Mining and Development of Novel SSR Markers Using Next Generation Sequencing (NGS) Data in Plants

 , ,
, , 
Abstract
:1. Introduction
1.1. Importance of Microsatellites and Their Use as Genetic Markers
1.2. Next-Generation Sequencing (NGS)
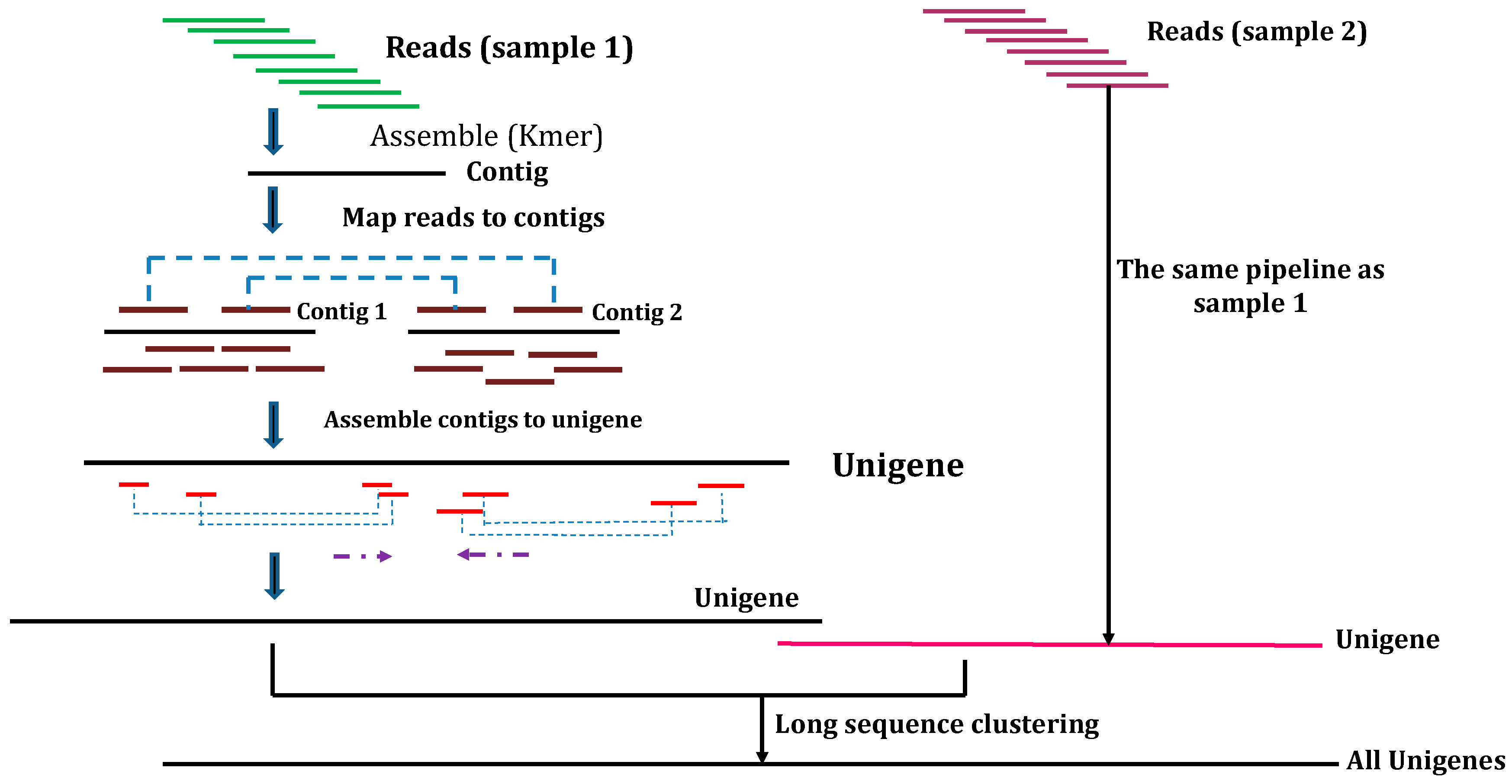
1.3. SSR Discovery by Transcriptome Sequencing (RNA-Seq)
2. Overview of the Process of SSR Development through Transcriptome de Novo Assembly Using the Illumina Platform
2.1. de Novo Assembly
- Inchworm: assembles the reads set into the unique sequences of transcripts by extending the sequences with the most abundant k-mers and then only reports the unique portions of differently spliced transcripts.
- Chrysalis: groups the overlapping Inchworm contigs by overlaps of k − 1 into clusters to construct de Bruijn graph components for each cluster, representing the full transcriptional complexity of a given gene or genes with the common sequence. Next, chrysalis partitions the full read set between clusters.
- Butterfly: resolves spliced and paralogous transcripts independently in parallel, ultimately reporting full-length transcripts.
2.2. Unigene Functional Annotation
2.3. Microsatellites Mining and Identification Tools
2.4. DNA Isolation, PCR Amplification, and SSR Validation
2.5. Genotyping STRs in Next-Generation Data: Challenges and Solutions
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, V.K.; Singh, A.K.; Singh, S.; Singh, B.D. Next-Generation Sequencing (NGS) Tools and Impact in Plant Breeding. In Advances in Plant Breeding Strategies: Breeding, Biotechnology and Molecular Tools; Springer: Cham, Switzerland, 2015; pp. 563–612. [Google Scholar]
- Punia, A.; Yadav, R.; Arora, P.; Chaudhury, A. Molecular and morphophysiological characterization of superior cluster bean (Cymopsis tetragonoloba) varieties. J. Crop Sci. Biotechnol. 2009, 12, 143–148. [Google Scholar] [CrossRef]
- Pathak, R.; Singh, S.; Singh, M.; Henry, A. Molecular assessment of genetic diversity in cluster bean (Cyamopsis tetragonoloba) genotypes. J. Genet. 2010, 89, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Kuravadi, N.A.; Tiwari, P.B.; Tanwar, U.K.; Tripathi, S.K.; Dhugga, K.S.; Gill, K.S.; Randhawa, G.S. Identification and Characterization of EST-SSR Markers in Cluster Bean (spp.). Crop Sci. 2014, 54, 1097–1102. [Google Scholar] [CrossRef]
- Kuravadi, N.A.; Yenagi, V.; Rangiah, K.; Mahesh, H.; Rajamani, A.; Shirke, M.D.; Russiachand, H.; Loganathan, R.M.; Lingu, C.S.; Siddappa, S. Comprehensive analyses of genomes, transcriptomes and metabolites of neem tree. PeerJ 2015, 3, e1066. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R. Genetic Markers and Biotechnology. In Clusterbean: Physiology, Genetics and Cultivation; Springer: Singapore, 2015; pp. 125–143. [Google Scholar]
- Kumar, S.; Parekh, M.J.; Patel, C.B.; Zala, H.N.; Sharma, R.; Kulkarni, K.S.; Fougat, R.S.; Bhatt, R.K.; Sakure, A.A. Development and validation of EST-derived SSR markers and diversity analysis in cluster bean (Cyamopsis tetragonoloba). J. Plant Biochem. Biotechnol. 2016, 25, 263–269. [Google Scholar] [CrossRef]
- Tanwar, U.K.; Pruthi, V.; Randhawa, G.S. RNA-Seq of Guar (Cyamopsis tetragonoloba, L. Taub.) Leaves: De novo Transcriptome Assembly, Functional Annotation and Development of Genomic Resources. Front. Plant Sci. 2017, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, N.S.; Ramos, H.C.C.; Caixeta, E.T.; Pereira, M.G. Plant breeding with marker-assisted selection in Brazil. Crop Breed. Appl. Biotechnol. 2014, 14, 54–60. [Google Scholar] [CrossRef]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Goel, R.; Pande, V.; Asif, M.H.; Mohanty, C.S. De novo sequencing and comparative analysis of leaf transcriptomes of diverse condensed tannin-containing lines of underutilized Psophocarpus tetragonolobus (L.) DC. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Rosazlina, R.; Jacobsen, N.; Ørgaard, M.; Othman, A.S. Utilizing next generation sequencing to characterize microsatellite loci in a tropical aquatic plant species Cryptocoryne cordata var. cordata (Araceae). Biochem. Syst. Ecol. 2015, 61, 385–389. [Google Scholar] [CrossRef]
- Zhao, D.-W.; Yang, J.-B.; Yang, S.-X.; Kato, K.; Luo, J.-P. Genetic diversity and domestication origin of tea plant Camellia taliensis (Theaceae) as revealed by microsatellite markers. BMC Plant Biol. 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Abdullah, T.L.; Ahmad, Z.; Abdullah, N.A.P. Effect of acute gamma irradiation on Curcuma alismatifolia varieties and detection of DNA polymorphism through SSR Marker. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, E.; Gemmell, N.J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, Y.D.; Tyekucheva, S.; Chiaromonte, F.; Makova, K.D. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008, 18, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Phumichai, C.; Phumichai, T.; Wongkaew, A. Novel chloroplast microsatellite (cpSSR) markers for genetic diversity assessment of cultivated and wild Hevea rubber. Plant Mol. Biol. Rep. 2015, 33, 1486–1498. [Google Scholar] [CrossRef]
- Lawson, M.J.; Zhang, L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.J.; Pádua, J.G.; Zucchi, M.I.; Vencovsky, R.; Vieira, M.L.C. Origin, evolution and genome distribution of microsatellites. Genet. Mol. Biol. 2006, 29, 294–307. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Zhang, M.-Y.; Liu, Q.-Z.; Li, L.-T.; Song, Y.; Wang, L.-F.; Zhang, S.-L.; Wu, J. Transferability of newly developed pear SSR markers to other Rosaceae species. Plant Mol. Biol. Rep. 2013, 31, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.S. SSR genotyping. In Plant Genotyping. Methods in Molecular Biology (Methods and Protocols); Batley, J., Ed.; Humana Press: New York, NY, USA, 2015; pp. 77–89. [Google Scholar]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Zargar, S.M.; Raatz, B.; Sonah, H.; Bhat, J.A.; Dar, Z.A.; Agrawal, G.K.; Rakwal, R. Recent advances in molecular marker techniques: Insight into QTL mapping, GWAS and genomic selection in plants. J. Crop Sci. Biotechnol. 2015, 18, 293–308. [Google Scholar] [CrossRef]
- Gao, H.; Jiang, K.; Geng, Y.; Chen, X.-Y. Development of microsatellite primers of the largest seagrass, Enhalus acoroides (Hydrocharitaceae). Am. J. Bot. 2012, 99, e99–e101. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.M.; Brar, D.S.; Ahloowalia, B. Molecular Techniques in Crop Improvement; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Antiqueira, L.M.O.R. Application of Microsatellite Molecular Markers in Studies of Genetic Diversity and Conservation of Plant Species of Cerrado. J. Plant Sci. 2013, 1, 1–5. [Google Scholar]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; Munhoz, C.D.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N. DNA molecular markers in plant breeding: Current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equipment 2017, 1–25. [Google Scholar] [CrossRef]
- Zheng, X.; Pan, C.; Diao, Y.; You, Y.; Yang, C.; Hu, Z. Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae). BMC Genom. 2013, 14, 490. [Google Scholar] [CrossRef] [PubMed]
- Nicot, N.; Chiquet, V.; Gandon, B.; Amilhat, L.; Legeai, F.; Leroy, P.; Bernard, M.; Sourdille, P. Study of simple sequence repeat (SSR) markers from wheat expressed sequence tags (ESTs). Theor. Appl. Genet. 2004, 109, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Röder, M.S.; Plaschke, J.; König, S.U.; Börner, A.; Sorrells, M.E.; Tanksley, S.D.; Ganal, M.W. Abundance, variability and chromosomal location of microsatellites in wheat. Mol. Gen. Genet. 1995, 246, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Ronning, C.M.; Stegalkina, S.S.; Ascenzi, R.A.; Bougri, O.; Hart, A.L.; Utterbach, T.R.; Vanaken, S.E.; Riedmuller, S.B.; White, J.A.; Cho, J. Comparative analyses of potato expressed sequence tag libraries. Plant Physiol. 2003, 131, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Kurata, N.A.; Nagamura, Y.; Yamamoto, K.; Harushima, Y.; Sue, N.; Wu, J.; Antonio, B.; Shomura, A.; Shimizu, T.; Lin, S.Y. A 300 kilobase interval genetic map of rice including 883 expressed sequences. Nat. Genet. 1994, 8, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Echalier, B.; Chao, S.; Lazo, G.; Butler, G.; Anderson, O.; Akhunov, E.; Dvořák, J.; Linkiewicz, A.; Ratnasiri, A. A chromosome bin map of 16,000 expressed sequence tag loci and distribution of genes among the three genomes of polyploid wheat. Genetics 2004, 168, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Burke, J. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Jo, K.M.; Jo, Y.; Chu, H.; Lian, S.; Cho, W.K. Development of EST-derived SSR markers using next-generation sequencing to reveal the genetic diversity of 50 chrysanthemum cultivars. Biochem. Syst. Ecol. 2015, 60, 37–45. [Google Scholar] [CrossRef]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, L.; Wang, L.; Wang, S.; Somta, P.; Cheng, X. Development and validation of EST-SSR markers from the transcriptome of adzuki bean (Vigna angularis). PLoS ONE 2015, 10, e0131939. [Google Scholar] [CrossRef] [PubMed]
- Temnykh, S.; DeClerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; McCouch, S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Eujayl, I.; Sorrells, M.; Baum, M.; Wolters, P.; Powell, W. Assessment of genotypic variation among cultivated durum wheat based on EST-SSRs and genomic SSRs. Euphytica 2001, 119, 39–43. [Google Scholar] [CrossRef]
- Yu, J.-K.; Dake, T.M.; Singh, S.; Benscher, D.; Li, W.; Gill, B.; Sorrells, M.E. Development and mapping of EST-derived simple sequence repeat markers for hexaploid wheat. Genome 2004, 47, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Ramu, P.; Kassahun, B.; Senthilvel, S.; Kumar, C.A.; Jayashree, B.; Folkertsma, R.; Reddy, L.A.; Kuruvinashetti, M.; Haussmann, B.; Hash, C. Exploiting rice–sorghum synteny for targeted development of EST-SSRs to enrich the sorghum genetic linkage map. Theor. Appl. Genet. 2009, 119, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Areshchenkova, T.; Ganal, M. Comparative analysis of polymorphism and chromosomal location of tomato microsatellite markers isolated from different sources. Theor. Appl. Genet. 2002, 104, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Poncet, V.; Rondeau, M.; Tranchant, C.; Cayrel, A.; Hamon, S.; De Kochko, A.; Hamon, P. SSR mining in coffee tree EST databases: Potential use of EST–SSRs as markers for the Coffea genus. Mol. Genet. Genom. 2006, 276, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Yang, C.; Tian, B.; Yang, J.-B.; Liu, A. Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communis L.). BMC Plant Biol. 2010, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Hua, W.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Abdullah, T.L.; Jain, S.M.; Sahebi, M.; Azizi, P. TILLING, high-resolution melting (HRM), and next-generation sequencing (NGS) techniques in plant mutation breeding. Mol. Breed. 2017, 37, 40. [Google Scholar] [CrossRef]
- Squirrell, J.; Hollingsworth, P.; Woodhead, M.; Russell, J.; Lowe, A.; Gibby, M.; Powell, W. How much effort is required to isolate nuclear microsatellites from plants? Mol. Ecol. 2003, 12, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Zane, L.; Bargelloni, L.; Patarnello, T. Strategies for microsatellite isolation: A review. Mol. Ecol. 2002, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Senalik, D.; McCown, B.; Zeldin, E.; Speers, J.; Hyman, J.; Bassil, N.; Hummer, K.; Simon, P.; Zalapa, J. Mining and validation of pyrosequenced simple sequence repeats (SSRs) from American cranberry (Vaccinium macrocarpon Ait.). Theor. Appl. Genet. 2012, 124, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 2010, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Csencsics, D.; Brodbeck, S.; Holderegger, R. Cost-effective, species-specific microsatellite development for the endangered dwarf bulrush (Typha minima) using next-generation sequencing technology. J. Hered. 2010, 101, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Reger, J.; Feulner, P.G.; Smadja, C.; Galindo, J.; Ekblom, R.; Bennison, C.; Ball, A.D.; Beckerman, A.P.; Slate, J. Adaptation genomics: The next generation. Trends Ecol. Evol. 2010, 25, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Wang, K.; Su, S.; Tian, R.; Li, Y.; Chen, M. De novo transcriptome analysis and microsatellite marker development for population genetic study of a serious insect pest, Rhopalosiphum padi (L.) (Hemiptera: Aphididae). PLoS ONE 2017, 12, e0172513. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.N.; Schlueter, J.; Spooner, D.M. Applications of next-generation sequencing in plant biology. Am. J. Bot. 2012, 99, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Chiu, Y.-C.; Wang, L.-B.; Kuo, Y.-L.; Chuang, E.Y.; Lai, L.-C.; Tsai, M.-H. Common applications of next-generation sequencing technologies in genomic research. Transl. Cancer Res. 2013, 2, 33–45. [Google Scholar]
- Grohme, M.A.; Soler, R.F.; Wink, M.; Frohme, M. Microsatellite marker discovery using single molecule real-time circular consensus sequencing on the Pacific Biosciences RS. BioTechniques 2013, 55, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Ambardar, S.; Gupta, R.; Trakroo, D.; Lal, R.; Vakhlu, J. High Throughput Sequencing: An Overview of Sequencing Chemistry. Indian J. Microbiol. 2016, 56, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Satya, P. Next generation sequencing technologies for next generation plant breeding. Front. Plant Sci. 2014, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Addisalem, A.; Esselink, G.D.; Bongers, F.; Smulders, M. Genomic sequencing and microsatellite marker development for Boswellia papyrifera, an economically important but threatened tree native to dry tropical forests. AoB Plants 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Blanca, J.; Cañizares, J.; Roig, C.; Ziarsolo, P.; Nuez, F.; Picó, B. Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genom. 2011, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, P.J.; Farmer, A.; Cannon, S.B.; Woodward, J.; Kudapa, H.; Tuteja, R.; Kumar, A.; BhanuPrakash, A.; Mulaosmanovic, B.; Gujaria, N. Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol. J. 2011, 9, 922–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.H.; Yoon, M.Y.; Cho, Y.I.; Chung, J.W.; Kim, K.T.; Cho, M.C.; Cheong, S.R.; Park, Y.J. Transcriptome analysis and SNP/SSR marker information of red pepper variety YCM334 and Taean. Scientia Horticulturae 2011, 129, 38–45. [Google Scholar] [CrossRef]
- Severin, A.J.; Woody, J.L.; Bolon, Y.-T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Zenoni, S.; Ferrarini, A.; Giacomelli, E.; Xumerle, L.; Fasoli, M.; Malerba, G.; Bellin, D.; Pezzotti, M.; Delledonne, M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010, 152, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.A.; Swain, M.T.; Hegarty, M.J.; Chernukin, I.; Lowe, M.; Allison, G.G.; Ruttink, T.; Abberton, M.T.; Jenkins, G.; Skøt, L. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Patel, R.K.; Tyagi, A.K.; Jain, M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011, 18, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Patel, R.K.; Jhanwar, S.; Priya, P.; Bhattacharjee, A.; Yadav, G.; Bhatia, S.; Chattopadhyay, D.; Tyagi, A.K.; Jain, M. Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol. 2011, 156, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, S.; Duan, J.; Wang, J.; Chen, S.; Cheng, Z.; Zhang, Q.; Liang, X.; Li, Y. De novo assembly and Characterization of the Transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.). BMC Genom. 2012, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Sun, Z.; Cui, B.; Zhang, Q.; Xiong, M.; Wang, X.; Zhou, D. Transcriptome analysis of colored calla lily (Zantedeschia rehmannii Engl.) by Illumina sequencing: De novo assembly, annotation and EST-SSR marker development. PeerJ 2016, 4, e2378. [Google Scholar] [CrossRef] [PubMed]
- Simsek, O.; Donmez, D.; Kacar, Y.A. RNA-Seq Analysis in Fruit Science: A Review. Am. J. Plant Biol. 2017, 2, 1–7. [Google Scholar]
- Li, S.; Tighe, S.W.; Nicolet, C.M.; Grove, D.; Levy, S.; Farmerie, W.; Viale, A.; Wright, C.; Schweitzer, P.A.; Gao, Y. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol. 2014, 32, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genom. 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Vijay, N.; Poelstra, J.W.; Künstner, A.; Wolf, J.B. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol. Ecol. 2013, 22, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yan, H.-D.; Zhang, X.-Q.; Zhang, J.; Frazier, T.P.; Huang, D.-J.; Lu, L.; Huang, L.-K.; Liu, W.; Peng, Y. De novo Transcriptome Analysis and Molecular Marker Development of Two Hemarthria Species. Front. Plant Sci. 2016, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Seco, D.; Zhang, Y.; Gutierrez-Mañero, F.J.; Martin, C.; Ramos-Solano, B. RNA-Seq analysis and transcriptome assembly for blackberry (Rubus sp. Var. Lochness) fruit. BMC Genom. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.A.; Zhai, J.; Nandety, R.S.; McCormick, K.P.; Zeng, J.; Mejia, D.; Meyers, B.C. Short-read sequencing technologies for transcriptional analyses. Annu. Rev. Plant Biol. 2009, 60, 305–333. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.B. Principles of transcriptome analysis and gene expression quantification: An RNA-seq tutorial. Mol. Ecol. Resour. 2013, 13, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.; Grosse, I.; Hähnel, U.; Siefken, R.; Prasad, M.; Stein, N.; Langridge, P.; Altschmied, L.; Graner, A. Genetic mapping and BAC assignment of EST-derived SSR markers shows non-uniform distribution of genes in the barley genome. Theor. Appl. Genet. 2006, 113, 239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, J.; Luo, Z.; Huang, L.; Chen, X.; Fang, B.; Li, Y.; Chen, J.; Zhang, X. Characterization and development of EST-derived SSR markers in cultivated sweetpotato (Ipomoea batatas). BMC Plant Biol. 2011, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Senalik, D.A.; Grzebelus, D.; Bowman, M.; Cavagnaro, P.F.; Matvienko, M.; Ashrafi, H.; Van Deynze, A.; Simon, P.W. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genom. 2011, 12, 389. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhang, Y.; Zhang, C.; Qi, F.; Li, X.; Mu, S.; Peng, Z. Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-seq analysis. PLoS ONE 2014, 9, e98910. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Wang, Y.; Zhang, X.; Li, H.; Lu, X.; Zhang, J.; Zhang, W.; Chen, S. De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PLoS ONE 2013, 8, e73767. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Pembleton, L.W.; Cogan, N.O.; Savin, K.W.; Leonforte, T.; Paull, J.; Materne, M.; Forster, J.W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genom. 2012, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, L.; Li, L.; Wang, S. De novo assembly of the common bean transcriptome using short reads for the discovery of drought-responsive genes. PLoS ONE 2014, 9, e109262. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fan, B.; Cao, Z.; Su, Q.; Wang, Y.; Zhang, Z.; Wu, J.; Tian, J. A deep sequencing analysis of transcriptomes and the development of EST-SSR markers in mungbean (Vigna radiata). J. Genet. 2016, 95, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Paudel, D.; Vendrame, W.; Wang, J. Enriching Genomic Resources and Marker Development from Transcript Sequences of Jatropha curcas for Microgravity Studies. Int. J. Genom. 2017, 2017. [Google Scholar] [CrossRef]
- Kovi, M.R.; Amdahl, H.; Alsheikh, M.; Rognli, O.A. De novo and reference transcriptome assembly of transcripts expressed during flowering provide insight into seed setting in tetraploid red clover. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Vatanparast, M.; Shetty, P.; Chopra, R.; Doyle, J.J.; Sathyanarayana, N.; Egan, A.N. Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae). Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Yang, H.; Sun, P.; Li, J.; Zhang, J.; Guo, Y.; Han, X.; Zhang, G.; Lu, M.; Hu, J. De novo transcriptome assembly, development of EST-SSR markers and population genetic analyses for the desert biomass willow, Salix psammophila. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Mora-Ortiz, M.; Swain, M.T.; Vickers, M.J.; Hegarty, M.J.; Kelly, R.; Smith, L.M.; Skøt, L. De novo transcriptome assembly for gene identification, analysis, annotation, and molecular marker discovery in Onobrychis viciifolia. BMC Genom. 2016, 17, 756. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Deng, M.; Zheng, S.-S.; Song, Y.-G. De novo transcriptome assembly and development of SSR markers of oaks Quercus austrocochinchinensis and Q. kerrii (Fagaceae). Tree Genet. Genom. 2016, 12, 103. [Google Scholar] [CrossRef]
- Zhou, T.; Li, Z.-H.; Bai, G.-Q.; Feng, L.; Chen, C.; Wei, Y.; Chang, Y.-X.; Zhao, G.-F. Transcriptome sequencing and development of genic SSR markers of an endangered Chinese endemic genus Dipteronia Oliver (Aceraceae). Molecules 2016, 21, 166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Luo, D.; Ma, L.; Xie, W.; Wang, Y.; Wang, Y.; Liu, Z. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- White, O.W.; Doo, B.; Carine, M.A.; Chapman, M.A. Transcriptome sequencing and simple sequence repeat marker development for three Macaronesian endemic plant species. Appl. Plant Sci. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, K.; Bi, D.; Zhou, B.S.; Shao, W.J. Characterization of the transcriptome and EST-SSR development in Boea clarkeana, a desiccation-tolerant plant endemic to China. PeerJ 2017, 5, e3422. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.K.; Wang, H.F.; Sakaguchi, S.; Landrein, S.; Isagi, Y.; Maki, M.; Zhu, Z.X. Development and characterization of EST-SSR markers in an East Asian temperate plant genus Diabelia (Caprifoliaceae). Plant Species Biol. 2017, 32, 247–251. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Y.; Zhao, Y.; Yang, S.; Udikeri, S.; Liu, T. De Novo Characterization of the Root Transcriptome and Development of EST-SSR Markers in Paris polyphylla Smith var. yunnanensis, an Endangered Medical Plant. J. Agric. Sci. Technol. 2016, 18, 437–452. [Google Scholar]
- Liang, M.; Yang, X.; Li, H.; Su, S.; Yi, H.; Chai, L.; Deng, X. De novo transcriptome assembly of pummelo and molecular marker development. PLoS ONE 2015, 10, e0120615. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Liu, Z.X.; Chen, X.; Zhang, T.; Zhou, H.J.; Hu, Y.H.; Zhao, P. Identification, development, and application of 12 polymorphic EST-SSR markers for an endemic Chinese walnut (Juglans cathayensis L.) using next-generation sequencing technology. Biochem. Syst. Ecol. 2015, 60, 74–80. [Google Scholar] [CrossRef]
- Ding, Q.; Li, J.; Wang, F.; Zhang, Y.; Li, H.; Zhang, J.; Gao, J. Characterization and development of EST-SSRs by deep transcriptome sequencing in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Int. J. Genom. 2015, 2015. [Google Scholar] [CrossRef]
- Zheng, X.; You, Y.; Diao, Y.; Zheng, X.; Xie, K.; Zhou, M.; Hu, Z.; Wang, Y. Development and characterization of genic-SSR markers from different Asia lotus (Nelumbo nucifera) types by RNA-seq. Gen. Mol. Res. 2015, 14, 11171–11184. [Google Scholar] [CrossRef] [PubMed]
- Ambreen, H.; Kumar, S.; Variath, M.T.; Joshi, G.; Bali, S.; Agarwal, M.; Kumar, A.; Jagannath, A.; Goel, S. Development of genomic microsatellite markers in Carthamus tinctorius L.(safflower) using next generation sequencing and assessment of their cross-species transferability and utility for diversity analysis. PLoS ONE 2015, 10, e0135443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.C.; Shih, H.C.; Wang, H.V.; Lin, Y.S.; Chang, C.H.; Chiang, Y.C.; Chou, C.H. RNA-seq SSRs of moth orchid and screening for molecular markers across genus Phalaenopsis (Orchidaceae). PLoS ONE 2015, 10, e0141761. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Cao, Y.N.; Yuan, N.; Nakamura, K.; Wang, G.M.; Qiu, Y.X. Characterization of transcriptome and development of novel EST-SSR makers based on next-generation sequencing technology in Neolitsea sericea (Lauraceae) endemic to East Asian land-bridge islands. Mol. Breed. 2015, 35, 1–15. [Google Scholar] [CrossRef]
- Ravishankar, K.; Dinesh, M.; Nischita, P.; Sandya, B. Development and characterization of microsatellite markers in mango (Mangifera indica) using next-generation sequencing technology and their transferability across species. Mol. Breed. 2015, 35, 1–13. [Google Scholar] [CrossRef]
- Torre, S.; Tattini, M.; Brunetti, C.; Fineschi, S.; Fini, A.; Ferrini, F.; Sebastiani, F. RNA-seq analysis of Quercus pubescens Leaves: De novo transcriptome assembly, annotation and functional markers development. PLoS ONE 2014, 9, e112487. [Google Scholar] [CrossRef] [PubMed]
- Izzah, N.K.; Lee, J.; Jayakodi, M.; Perumal, S.; Jin, M.; Park, B.-S.; Ahn, K.; Yang, T.-J. Transcriptome sequencing of two parental lines of cabbage (Brassica oleracea L. var. capitata L.) and construction of an EST-based genetic map. BMC Genom. 2014, 15, 149. [Google Scholar] [CrossRef] [PubMed]
- Salgado, L.R.; Koop, D.M.; Pinheiro, D.G.; Rivallan, R.; Le Guen, V.; Nicolás, M.F.; De Almeida, L.G.P.; Rocha, V.R.; Magalhães, M.; Gerber, A.L. De novo transcriptome analysis of Hevea brasiliensis tissues by RNA-seq and screening for molecular markers. BMC Genom. 2014, 15, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yu, G.; Shi, B.; Wang, X.; Qiang, H.; Gao, H. Development and characterization of simple sequence repeat (SSR) markers based on RNA-sequencing of Medicago sativa and in silico mapping onto the M. truncatula genome. PLoS ONE 2014, 9, e92029. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Cogan, N.O.; Kaur, S.; Drayton, M.; Mouradov, A.; Panter, S.; Schrauf, G.E.; Mason, J.G.; Spangenberg, G.C. Gene discovery and molecular marker development, based on high-throughput transcript sequencing of Paspalum dilatatum Poir. PLoS ONE 2014, 9, e85050. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Chen, X.; Zou, D. Sequencing, de novo assembly, annotation and SSR and SNP detection of sabaigrass (Eulaliopsis binata) transcriptome. Genomics 2013, 102, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Kim, T.S.; Suresh, S.; Lee, S.Y.; Cho, G.T. Development of 65 novel polymorphic cDNA-SSR markers in common vetch (Vicia sativa subsp. sativa) using next generation sequencing. Molecules 2013, 18, 8376–8392. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Park, J.H.; Cho, G.T.; Lee, H.S.; Baek, H.J.; Lee, S.Y.; Chung, J.W. Development and molecular characterization of 55 novel polymorphic cDNA-SSR markers in faba bean (Vicia faba L.) using 454 pyrosequencing. Molecules 2013, 18, 1844–1856. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Shah, N.; Bhatia, S. Development of an expressed gene catalogue and molecular markers from the de novo assembly of short sequence reads of the lentil (Lens culinaris Medik.) transcriptome. Plant Biotechnol. J. 2013, 11, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-Q.; Wang, L.-Y.; Wei, K.; Zhang, C.-C.; Wu, L.-Y.; Qi, G.-N.; Cheng, H.; Zhang, Q.; Cui, Q.-M.; Liang, J.-B. Floral transcriptome sequencing for SSR marker development and linkage map construction in the tea plant (Camellia sinensis). PLoS ONE 2013, 8, e81611. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, D.; Li, J.; Yu, B.; Qiao, X.; Huang, H.; He, Y. De novo characterization of leaf transcriptome using 454 sequencing and development of EST-SSR markers in tea (Camellia sinensis). Plant Mol. Biol. Rep. 2013, 31, 524–538. [Google Scholar] [CrossRef]
- Pazos-Navarro, M.; Dabauza, M.; Correal, E.; Hanson, K.; Teakle, N.; Real, D.; Nelson, M.N. Next generation DNA sequencing technology delivers valuable genetic markers for the genomic orphan legume species, Bituminaria bituminosa. BMC Genet. 2011, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Cogan, N.O.; Pembleton, L.W.; Shinozuka, M.; Savin, K.W.; Materne, M.; Forster, J.W. Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genom. 2011, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Triwitayakorn, K.; Chatkulkawin, P.; Kanjanawattanawong, S.; Sraphet, S.; Yoocha, T.; Sangsrakru, D.; Chanprasert, J.; Ngamphiw, C.; Jomchai, N.; Therawattanasuk, K. Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res. 2011, 18, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Surget-Groba, Y.; Montoya-Burgos, J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Bruno, V.M.; Fang, Z.; Meng, X.; Blow, M.; Zhang, T.; Sherlock, G.; Snyder, M.; Wang, Z. Rnnotator: An automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genom. 2010, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Cameron, M.; Williams, H.E.; Cannane, A. Improved gapped alignment in BLAST. IEEE/ACM Trans. Comput. Biol. Bioinform. 2004, 1, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Group, W.P.W. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Da Maia, L.C.; Palmieri, D.A.; De Souza, V.Q.; Kopp, M.M.; de Carvalho, F.I.F.; Costa de Oliveira, A. SSR locator: Tool for simple sequence repeat discovery integrated with primer design and PCR simulation. Int. J. Plant Genom. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, P.; Luo, Z. GMATo: A novel tool for the identification and analysis of microsatellites in large genomes. Bioinformation 2013, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Kumar, R.; Srivastava, P.; Agarwal, S.; Srivastava, S.; Nagpure, N.S.; Jena, J.K.; Kushwaha, B. WGSSAT: A High-Throughput Computational Pipeline for Mining and Annotation of SSR Markers From Whole Genomes. J. Hered. 2017. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, K.J.; Jansen, A.; Lewitter, F.; Fink, G.R. Intragenic tandem repeats generate functional variability. Nat. Genet. 2005, 37, 986. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2012, 13, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Fungtammasan, A.; Ananda, G.; Hile, S.E.; Su, M.S.-W.; Sun, C.; Harris, R.; Medvedev, P.; Eckert, K.; Makova, K.D. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. 2015, 25, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Golan, D.; Rosset, S.; Erlich, Y. lobSTR: A short tandem repeat profiler for personal genomes. Genome Res. 2012, 22, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Highnam, G.; Franck, C.; Martin, A.; Stephens, C.; Puthige, A.; Mittelman, D. Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res. 2012, 41, e32. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Tasker, E.; Willadsen, K.; Imelfort, M.; Vishwanathan, S.; Sureshkumar, S.; Balasubramanian, S.; Bodén, M. Inferring short tandem repeat variation from paired-end short reads. Nucleic Acids Res. 2013, 42, e16. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, C.; D’Agostino, N. PSR: Polymorphic SSR retrieval. BMC Res. Notes 2015, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Buckler, E.S.; Ilut, D.C.; Wang, X.; Kretzschmar, T.; Gore, M.A.; Mitchell, S.E. rAmpSeq: Using repetitive sequences for robust genotyping. BioRxiv 2016. [Google Scholar] [CrossRef]
- Tang, H.; Nzabarushimana, E. STRScan: Targeted profiling of short tandem repeats in whole-genome sequencing data. BMC Bioinform. 2017, 18, 398. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.P. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [Green Version]



{kind=link}
{kind=link}
| Species | SSR Type | No. Unigenes | NGS Technology | Total No. of Discovered SSRs | Total No. of SSR Primer Designed | Total Polymorphic SSR Primers | Reference |
|---|---|---|---|---|---|---|---|
| Jatropha curcas | SSR | 115,611 | Roche 454 Genome Sequencer | 9798 | 262 | 33 | [102] |
| Guar (Cyamopsis tetragonoloba, L. Taub.) | SSR | 62,146 | Illumina HiSeq 2000 sequencing platform | 5773 | 20 | 13 | [8] |
| Red clover (Trifolium pratense L.) | SSR | 80,328/83,489/; 84,545/84,442 | Illumina HiSeq 2000 sequencing platform | 15 | n/a | 15 | [103] |
| Winged bean (Psophocarpus tetragonolobus) | EST-SSR | 97,241 | Roche 454 Genome Sequencer FLX (Titanium chemistry) | 12,956 | 2994 | n/a | [104] |
| Colored calla lily (Zantedeschia rehmannii Engl.) | EST-SSR | 39,298 | Illumina HiSeq 2000 sequencing platform | 9933 | 200 | 58 | [80] |
| Salix psammophila | EST-SSR | 71,458 | Illumina HiSeq2500 platform | 6346 | 168 | 27 | [105] |
| Sainfoin (Onobrychis viciifolia) | SSR | 92,772 | Illumina Hiseq 2000 sequencing platform | 3823 | 100 | n/a | [106] |
| Two Hemarthria Species | SSR | 137,142/77,150 | Illumina HiSeqTM 2500 sequencing platform | 10,888 | 4846 | 34 | [89] |
| Oak (Quercus austrocochinchinensis) & (Q. kerrii) | SSR | 49,845/50,767 | Illumina MiSeq sequencing platform | 13,762/13,430 | 5196/5021 | 18 | [107] |
| Dipteronia oliver (Aceraceae) | SSR | 99,358 | Illumina Hiseq 2000 sequencing platform | 12,377 | 4179 | 97 | [108] |
| Elymus sibiricus L. | EST-SSR | 94,458 | Illumina HiSeq2000 sequencing platform | 8769 | 500 | 112 | [109] |
| Argyranthemum broussonetii, Echium wildpretii, Descurainia bourgaeana | SSR | 80,620 | Illumina MiSeq sequencing platform | 2282 | 30 | 8 | [110] |
| 58,526 | 1284 | n/a | n/a | ||||
| 44,287 | 1972 | n/a | n/a | ||||
| Boea clarkeana Hemsl. (Boea, Gesneriaceae) | EST-SSR | 91,449 | Illumina HiSeqTM 2000 sequencing platform | 8563 | 436 | 17 | [111] |
| Diabelia (Caprifoliaceae) | EST-SSR | 58669 | Illumina HiSeqTM 2000 sequencing platform | n/a | 2746 | 13 | [112] |
| Paris polyphylla Smith | EST-SSR | 56,095 | Illumina HiSeq2000 sequencing platform | 3853 | 80 | 9 | [113] |
| Pummelo (Citrus grandis (L.) Osbeck) | SSR | 57,212 | Illumina HiSeq2000 sequencing platform | 10,276 | 1174 | 29 | [114] |
| Chinese walnut (Juglans cathayensis L.) | EST-SSR | 116814 | Illumina HiSeq2000 sequencing platform | 22,484 | 62 | 12 | [115] |
| Chinese cabbage (Brassica rapa L. ssp. pekinensis) | EST-SSR | 51,694 | Solexa/Illumina | 10,420 | 24 | 17 | [116] |
| Lotus (Nelumbo nucifera) | SSR | 105,834 | Illumina HiSeqTM 2000 sequencing platform | 11,178 | 6568 | 80 | [117] |
| Carthamus tinctorius L. (Safflower) | SSR | 2,043,956 | Illumina HiSeqTM 2000 sequencing platform | 23,067 | 325 | 93 | [118] |
| Phalaenopsis aphrodite subsp. formosana | EST-SSR | 22,598 | Illumina HiSeqTM 2000 sequencing platform | 1439 | 1051 | 10 | [119] |
| Neolitsea sericea (Lauraceae) | EST-SSR | 68,624 | Illumina HiSeqTM 2000 sequencing platform | 13,213 | 1191 | 13 | [120] |
| Mango (Mangifera indica) | SSR | 66,288 | Illumina HiSeq 2000 sequencing platform | 106,049 | 84,118 | 90 | [121] |
| Adzuki bean (Vigna angularis) | EST-SSR | 112 million | Illumina HiSeq2000 sequencing platform | 7947 | 296 | 38 | [41] |
| Quercus pubescens | SSR | 96,006 | Illumina HiSeq 2000 sequencing platform | 14,202 | 10,864 | 20 | [122] |
| Brassica oleracea L. var. capitate L. | EST-SSR | 34,688 and 40,947 | 454 GS FLX Titanium Sequencer | 2405 | 937 | 116 | [123] |
| Hevea brasiliensis | SSR | 19,708 | Roche 454 sequencing platform | 1397 | n/a | n/a | [124] |
| Medicago sativa | EST-SSR | 54,278 | Illumina HiSeqTM 2000 sequencing platform | 4493 | 837 | 372 | [125] |
| Paspalum dilatatum Poir. | EST-SSR | 20169 | GS FLX Titanium technology | 2339 | 96 | 32 | [126] |
| Red clover (Trifolium pratense L.) | SSR | 45181 | Illumina HiSeq2000 sequencing platform | 3127 | 2193 | n/a | [76] |
| Eulaliopsis binata | SSR | 59,134 | Illumina HiSeq 2000 sequencing platform | 6681 | 5,723 | 24 | [127] |
| Common vetch (Vicia sativa subsp. sativa) | cDNA-SSR (cSSR) | n/a | 454 Pyrosequencing platform | 3811 | 300 | 65 | [128] |
| Faba bean (Vicia faba L.) | cDNA-SSR (cSSR) | n/a | 454 Pyrosequencing platform | 1729 | 240 | 55 | [129] |
| lentil (Lens culinaris Medik.) | SSR | 55,463 | Illumina Genome Analyzer II platform | 8722 | 5,673 | 23 | [130] |
| Amorphophallus (Araceae) | SSR | 135,822 | Illumina HiSeq™ 2000 sequencing platform | 19,596 | 10,754 | 205 | [31] |
| Tea (Camellia sinensis) | SSR | 75,531 | Illumina HiSeq™ 2000 platform | 12,582 | 2439 | 431 | [131] |
| Faba bean (Vicia faba L.) | cDNA-SSR (cSSR) | n/a | 454 Pyrosequencing platform | 1729 | 240 | 55 | [129] |
| Tea (Camellia sinensis) | EST-SSR | 25,637 | Roche/454 Genome Sequencer FLX Instrument | 3767 | 100 | 36 | [132] |
| Rubber tree (Hevea brasiliensis Muell. Arg.) | EST-SSR | 22,756 | Illumina HiSeqTM 2000 sequencing platform | 39,257 | 110 | 61 | [49] |
| Peanut (Arachis hypogaea L.) | SSR | 59,077 | Solexa HiSeq™ 2000 sequencing platform | 3919 | 160 | 65 | [79] |
| Bituminaria bituminosa | SSR | 3838 | Roche 454 sequencing platform | 3419 | 240 | 21 | [133] |
| (Sesamum indicum L.) | EST-SSR | 86,222 | Illumina HiSeq2000 sequencing platform | 7702 | 50 | 40 | [51] |
| Pigeonpea [Cajanus cajan (L.) Millspaugh] | SSR | 43,324 | 454 GS-FLX sequencing platform | 3771 | 2877 | 20 | [72] |
| Chickpea (Cicer arietinum L.) | SSR and SNP | 103,215 | Roche⁄454 and Illumina⁄Solexa | 26,252 | 3172 | 42 | [71] |
| Lentil (Lens culinaris Medik.) | EST-SSR | 25,592 | Roche 454 GS-FLX Titanium platform | 1.38 × 106 | 2393 | 51 | [134] |
| Hevea brasiliensis | EST-SSR | 113,313 | 454 pyrosequencing platform | 17,819 | 430 | 47 | [135] |
| Sweet potato (Ipomoea batatas) | cDNA SSR (cSSR) | 56,516 | Illumina paired-end sequencing platform | 4114 | 100 | 92 | [86] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taheri, S.; Lee Abdullah, T.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and Development of Novel SSR Markers Using Next Generation Sequencing (NGS) Data in Plants. Molecules 2018, 23, 399. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020399
Taheri S, Lee Abdullah T, Yusop MR, Hanafi MM, Sahebi M, Azizi P, Shamshiri RR. Mining and Development of Novel SSR Markers Using Next Generation Sequencing (NGS) Data in Plants. Molecules. 2018; 23(2):399. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020399
Chicago/Turabian StyleTaheri, Sima, Thohirah Lee Abdullah, Mohd Rafii Yusop, Mohamed Musa Hanafi, Mahbod Sahebi, Parisa Azizi, and Redmond Ramin Shamshiri. 2018. "Mining and Development of Novel SSR Markers Using Next Generation Sequencing (NGS) Data in Plants" Molecules 23, no. 2: 399. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020399