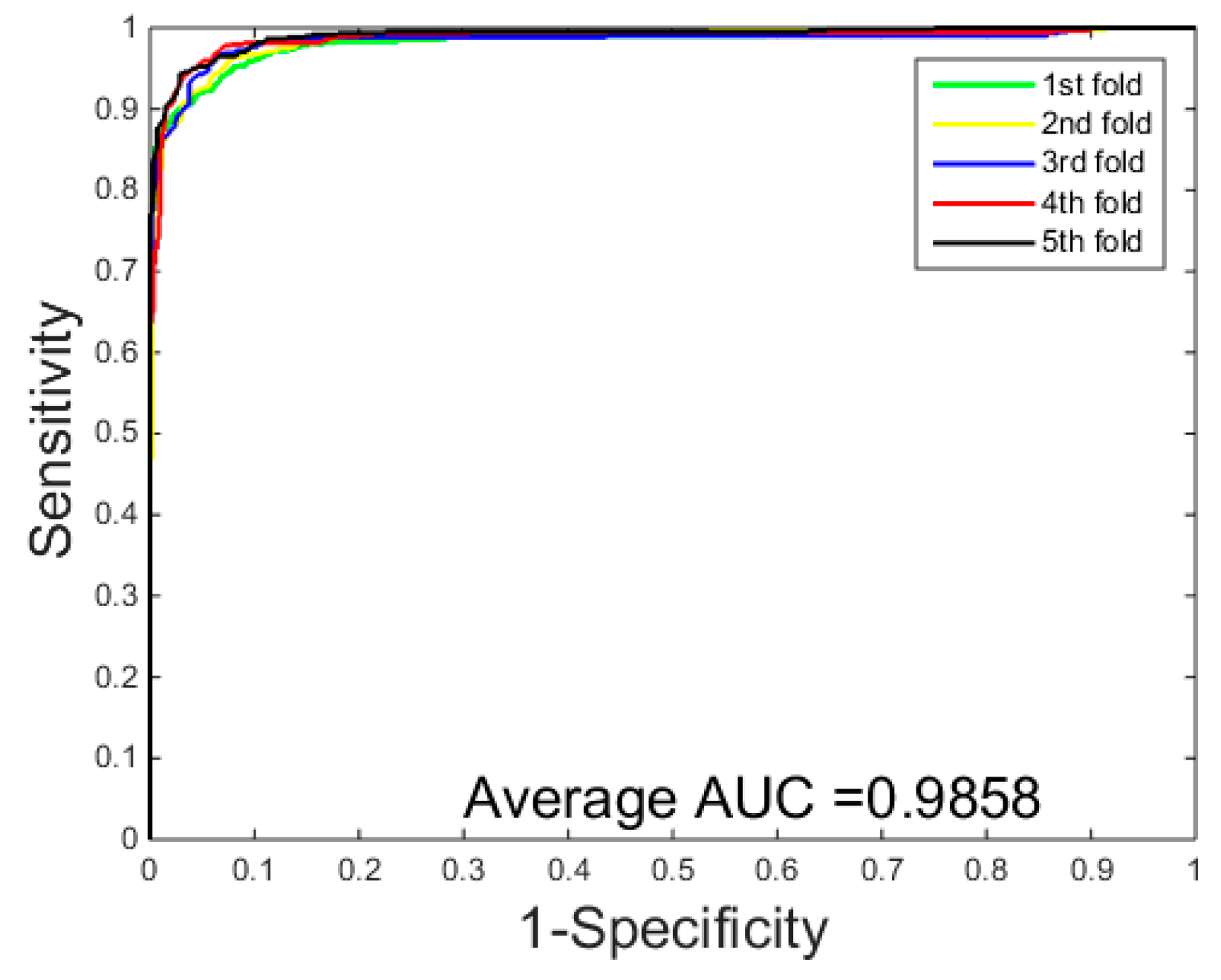
3.1. Performance Evaluation
3.2. Assessment of Prediction
3.3. Comparison the Proposed Method with the SVM-Based Approach
3.4. Performance on Independent Dataset
3.5. Comparison with Other Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rüetschi, U.; Rosén, A.; Karlsson, G.; Zetterberg, H.; Rymo, L.; Hagberg, H.; Jacobsson, B. Proteomic analysis using protein chips to detect biomarkers in cervical and amniotic fluid in women with intra-amniotic inflammation. J. Proteome Res. 1900, 4, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Niranjanakumari, S.; Lasda, E.; Brazas, R.; Garcia-Blanco, M.A. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 2002, 26, 182–190. [Google Scholar] [CrossRef]
- Kuo, M.H.; Allis, C.D. In vivo cross-linking and immunoprecipitation for studying dynamic protein: DNA associations in a chromatin environment. Methods 1999, 19, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Boldin, M.P.; Kovalenko, A.V.; Malinin, N.L.; Mett, I.L.; Camonis, J.H. The yeast two-hybrid screening technique and its use in the study of protein-protein interactions in apoptosis. Curr. Opin. Immunol. 1998, 10, 131–136. [Google Scholar] [CrossRef]
- Fang, Y.; Macool, D.; Xue, Z.; Heppard, E.; Hainey, C.; Tingey, S.; Miao, G.H. Development of a high-throughput yeast two-hybrid screening system to study protein-protein interactions in plants. Mol. Genet. Genom. 2002, 267, 142–153. [Google Scholar]
- Sun, J.; Xu, J.; Liu, Z.; Zhao, A.; Zhao, A.; Shi, T.; Li, Y. Refined phylogenetic profiles method for predicting protein–protein interactions. Bioinformatics 2005, 21, 3409–3415. [Google Scholar] [CrossRef] [PubMed]
- Weiller, G.F. Phylogenetic profiles: A graphical method for detecting genetic recombinations in homologous sequences. Mol. Biol. Evol. 1998, 15, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.H.; Yu, H.C. Literature extraction of protein functions using sentence pattern mining. IEEE Trans. Knowl. Data Eng. 2005, 17, 1088–1098. [Google Scholar] [CrossRef]
- Ma, X.; Lee, H.; Wang, L.; Sun, F. CGI: A new approach for prioritizing genes by combining gene expression and protein-protein interaction data. Bioinformatics 2007, 23, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.; Pavlovic, V. Protein classification using probabilistic chain graphs and the gene ontology structure. Bioinformatics 2006, 22, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Iliopoulos, I.; Kyrpides, N.C.; Ouzounis, C.A. Protein interaction maps for complete genomes based on gene fusion events. Nature 1999, 402, 86–90. [Google Scholar] [PubMed]
- Tsoka, S.; Ouzounis, C.A. Prediction of protein interactions: Metabolic enzymes are frequently involved in gene fusion. Nat. Genet. 2000, 26, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 2009, 5, e1000585. [Google Scholar] [CrossRef] [PubMed]
- Aytuna, A.S.; Gursoy, A.; Keskin, O. Prediction of protein-protein interactions by combining structure and sequence conservation in protein interfaces. Bioinformatics 2005, 21, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.R.; Gough, D.A. Whole-proteome interaction mining. Bioinformatics 2003, 19, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Cai, Y.D. Predicting protein-protein interactions from sequences in a hybridization space. J. Proteome Res. 2006, 5, 316–322. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Lei, Y.K.; Zhu, L.; Xia, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Yin, Z.; Han, K.; Huang, D.S.; Zhou, X. A semi-supervised learning approach to predict synthetic genetic interactions by combining functional and topological properties of functional gene network. BMC Bioinform. 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Li, S.; Gao, X.; Luo, X.; Ji, Z. Large-scale protein-protein interactions detection by integrating big biosensing data with computational model. BioMed Res. Int. 2014, 2014, 598129. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Zhu, L.; Zheng, C.H.; Yu, H.J.; Deng, S.P.; Ji, Z. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinform. 2014, 15, S9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, Z.; Li, X.; Chen, X.; Jiang, T.; Zhang, J. PCVMZM: Using the probabilistic classification vector machines model combined with a Zernike moments descriptor to predict protein-protein interactions from protein sequences. Int. J. Mol. Sci. 2017, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xia, J.F.; Gui, J. Prediction of protein-protein interactions from protein sequence using local descriptors. Protein Peptide Lett. 2010, 17, 1085–1090. [Google Scholar] [CrossRef]
- Wong, L.; You, Z.H.; Ming, Z.; Li, J.; Chen, X.; Huang, Y.A. Detection of interactions between proteins through rotation forest and local phase quantization descriptors. Int. J. Mol. Sci. 2016, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A. An ensemble of K-local hyperplanes for predicting protein-protein interactions. Bioinformatics 2006, 10, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L. Fusion of classifiers for predicting protein–protein interactions. Neurocomputing 2005, 68, 289–296. [Google Scholar] [CrossRef]
- Nanni, L. Letters: Hyperplanes for predicting protein-protein interactions. Neurocomputing 2005, 69, 257–263. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iPPBS-Opt: A sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules 2016, 21, 95. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition. J. Biomol. Struct. Dyn. 2015, 34, 1946–1961. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iPPI-Esml: An ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J. Theor. Biol. 2015, 377, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, B.; Revote, J.; Leier, A.; Marquez-Lago, T.T.; Webb, G.; Song, J.; Chou, K.C.; Lithgow, T. POSSUM: A bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017, in press. [Google Scholar] [CrossRef]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The Database of Interacting Proteins: 2001 update. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.C.; Lin, X.; Chen, X.W. On Position-Specific Scoring Matrix for Protein Function Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.-H.; Huang, H.Y.; Syu, Y.T.; Wu, C.P. Real value prediction of protein solvent accessibility using enhanced PSSM features. BMC Bioinform. 2008, 9 (Suppl. S12), 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyons, J.; Dehzangi, A.; Paliwal, K.K. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition. IEEE Trans. Nanobiosci. 2013, 320, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Maccallum, R.M.; Sternberg, M.J.E. Recognition of remote protein homologies using three-dimensional information to generate a position specific scoring matrix in the program 3D-PSSM. In Proceedings of the Third Annual International Conference on Computational Molecular Biology, Lyon, France, 11–14 April 1999; pp. 218–225. [Google Scholar]
- Zhang, H.; Shu, H.; Han, G.N.; Coatrieux, G.; Luo, L.; Coatrieux, J.L. Blurred Image Recognition by Legendre Moment Invariants. IEEE Trans. Image Process. A Publ. IEEE Signal Process. Soc. 2010, 19, 596–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, C.W.; Raveendran, P.; Mukundan, R. Translation and scale invariants of Legendre moments. Pattern Recognit. 2004, 37, 119–129. [Google Scholar] [CrossRef]
- Hosny, K.M. Exact Legendre moment computation for gray level images. Pattern Recognit. 2007, 40, 3597–3605. [Google Scholar] [CrossRef]
- Zhang, H.; Shu, H.; Coatrieux, G.; Zhu, J.; Wu, Q.M.J.; Zhang, Y.; Zhu, H.; Luo, L. Affine legendre moment invariants for image watermarking robust to geometric distortions. IEEE Trans. Image Process. 2011, 20, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhang, H.; Shu, H.; Luo, L. Image recognition by combined invariants of legendre moment. In Proceedings of the IEEE International Conference on Information and Automation, Colombo, Sri Lanka, 17–19 December 2010; pp. 1793–1798. [Google Scholar]
- Baldi, P.; Hornik, K. Neural networks and principal component analysis: Learning from examples without local minima. Neural Netw. 1989, 2, 53–58. [Google Scholar] [CrossRef]
- Moore, B. Principal component analysis in linear systems: Controllability, observability, and model reduction. IEEE Trans. Autom. Control 2003, 26, 17–32. [Google Scholar] [CrossRef]
- Braak, C.J.F.T. CANOCO- A FORTRAN Program for Canonical Community Ordination by Canonical Correspondence Analysis, Principal Component Analysis and Redundancy Analysis; Version 2.1; Ministerie van Landbouw en Visserij: Wageningen, The Netherlands, 1988. [Google Scholar]
- Wang, Y.-B.; You, Z.-H.; Li, X.; Jiang, T.-H.; Chen, X.; Zhou, X.; Wang, L. Predicting protein-protein interactions from protein sequences by a stacked sparse autoencoder deep neural network. Mol. Biosyst. 2017, 13, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tino, P.; Yao, X. Efficient probabilistic classification vector machine with incremental basis function selection. IEEE Trans. Neural Netw. Learn. Syst. 2014, 25, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tino, P.; Yao, X. 1 Probabilistic classification vector machines. IEEE Trans. Neural Netw. 2009, 20, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.A.; Mcneil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Dodd, L.E.; Pepe, M.S. Partial AUC estimation and regression. Biometrics 2003, 59, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Deng, E.Z.; Ding, H.; Chen, W.; Chou, K.C. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2007, 2, 389–396. [Google Scholar] [CrossRef]
- Zou, Q.; Li, X.B.; Jiang, W.R.; Lin, Z.Y.; Li, G.L.; Chen, K. Survey of MapReduce frame operation in bioinformatics. Brief. Bioinform. 2014, 15, 637. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Zou, Q.; Mao, Y.; Hu, L.; Wu, Y.; Ji, Z. Mirclassify: An advanced web server for miRNA family classification and annotation. Comput. Biol. Med. 2014, 45, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Hu, Q.; Guo, M.; Wang, G. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics 2015, 31, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Long, R.; Chou, K.C. iDHS-EL: Identifying DNase I hypersensitive-sites by fusing three different modes of pseu-do nucleotide composition into an ensemble learning framework. Bioinformatics 2016, 32, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Fang, L.; Wang, X.; Chou, K.C. repDNA: A Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics 2015, 31, 1307. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, S.; Long, R.; Chou, K.C. iRSpot-EL: Identify recombination spots with an ensemble learning approach. Bioinformatics 2017, 33, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Sun, B.Q.; Xiao, X.; Xu, Z.C.; Chou, K.C. iHyd-PseCp: Identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget 2016, 7, 44310–44321. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Xiao, X.; Xu, Z.C.; Chou, K.C. iPhos-PseEn: Identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget 2016, 7, 51270–51283. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Sun, B.Q.; Xiao, X.; Xu, D.; Chou, K.C. iPhos-PseEvo: Identifying Human Phosphorylated Proteins by Incorporating Evolutionary Information into General PseAAC via Grey System Theory. Mol. Inform. 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Jiang, S.Y.; Sun, B.Q.; Xiao, X.; Cheng, X.; Chou, K.C. iRNA-2methyl: Identify RNA 2’-O-methylation sites by incorporating sequence-coupled effects into general PseKNC and ensemble classifier. Med. Chem. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr. Top. Med. Chem. 2017, 17, 2337–2358. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Testing Set | Acc (%) | Sn (%) | Pe (%) | Mcc (%) |
|---|---|---|---|---|
| 1 | 96.67 | 97.52 | 95.93 | 93.55 |
| 2 | 96.33 | 96.59 | 95.93 | 92.93 |
| 3 | 96.25 | 96.40 | 96.24 | 92.78 |
| 4 | 96.42 | 95.78 | 96.93 | 93.09 |
| 5 | 96.17 | 96.69 | 95.74 | 92.63 |
| Average | 96.37 ± 0.2 | 96.60 ± 0.6 | 96.15 ± 0.5 | 93.00 ± 0.4 |
| Testing Set | Acc (%) | Sn (%) | Pe (%) | Mcc (%) |
|---|---|---|---|---|
| 1 | 93.48 | 94.74 | 92.15 | 87.81 |
| 2 | 93.48 | 89.72 | 96.56 | 87.75 |
| 3 | 93.14 | 89.97 | 96.42 | 87.20 |
| 4 | 93.65 | 89.94 | 97.88 | 88.09 |
| 5 | 93.66 | 92.61 | 94.27 | 88.12 |
| Average | 93.48 ± 0.2 | 94.40 ± 2.2 | 95.46 ± 2.3 | 87.79 ± 0.4 |
| Testing Set | Acc (%) | Sn (%) | Pe (%) | Mcc (%) |
|---|---|---|---|---|
| 1 | 92.83 | 96.20 | 90.23 | 86.66 |
| 2 | 92.67 | 97.10 | 88.91 | 86.38 |
| 3 | 92.25 | 85.60 | 99.05 | 85.60 |
| 4 | 92.25 | 98.15 | 87.65 | 85.62 |
| 5 | 92.34 | 85.45 | 99.23 | 85.73 |
| Average | 92.47 ± 0.3 | 92.50 ± 6.4 | 93.01 ± 5.7 | 86.00 ± 0.5 |
| Testing Set | Acc (%) | Sn (%) | Pe (%) | Mcc (%) |
|---|---|---|---|---|
| 1 | 90.74 | 99.65 | 84.27 | 82.99 |
| 2 | 90.22 | 99.65 | 83.38 | 82.14 |
| 3 | 90.74 | 81.94 | 100.00 | 82.98 |
| 4 | 90.39 | 82.47 | 99.22 | 82.48 |
| 5 | 90.41 | 100.00 | 83.53 | 82.42 |
| Average | 90.50 ± 0.2 | 92.74 ± 9.6 | 90.08 ± 8.7 | 82.60 ± 0.4 |
| Species | Test Pairs | Accuracy |
|---|---|---|
| Mix_Celeg | 4013 | 92.60% |
| Mix_Ecoli | 6954 | 92.80% |
| Mix_Hsapi | 1412 | 80.10% |
| Mix_Mmusc | 313 | 89.14% |
| Model | Testing Set | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|---|
| Guo [22] | ACC | 89.33 ± 2.67 | 89.93 ± 3.68 | 88.87 ± 6.16 | N/A |
| AC | 87.36 ± 1.38 | 87.30 ± 4.68 | 87.82 ± 4.33 | N/A | |
| Yang [23] | Cod1 | 75.08 ± 1.13 | 75.81 ± 1.20 | 74.75 ± 1.23 | N/A |
| Cod2 | 80.04 ± 1.06 | 76.77 ± 0.69 | 82.17 ± 1.35 | N/A | |
| Cod3 | 80.41 ± 0.47 | 78.14 ± 0.90 | 81.66 ± 0.99 | N/A | |
| Cod4 | 86.15 ± 1.17 | 81.03 ± 1.74 | 90.24 ± 1.34 | N/A | |
| You [17] | PCA-EELM | 87.00 ± 0.29 | 86.15 ± 0.43 | 87.59 ± 0.32 | 77.36 ± 0.44 |
| Wong [24] | RF-PR-LPQ | 93.92 ± 0.36 | 91.10 ± 0.31 | 96.45 ± 0.45 | 88.56 ± 0.63 |
| Proposed Method | PCVM | 96.37 ± 0.20 | 96.60 ± 0.6 | 96.15 ± 0.5 | 93.00 ± 0.4 |
| Model | Acc (%) | Sen (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|
| Nanni [25] | 83.00 | 86.00 | 85.10 | N/A |
| Nanni [26] | 84.00 | 86.00 | 84.00 | N/A |
| Nanni and Lumini [27] | 86.60 | 86.70 | 85.00 | N/A |
| Z-H You [17] | 87.50 | 88.95 | 86.15 | 78.13 |
| L Nanni [26] | 84.00 | 84.00 | 84.00 | N/A |
| Proposed Method | 93.48 | 94.40 | 95.46 | 87.79 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-B.; You, Z.-H.; Li, L.-P.; Huang, Y.-A.; Yi, H.-C. Detection of Interactions between Proteins by Using Legendre Moments Descriptor to Extract Discriminatory Information Embedded in PSSM. Molecules 2017, 22, 1366. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22081366
Wang Y-B, You Z-H, Li L-P, Huang Y-A, Yi H-C. Detection of Interactions between Proteins by Using Legendre Moments Descriptor to Extract Discriminatory Information Embedded in PSSM. Molecules. 2017; 22(8):1366. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22081366
Chicago/Turabian StyleWang, Yan-Bin, Zhu-Hong You, Li-Ping Li, Yu-An Huang, and Hai-Cheng Yi. 2017. "Detection of Interactions between Proteins by Using Legendre Moments Descriptor to Extract Discriminatory Information Embedded in PSSM" Molecules 22, no. 8: 1366. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22081366