
Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy


1
Department of Chemistry, Graduate School of Science Kyoto University, Kitashirakawa-Oiwakecho, Sakyo, Kyoto, 606-8502, Japan
2
Institute for Integrated Cell-Material Science (WPI-iCeMS) Kyoto University, Yoshida-Ushinomiyacho, Sakyo, Kyoto, 606-8501, Japan
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(3), 429; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030429
Submission received: 3 December 2018
/
Revised: 22 January 2019
/
Accepted: 23 January 2019
/
Published: 24 January 2019
(This article belongs to the Special Issue G-Quadruplex Ligands and Cancer)
Abstract
:A G-quadruplex (G4) is a well-known nucleic acid secondary structure comprising guanine-rich sequences, and has profound implications for various pharmacological and biological events, including cancers. Therefore, ligands interacting with G4s have attracted great attention as potential anticancer therapies or in molecular probe applications. To date, a large variety of DNA/RNA G4 ligands have been developed by a number of laboratories. As protein-targeting drugs face similar situations, G-quadruplex-interacting drugs displayed low selectivity to the targeted G-quadruplex structure. This low selectivity could cause unexpected effects that are usually reasons to halt the drug development process. In this review, we address the recent research on synthetic G4 DNA-interacting ligands that allow targeting of selected G4s as an approach toward the discovery of highly effective anticancer drugs.
1. Introduction
A G-quadruplex (G4) is an alternative form of DNA or RNA comprising several planar layers of four guanines (G-tetrads) held together via Hoogsteen hydrogen bonding (Figure 1) [1,2,3,4,5]. The G4 DNA has long been studied from various aspects and is now considered to be an important player in biological and biomedical events, including the control of promoter activity [6,7,8,9], genome instability [10,11,12,13], benchmarks for specified chromatin remodeling and replication [14,15], and epigenetic alterations [16,17]. The sequences, 5′-G≥3N1–7G≥3N1–7G≥3N1–7G≥3-3′, are advocated as consensus sequences that have the ability to form intramolecular G4s [18,19], although several exceptions have been reported at the present time [20,21,22,23,24]. Extensive physical characterizations of the G4 structure by means of a series of assays based on UV and CD spectroscopy have clearly demonstrated that the structure has extremely high stability when possessing one or two nucleotide(s) between the G-tracts (Tm = ca. 70 ~ 90 °C) [25]. In parallel, a large number of defined G4 structures were elucidated at the atomic level using nuclear magnetic resonance (NMR) spectroscopy and X-ray crystallography, opening a new avenue for the rational design of G4 ligands [26,27,28,29,30,31,32,33,34]. Such high thermal stability and availability of defined structures allow us to develop applications in the field of DNA nanotechnology [35], as well as biological and biomedical studies.
In particular, given the characteristic human genome sequence of telomere (TTAGGG)n that is located at the end of chromosomes and in a single-stranded context, the formation of G4 structures in this region is first assumed in the human genome [36,37]. Abundant evidence has accumulated during the past two decades that the G4 structure is truly formed in the telomere region and has an important role in telomere-end processing in cells [38]. More importantly, stabilization of telomere G4s and blockage of telomerase activities by small molecules, exemplified by telomestatin, has been shown to be a new strategy for antitumor therapy [39,40].
G4-forming sequences observed in the promoter of cancer-related genes have also received a great deal of attention as potential biomedical targets for antitumor therapy [8]. Quarfloxin, a ligand that interacts with G4, had completed Phase II trials as a candidate therapeutic agent against several tumors, including neuroendocrine tumors, carcinoid tumors, and lymphoma [41]. It is considered that quarfloxin disrupts the G4–nucleolin complexes of ribosomal DNA in the nucleolus, which in turn redistributes nucleolin into the nucleoplasm, where it binds to a G4 in the promoter region of the c-MYC proto-oncogene to inhibit its gene expression [8]. The Phase III trials for quarfloxin are currently not proceeding due to high albumin binding [42]. In addition to this report, other tumor-related genes, including hTERT [43], c-kit [44], KRAS [45,46], BCL2 [47], and VEGF [48], were identified as genes in which the formation of a G4 was involved in transcriptional regulation, and its stabilization by small molecules attenuated promoter activity, eventually inducing tumor apoptosis.
Relatively recent studies revealed that G4 also had an impact on differentiation- and neuron-related genes [20]. For instance, OCT4 expression may be governed, to some degree, by G4 formation at the proximal promoter in human embryonic stem cells (CCTL14) [49], whereas the excessive formation of repetitive G4 structures on an expandable (GGGGCC)n in C9orf72 gene or (CGG)n in FMR1 gene accounts for some neurogenetic disorders [50]. On the contrary, G4 can act positively in neurons, where G4 structures at the CpG island located in xl3b are recognized by ATRX, contributing to appropriate synaptic function [51].
Extensive studies of G4s and ligands that interact with them lead investigators to believe in the notion that G4s are able to widely form in guanine-rich regions of the genome [52], in the context of cellular dynamics as exemplified by transcription, duplication, and DNA repair processes, in which DNA strands are transiently dissociated to generate flexible DNA single strands. Although numerous investigators have made tremendous efforts to obtain highly active G4 ligands, and some of them have attained great success in the development of drugs in vivo [53,54,55,56]; however, these drugs are still only midway towards approval for clinical use.
One conceivable obstacle impeding the clinical application of G4-interacting molecules seems to rest with selectivity, although the global or multiple G4 targeting approaches may be effective [57,58,59,60,61], and in fact, CX-5461, a DNA G4 stabilizer, is currently at advanced phase I clinical trials for patients with BRCA1/2 deficient tumors [57,58]. Bioinformatics and next-generation sequencing (NGS) analysis estimated that 376,000 or more putative G4-forming sequences exist in the human genome [18,62]. A growing number of G4-driven genes have also been reported, suggesting the high importance of the expanded variety of G4-interacting ligands that possess differential binding profiles [8,53,54,55,56]. However, poor ligand designability arising from the topological similarity of the skeleton of diverse G4s has remained a bottleneck for obtaining specificity toward individual G4s. Very recently, investigators have entered a new phase of the development of next-generation ligands that interact with G4, in which they consider the ligand selectivity to a particular G4 to be targeted, potentially leading to the development of molecules with high antitumor activity and bioactivity with minimal antitumor therapy side effects [56]. In this review, we address the recent research progress toward developing G4-interacting molecules that exhibit antitumor activities by affecting a particular cancer-related gene with reduced off-target effects that likely rely on a clear selectivity for targeted G4s.
2. Global G-Quadruplex-Selective Ligands
Since G4-interacting molecules were developed based on duplex DNA-binding molecules, investigators have initially endeavored to develop G4 ligands that have a clear selectivity for G4 structures over the duplex DNA [63,64,65]. Molecules interacting with telomere G4s, the 2,6-diamidoanthraquinone derivatives, were first found to act as telomerase inhibitors by Neidle and Hurley and their group [36]. The cationic porphyrin, TMPyP4, whose planar skeleton and cationic propensity would facilitate G4 binding, was also identified as a G4 binder [66]. Telomestatin, a macrocycle naturally occurring in Streptomyces annulatus, was found to exhibit telomerase-inhibiting activity by binding to telomeric G4 structures, causing telomere dysfunction, including telomere length shortening and delocalization of telomere-related proteins [39,40]. That discovery was noteworthy in that it suggested the existence of G4s in vivo. These pioneering works accelerated the development of G4-selective synthetic molecules, along with the advance in rigid methods and techniques to characterize the binding profiles of such G4 ligands [21b]. In fact, several commercially available G4 ligands, such as BRACO19 [67], pyridostatin [68], Phen-DC3 [69], L2H2-6OTD [70], and L1H1-7OTD [71], all of which have negligible binding affinities to duplex DNAs, are indispensable to biochemical, biophysical, and chemical biology studies of G4s.
3. Cancer-Related G-Quadruplexes and Their Interacting Molecules
3.1. Telomere
A telomere is a structure of the ends of a chromosome, in which a repeated microsatellite sequence and its various related proteins (called a shelterin complex), including POT1 and TRF2, which are necessary for telomere maintenance, protect the telomeric DNA from DNA repair mechanisms, and also regulates telomerase activity in mammals [72]. The human telomere region comprises a single microsatellite repeat sequence, (GGGTTA)n, with a 3′ overhang at its terminus (200 ± 75 nucleotides). The length of the telomere sequence gradually shortens along with DNA replication, which limits cell growth and proliferation. The G4 structures observed in this region initially received great attention because a single-stranded background offers a greater likelihood of G4 formation. Interestingly, ATRX, POT1, TPP1, and other G4-binding proteins function in the telomere processing-like recruitment of a specific protein in a G4-involved manner, whereas varied G4 helicases, including Pif1 and BLM, act to unwind the telomere G4 structures to maintain the shelterin complex [54,73]. Furthermore, because telomerase, a protein complex for elongating the telomere sequences, is highly expressed in many tumor cells, and the abnormal processing of the telomere 3′ overhang region may confer cellular immortality, a telomere G4 could be thought of as a potential biomedical target for small molecules that inhibit telomerase activity in telomerase-positive tumor cells [36,37,39,40,74].
The 2,6-diamidoanthraquinone derivatives and Telomestatin were found to be telomerase inhibitors through their binding to telomere G4s at an early stage of G4-interacting ligand studies [36,39,40]. RHPS4 was also shown to induce telomere dysfunction by disturbing the integrity of the shelterin complex in mammalian cancer cells [38]. Later relevant studies found that a large variety of alternative higher-order structures derived from the canonical telomere G4 might be adopted at the 3′ overhang region [20,21,75,76]. Those structures and their specific motifs are amenable to a gain of specificity for telomere G4s [56] (Figure 2a).
Dimeric G4 ligands target dimeric G4s. Tandemly aligned G4 ligands permit the favorable discrimination of a dimeric G4 from one that is monomeric. The dinickel salophen dimer [77], berberine dimer [78], and telomestatin derivative tetramer [79] are examples of such ligands that discriminate G4s successfully (Figure 2b). Binding to the side faces of dimeric G4s confers a preference for multimeric G4s over one that is monomeric, as exemplified by m-TMPipEOPP (Figure 2b) [80]. By contrast, chiral helical supramolecules, Ni-M, exhibit a binding preference for dimers over monomers, with a 200-fold selectivity, probably because two consecutive G4s offer a preferred binding site (Figure 2b) [81]. Conversely, another enantiomer, Ni-P, is capable of converting a monomeric antiparallel form to a monomeric hybrid form [82]. It is noteworthy that these two enantiomers display different abilities to affect telomere dysfunction in vivo; NiP, rather than NiM, preferentially inhibits cell growth in breast CSCs compared with bulk cancer cells. This inhibition relies on telomere uncapping with the delocalization of TRF2 and POT1 from telomeres, telomere DNA damage, and degradation of the 3′-overhang by G4-mediated binding of NiP [83].
The junction pocket between two G4 units also serves as a target for specific recognition. In early studies of selective G4-interacting ligands, our group proposed the possibility that a chiral helical molecule, helicene M1, allowed for the targeting of the junction cavity between two G4 units (Figure 2b) [84]. A triaryl-substituted imidazole molecule, IZNP1, was shown to be correctly positioned into the junction by molecular modeling and to exhibit a reduced binding affinity for TERRA multimeric RNA G4s (Figure 2b) [85]. Notably, this structure specificity was biologically validated, where IZNP1 caused telomeric DNA damage and telomere dysfunction without affecting several well-studied oncogenes that have monomeric G4s in their promoter regions.
Most recently, Yang and colleagues reported that a berberine derivative, EPI, discriminates a hybrid type 2 telomere G4 structure formed by a wtTel26 sequence 5′-(TTAGGG)4TT-3′ from the other adoptable topologies and promoter G4s (c-MYC, BCL2, and PDGFR), as elucidated by an NMR structural analysis (Figure 2c) [86]. In addition, EPI has the ability to convert the other conformations of the telomere G4 hybrid type 1, and antiparallel (basket type) G4s into the type 2 hybrid. Such an unprecedented propensity and selectivity relies on the stacking onto the terminal G-quartet hydrogen bonding formation with the flanking base, and the creation of a binding pocket by adaptively forming a TTA triad above the ligand. In this study, the biological impact of EPI on telomere function, including telomerase inhibition and delocalization of telomere-related proteins in cells, was not examined. However, it is conceived as that a 5′-flanking element observed in the genome context would likely serve as an adaptive and selective binding site of EPI to the telomere region, resulting in highly disruptive activity to telomere function with minimal side effects.
A long-loop DNA sequence arranged by a monomeric G4 was also amenable to molecular recognition by hybridization of the complementary strand, which was demonstrated by an atomic-level NMR analysis [20]. In particular, hybrid molecules constructed with a complementary sequence and a G4-interacting ligand would in future be added as candidates for selective recognition of telomere G4s.
3.2. c-MYC
c-MYC encodes a multifunctional transcription factor that can act as a transcription activator of some genes involved in cell proliferation, while acting as a transcription repressor of other genes involved in growth arrest [87,88]. There are a broad variety of c-MYC-responsive genes that engage in important cellular functions in concert, such as cell proliferation, metabolic transformation, and metastatic capacity [89]. In tumor cells, c-MYC protein function is almost always activated primarily through upstream oncogenic pathways. As the overexpression of the c-MYC is served in various human malignancies (particularly in 80% of solid tumors), downregulation of the gene is an effective approach to cancer therapy [87,88]. However, the c-MYC protein is generally considered to be an undruggable target by small molecules owing to its short half-life, large dimension, and unstructured nature [8].
The c-MYC promoter region contains the nuclease hypersensitive element (NHE) III1, which is located −142 to −115 base pairs upstream of the P1 promoter (Figure 3a). There is one putative G4-forming sequence (PQS) in this element, which is capable of forming a nonduplex species, possibly accompanied by local unwinding or melting of the duplex structure under the influence of negative supercoiling stress [90,91,92]. Structural dynamics in this region have also been considered to be a possible key mechanism in certain carcinomas, largely to govern c-MYC transcription, and the formation of a G4 is likely to act as a downregulator. Hence, G4-interacting ligands may contribute to suppression of downstream c-MYC expression by ligand-mediated G4 stabilization [7]. In this context, c-MYC targeting G4-interacting ligands have been studied during the past two decades with an aim toward drug applications for antitumor therapy.
An exon-specific RT-qPCR assay using two pairs of Burkitt’s lymphoma cell lines (CA46 and RAJI), devised by Brooks group, allowed us to evaluate whether G4 ligands directly act for c-MYC G4 or not in cells [93,94]. This assay relies on the unique reciprocal translocation of c-MYC gene locus in the two cell lines by the translocation in CA46, where exon 1 is separated from exons 2 and 3; the G4-driven transcriptional activity was only maintained for exon 1, whereas these three exons and the PQS remained in tandem after the translocation in RAJI. That is why the use of primers specific for the exons 1 and 2 in a qPCR permits the demonstration of the c-MYC G4-mediated control by G4 ligands in the cellular context. In fact, a G4-interacting ligand, GQC-05 (NSC338258), was shown to directly suppress gene expression in a c-MYC G4-mediated manner using this assay [93]. This system is now a standard method for studying the intracellular activities of the c-MYC G4 targeting small molecules.
Recently, G4-interacting ligands that have a clear selectivity for c-MYC G4 were reported, and some of these proved to have effective antitumor activities, probably by reducing off-target effects as seen in biological experiments.
Sequence-selective recognition of duplex DNAs adjacent to an intended G4 serves to gain specificity in the genomic context. Our group has developed hybrid molecules, where a cIKP as a G4-binding motif is covalently linked to a sequence-selective duplex-binding molecule, pyrrole-imidazole polyamide (PIP) [95]. These hybrid molecules display simultaneous and synergistic recognition of both the c-MYC G4 and its proximal duplex (Figure 3b). Among these, hybrid 3 exhibits a 4.6-fold selectivity for a G4/duplex substrate that was predicted to be the most likely off target (KD = 3.7 × 10−9 versus 17 × 10−9 M) [95]. The PIP part of the hybrid molecules accounts for their selectivity—PIPs are able to recognize a G/C base pair by antiparallel pairing of an imidazole/pyrrole moiety and an A/T or a T/A base pair by antiparallel pairing of a P/P pair. A β-alanine/β-alanine pair reads an A/T or a T/A base pair in a way similar to a P/P pair [96]. This ligand-design methodology is, in theory, applicable to the selective targeting of a broad variety of designated G4s in the genome.
Dash and colleagues reported that a crescent-shaped thiazole peptide, TH3, preferentially stabilized c-MYC G4 over G4s in other promoters (c-kit 1, c-kit 2, and BCL2) and duplex DNA (Figure 3b) [97]. The binding preference was confirmed by KD values calculated using fluorimetric titration assays, in which TH3 showed four- to fivefold higher binding affinity to c-MYC G4 (KD ~ 0.3 μM). Importantly, the preference in vitro was confirmed biologically. Western blotting, qRT-PCR, and luciferase reporter assays using cancer cell lines (HeLa and A549) revealed that TH3 is able to repress the transcription of c-MYC at a protein level without affecting BCL2 expression, suggesting the preferential recognition and stabilization of the c-MYC G4 by TH3. Such preferential targeting by TH3 might result in selective antiproliferative effects against the cancer cells over normal human cells (NKE). Molecular insight from an NMR analysis suggested that TH3 interacted with an AT-rich capping structure at both 5′ and 3′ ends that was unique to the c-MYC G4 structure.
Tan and colleagues developed a new four-leaf-clover-like molecule, IZCZ-3, that preferentially binds to, and stabilizes, c-MYC G4 (Figure 3b) [98]. This compound is a conjugate of diaryl-substituted imidazole with a carbazole moiety. Previously, a triaryl-substituted imidazole unit was shown to be a G4-selective ligand, and its derivatives were shown to be distinct probes that were able to discern different G4s, while the carbazole moiety and its derivatives showed significant binding affinities toward the c-MYC G4 [98,99,100,101]. By combining these features, these investigators expected that the conjugation of a triaryl-substituted imidazole scaffold with a carbazole moiety might be a suitable way to create c-MYC G4 ligands with high affinity and selectivity. Among the conjugates designed, IZCZ-3 was found to have about eightfold preference for the c-MYC G4 (KD ~ 0.1 μM) over the G4s in the promoters for other genes (VEGF, BCL2, c-kit1, and KRAS), as demonstrated by fluorescence titration experiments. They further confirmed its biological activities extensively using western blotting, exon-specific RT-qPCR [93,94], and a luciferase reporter assay, demonstrating that IZCZ-3 was able to repress the expression of c-MYC at the protein level in a c-MYC G4-mediated manner. More importantly, this ligand showed cytotoxicity against cancer cell lines overexpressing c-MYC but not against human normal cells or primary mouse cells, suggesting reduced side effects based on the G4 selectivity. The investigators confirmed that the cytotoxicity induced by IZCZ-3 likely originated from the downregulation of c-MYC expression by checking the expression of certain cell cycles and apoptosis regulators associated with c-MYC. Moreover, the antitumor activity of this molecule was demonstrated in vivo using xenograft mouse models, showing an inhibition of the tumor growth similar to an anticancer drug, doxorubicin. Collectively, IZCZ-3 showed a high affinity and discriminating capability for c-MYC distinct from other parallel G4s, as well as antiparallel or hybrid G4s, whose differential binding profile conferred a selective inhibitory ability against c-MYC-driven tumors.
A new approach, using a small molecule microarray screen of 20,000 compounds using fluorescently labeled c-MYC G4 DNA, successfully identified a c-MYC G4-selective ligand (Figure 3b) [102]. The direct interaction between c-MYC G4 and its G4-meditated influence over DNA replication was validated by an SPR-binding assay and a PCR stop assay. Importantly, the ligand inhibited c-MYC transcription and reduced cell viability in a panel of myeloma cell lines, whereas it exhibited minimal effects for a cell that harbors a c-MYC translocation, deleting the G4-forming element in the promoter and normal blood mononucleocytes. Furthermore, gene expression analysis and RT-qPCR demonstrated that the ligand did not change other G4-driven gene expression, including genes for BCL2, KRAS, HIFA, VEGF, and, Rb1, indicative of excellent selectivity. Similarly, another screening approach permitted the selection of a c-MYC-G4-interacting ligand [103]. Although the detailed selectivity to c-MYC G4 over the others was not mentioned in the report, the identified compound (Tz 1) allowed for an excellent repression of c-MYC at low micromolar concentrations in cultures of colorectal carcinoma (HCT116) cells, and the G4-mediated action was biologically validated by an exon-specific RT-qPCR assay [93,94] in Burkitt’s lymphoma (CA46) cells (Figure 3b).
3.3. VEGF
Tumor progression and metastasis render the tumors more mature and malignant than undeveloped neoplasms, eventually resulting in deterioration and immortality. Overexpressed VEGF (Vascular Endothelial Growth Factor) proteins, including VEGFA, VEGFB, VEGFC, VEGFD, VEGFE, and PIGF in tumor cells, are responsible for induced neovascularization. The expression of human VEGF, which is frequently elevated in many types of cancers, is regulated mainly at the transcriptional level [104,105]. In a reporter assay system using several cancer cell lines, regulation of VEGF was regulated basically by a sequence from –85 to –50 relative to the transcription start site containing five arrays of more than three consecutive G-tracts, which are likely to adopt the G4 form of DNAs [106,107]. VEGF is an attractive target molecule for malignant tumor therapy, and antibody drugs targeting it have been approved for solid tumor treatment [108,109]. Interestingly, VEGF has a promoter region, in which the G4-forming sequences are located [48]. The sequences are also consensus sequences for transcription factors, such as Egr-1 and Sp1, suggesting that the dynamic equilibrium of DNA forms in this region regulate VEGF expression (Figure 4a) [106,107]. In addition, hormone response element (HRE, 5′-ATACGTG-3′), situated 969~975 nucleotides upstream of the transcription start site, also regulate the transcription of VEGF gene (Figure 4a), and pyrrole-imidazole polyamides (PIPs) targeted to this element were shown to repress the VEGF gene expression by interfering with the binding of HRE-binding transcription factor (HIF-α) [110].
Initially, the interaction of TMPyP4 and telomestatin with G4 oligonucleotides was proven to unwind the duplex DNA oligomer into an ssDNA oligomer and to stabilize the G4 structure [48], and Se2SAP, a global G4-interacting ligand, efficiently suppressed VEGF expression in two adenocarcinoma cell lines (HEC1A and MDA-MB-231) [111]. These data offer the possibility that the transcription regulation of VEGF is controllable by ligand-mediated G4 stabilization and led to the application of G4-interacting ligands for cancer therapy. Similarly, a perylene monoimide derivative, PM2, was found to be a VEGF downregulator, likely by direct interaction with the G4 structure [112] (Figure 4b). A quindoline derivative, SYUIQ-FM05, also demonstrated strong interactions with a VEGF G4 and exhibited potential antiangiogenic and antitumor activities [113] (Figure 4b). On the basis of these successful studies, several VEGF G4-preferred ligands have been developed through low-volume screening by means of docking and spectroscopic approaches [114,115]. Biological activities of these ligands have not been examined thus far, and therefore we await a future study for their determination.
3.4. BCL2
BCL2 gene is recognized as an apoptosis-related gene whose translated product resides on the cytoplasmic face of the mitochondrial outer membrane and acts to suppress the movability of apoptosis-induced proteins by controlling mitochondrial membrane permeability [116]. Overexpressed BCL2 is associated with aberrant carcinoma growth in various human diseases, particularly with solid tumors, such as lymphomas, non-small-cell lung cancer, myeloma, and melanoma, having been recognized as targets for cancer therapy in the past three decades [117]. Several approaches have been made to downregulate the BCL2 expression in cancer cells toward cancer therapy, including using small molecules to disrupt protein–protein interactions [118], antisense oligonucleotides [119], and peptidomimetics [120]. Overexpression of BCL2 is also indicated to be a principal element of chemoresistance, particularly for lymphocytic cancers [121,122]. For instance, transfection of BCL2 into A549 cells induced a resistance to the apoptotic effect triggered by triazine derivative 12459, a G4-interacting ligand that inhibits telomerase activity [123]. As another approach, the molecular decay effect by the guanine-rich AS1411 aptamer that can be stably folded into a G4 structure causes the destabilization of BCL2 mRNA and degradation with RNase by interfering with the binding of nucleolin to the AU-rich element of BCL2 mRNA, eventually inducing apoptosis [124]. This approach is reminiscent of the involvement of G4 formation in gene expression.
Amplification and translocation of BCL2 genes are shown to be equally common mechanisms that cause its overexpression in human cancer cells [125]. The human gene for BCL2 includes P1 and P2 promoters and has multiple transcription start sites (Figure 5a). The major transcription regulation is less driven by a TATA-box in promoter 2, while the P1 promoter that is situated 1386–1423 nucleotides upstream of the translation start site has been largely implicated in the control of BCL2 transcription [126,127]. The GC-rich element exists 1490–1451 nucleotides upstream of the P1 promoter, where multiple transcription factors have been said to be implicated in BCL2 gene expression, including Sp1 [126], WT1 [128], E2F [129], and NGF [130]. The multiple G4 structures in this region were well elucidated by Hurley and Yang [47,131,132]. The regulatory effect of the G4s was suggested by luciferase reporter assays, in which mutation or deletion of this region resulted in an increase in promoter activity in B lymphocytes (DHL-4) [127] or human promyelocytic leukemia (HL-60) cells [133]. More recently, Onel, Yang, and coworkers demonstrated by a luciferase reporter assay using BCL2 promoter and mutated sequences that the formation of another G4 situated almost on the upper region of the P1 promoter attenuated the promoter activity (Figure 5a) [134]. Based on these studies, an approach to stabilizing the G4s formed in the regulatory element and attenuating the promoter activity by ligands has also been studied for cancer therapy, similar to the small molecule targeting of c-MYC G4.
Proof-of-concept studies were performed by Huang, Gu, and colleagues, in which SYUIQ-FM05, as mentioned in the last section, was able to repress BCL2 transcription with a negligible influence on the case, using a promoter mutated to abolish its ability to form G4 in a reporter assay [133]. Moreover, these investigators demonstrated that the ligands induced apoptosis of HL-60 cells. Very recently, furo[2,3-d]pyridazin-4(5H)-one derivatives were screened as a new class of G4-interacting ligands for BCL2-targeted therapeutics, and two hit compounds identified were found to bind to BCL2 G4 structures with clear preferences over c-kit, c-MYC, and telomere G4s, as well as dsDNA (Figure 5b) [135]. Importantly, one of the two compounds repressed BCL2 expression, showing a remarkable cytotoxicity to Jurkat (human acute T cell leukemia) cell lines. Additionally, we introduce a probe preferentially targeting BCL2 G4, carbazole TO, whose fluorescence intensity is more greatly enhanced in the presence of BCL2 G4 than it is in the presence of G4s of other promoters, telomere G4, ssDNA, and dsDNA (by approximately 2.6–19 fold) (Figure 5b) [136]. Such a ligand having excellent selectivity to a particular G4 may be applicable to a potent facile light-up probe for BCL2 G4 in admixture contexts, such as cellular environments in diagnostics, therapeutics, and biosensors.
In a different way, invading PNA to directly hybridize the cytosine-rich strand and to indirectly stabilize the BCL2 G4 facilitates G4 formation [137]. This would be a versatile way to target and stabilize a particular G4.
In addition to the G4s, the i-motif, another form of DNA that forms in cytosine-rich sequences, is involved in transcriptional regulation, in which the binding of hnRNP LL to the i-motif structure likely activates BCL2 gene expression [138,139]. Moreover, a molecule interacting with the i-motif, IM-48, was identified modulating BCL2 gene expression by affecting the dynamic equilibrium of the i-motif and the flexible hairpin form [138,139], opening a new avenue to modulate the expression of BCL2 more precisely. Targeting such canonical DNAs formed in the regulatory element of the promoter may be an effective way to target a particular target to combat the tumor.
3.5. c-Kit
The c-kit proto-oncogene encodes a receptor tyrosine kinase that is bridged and activated by the binding of dimerized stem cell factors (SCF), and in turn stimulates proliferation, differentiation, and survival in hemopoietic precursor cells [140,141,142]. Malfunctions of Kit acquired by overexpression or mutations have been associated with several diseases, including gastrointestinal stromal tumors (GIST), mastocytosis, and acute myelogenous leukemia (AML) [143,144,145], and although the kinase inhibitor Imatinib (Glivec) has been successfully developed as an FDA-approved drug for GIST, long-term exposure often causes secondary mutations at exons 13, 14, or 17, which encode tyrosine kinase domains [146]. Notably, drug resistance derived from mutations at exon 17 is found to severely attenuate the therapeutic effect of imatinib [147]. A compelling approach to fundamentally suppress c-kit expression is highly desirable.
The human c-kit promoter is devoid of both TATA and CCAT boxes [148,149]. Instead, the region within 200 bp upstream from TSS is highly rich in GC content, where several transcription factors, including MAZ in human normal fibroblasts and SP1 in hematopoietic cells and carcinomas, are implicated (Figure 6a) [150,151]. Two well-defined G4 structures were resolved [44,152], and the three-dimensional structural dynamics are shown to be involved in the regulation of c-kit gene transcription, accelerating the development of c-kit G4-preferred ligands [153,154,155,156]. The modulation of such structural dynamics by small molecules is effective for suppressing gene expression and exhibits an apoptotic effect.
The pioneering work performed by Balasubramanian and colleagues warrants attention, where low-volume screening was conducted using six isoalloxazine ligands. Among the ligands, 1a and 1d, possessing three N,N-dimethyl amine-substituted and one N,N-dimethyl amine/2 fluorine-substituted tails, respectively, show a clear binding preference to c-kit G4s over a telomere G4, as demonstrated by an SPR-binding assay, and an inhibitory effect for the gene expression in two different cancer cell lines by RT-qPCR assays (Figure 6b) [157].
The feasibility of targeting the c-kit promoter G4 by small molecules was further confirmed in patient-derived GIST cells. Neidle and colleagues demonstrated that a naphthalene diimide derivative strongly stabilized c-kit G4 structures and almost completely reduced the level of protein expression, resulting in a strong effect on growth arrest in the GIST tumor cells (Figure 6b) [158].
To obtain a more bioactive c-kit G4-interacting ligand, several cell-based screenings of G4 ligands have been performed. This approach is expected to overcome the incompatibility between the outcomes of in vitro and cellular applications, which is often seen in ligand discovery based in vitro. To give examples, luciferase reporter assays performed on a 96-well plate using the human gastric carcinoma cell line (HGC-27) led to the discovery of two benzo[a]phenoxazine (BPO) derivatives as potent c-kit G4 ligands (Figure 6b) [159]. Subsequent RT–qPCR and SPR-binding analyses confirmed that these two molecules acted as endogenous c-kit suppressors in an HGC-27 cell line, probably through binding to c-kit promoter G4s. Similarly, two quinazolone derivatives were identified that could downregulate c-kit expression at the protein level (Figure 6b) [160].
3.6. Human Telomerase Reverse Transcriptase
Human telomerase reverse transcriptase (hTERT), which encodes the catalytic subunit of telomerase, has commanded considerable attention as a compelling biomedical target, particularly for cancer, as elevated TERT expression is observed in ~90% of human cancer cells, whereas it is normally silenced in most normal cells [161,162]. Aberrantly expressed TERT accelerates telomerase activity irregularly to maintain the telomere length [163]. Other than its canonical role in maintaining telomere length, TERT suppresses BCL2-dependent apoptosis [164] to regulate the chromatin state [165] and DNA damage responses [165,166], and to promote c-MYC and Wnt-driven cellular proliferation [167,168].
The mutations that were identified in >70% of melanomas partially account for the elevated level of TERT expression [169]. C to T mutations in the sense strand (G to A mutations in the antisense strand) in the TERT promoter highly activate transcription by creating a new consensus sequence for the binding of ETS/TCF (E-26/ternary complex factor) [170]. Patients who have tumors expressing elevated levels of TERT exhibit even worse survival rates than those who have tumors expressing relatively lower levels of TERT [171]. These observations clearly indicate that TERT promoter targeting based on the mutations might have a great impact on tumor therapeutics covering a wide range of tumors (Figure 7a).
We would like to highlight a unique approach to addressing the issue of hTERT downregulation on the basis of the mutations in a G4-mediated manner. The region located approximately -90 to -22 upstream of the transcription start site was abundant in GC base pairs and has multiple PQSs (Figure 7a) [43]. In particular, tandemly aligned G4s formed by the entire PQSs have been suggested to be a key mechanism in maintaining the normal transcriptional levels of the hTERT gene [43,172]. Interestingly, the involvement of the dynamic equilibrium of such three-dimensional structures of DNA upon the somatic mutations (G/C to T/A) in this region is attributable to activated TERT expression [172]. Hurley and colleagues developed a small molecule that binds to the higher-order G4 structure observed in the hTERT promoter via dual-motif targeting for the G4 and the mismatched duplex stem loop (GTC365, Figure 7b) [172]. The mutations situated at a mismatched duplex-stem loop within G4 structures alter the folding pattern of G4s to increase the gene expression, likely by inhibiting a certain transcription repressor. The interplay of GCT365 and the element was proposed to act like a chaperone to steer the correct folding seen in the wild-type G-rich element of the promoter, resulting in the reactivation of the silencing function.
3.7. KRAS
The RAS gene family, including HRAS, NRAS, and KRAS, was first discovered in human tumors as driver oncogenes and has long been recognized as an important therapeutic target. Mutation of KRAS is one of the most oncogenic driver mutations in pancreatic, colorectal, and lung cancers, and plays a role in acquiring and increasing the drug resistance [173,174]. Hence, direct targeting of active KRAS by small molecules was considered to be a compelling strategy for combating KRAS mutant tumors, yet it remains at an unsuccessful stage. Recently, our group has developed a novel approach that directly targets the mutant DNA using an alkylating pyrrole-imidazole polyamide (PIP) molecule, which is capable of selectively alkylating oncogenic codon 12 mutant DNA and causing strand cleavage, and consequent tumor growth suppression in a tumor xenograft model of cancer in mice [175].
G4-mediated promoter targeting is also reported. The NHE in the KRAS proximal promoter is highly abundant in G-rich sequences, and several transcription factors interact with a G4 structure formed in this region (Figure 8a) [176,177,178]. A polypurine G-rich element, located approximately –300 to –100 nucleotides upstream of the exon 0/intron 1 boundary in a murine or human genome, is likely to be a component of the promoter activity, and includes multiple PQSs [45,46,176,177,178,179,180]. Importantly, pyrene-modified oligonucleotides that were devised to be a more stable form of the KRAS G4 formed in the PQS1 were able to attract the factors essential for transcription and to exhibit a strong antiproliferative activity through a G4-decoy effect in pancreatic cancer cells [181].
Small molecule targeting for the KRAS G4 would also be a promising strategy for suppressing gene expression. Indeed, Paulo and colleges have prepared a small library of indolo[3,2-c]quinolines (IQc), and found that two triple-cation derivatives had a clear preference for KRAS G4 over a telomere G4 and duplex DNA, and acted as superior downregulators of KRAS expression and inhibited mutated KRAS expression in HCT116 and SW620 cells [182]. The data presented suggest that direct targeting of the KRAS G4 at the transcription level in KRAS mutant tumors by small molecules selective for G4 might be a promising therapeutic strategy for tumors. More recently, comprehensive studies on the human KRAS promoter, in terms of the possible G4 structures and their relevance to the promoter activity, which were performed by Brooks group, revealed that a newly discovered G4 formed in the PQS2 more critically affected the attenuation of the promoter (Figure 8a) [46]. These findings would contribute to the creation of more efficient and selective G4-mediated transcriptional repression by ligands in a future study.
3.8. c-Myb
c-Myb is largely expressed in an early stage of the differentiation of hematopoietic cells, and its expression is gradually decreased toward the end of their differentiation [183]. It encodes a transcription factor that plays a critical role in the proliferation, differentiation, and survival of hematopoietic progenitor cells; c-myb was identified by the discovery of v-myb found in avian myeloblastosis virus and E26 [184]. This gene is also recognized as a proto-oncogene, high expression of which is related to promoting the development of hematological cancers and adenocarcinomas [185,186,187,188,189] by a mechanism based on its canonical proliferative property.
The regulation of c-myb expression at a transcription level relies on multiple activating and repressing transcription factors in a cell-type-dependent fashion [190,191,192,193,194,195]. Notably, a region in the promoter with three (GGA)4 triplet repeats beginning 17 bp downstream of the transcription start site on the antisense strand is implicated in the promoter activity through its formation of thermally-stable higher-order parallel G4 structures [196,197,198]. Partial deletion of the (GGA)4 triplet repeats blocks the ability to form the dimerized G4 which enhances the promoter activity, suggesting that the G4 structure forms by utilizing the three (GGA)4 triplet repeats, which function as a negative regulator of the c-myb promoter activity [196]. Additionally, MAZ protein may bind to the c-myb G4 structure and negatively regulate the promoter activity.
The GC-rich promoter region in the DNase hypersensitive elements (to approximately 850 bp upstream of the transcription start site) is also reminiscent of the putative G4 formation and its involvement of transcription regulation [199,200]. Recently, the group led by Yuan used a reporter assay system to extensively examine the way that folded and unfolded G4s affect c-myb expression through their effect on c-myb promoter activity [201]. In this system, four PQSs in the c-myb promoter were selected as potential G4 formation elements, and the activity in MCF-7 cells was compared with that in cells with a promoter-containing plasmid, where mutations were made so as not to form G4s in the respective PQSs (Figure 9a). The promoter activity of the PQS1-mutated plasmid was markedly reduced, whereas the PQS1, −2, and −3-mutated plasmids exhibited no significant changes in these promoter activities. These data strongly implied that the transcription regulation on those G-rich sequences was considerably mediated by the formation of the G4 structures on the PQS1 element in MCF-7 cells, where the binding of a transcription suppresser was likely impeded. The newly discovered c-myb G4-interacting ligand, topotecan, increased the transcription level in the wild-type plasmid without affecting the use of the PQS1-mutated plasmid (Figure 9b). This downregulation effect was confirmed in the endogenous c-myb expression at the protein level. Since the binding specificity of topotecan to the c-myb G4s among the other G4s has never been mentioned, further studies are needed.
When the focus moves more specifically to diseases, c-myb is identified as a target in glioma stem cells for glioblastoma multiforme (GBM) therapy, in which expression was considerably elevated in GBM tissues relative to normal tissues [202]. Interestingly, telomestatin, a ligand interacting with G4, globally impairs the maintenance of the GSC stem cell state through an apoptotic pathway, largely by reducing a c-myb expression in vitro and in vivo. Although the direct interplay of telomestatin and c-myb G4s in the promoter has not been examined, these observations offer the possibility that direct targeting of c-myb G4 DNA is a compelling therapeutic approach to GBM treatment.
3.9. Others (PDGFR-β, PDGF-A, STAT3, FGFR2)
Other G4s formed in putative regulatory elements in the promoters of cancer-related genes have been reported, and are proposed as targetable by ligands interacting with G4 (in promotors in genes for PDGFR-β [203], PDGF-A [204], STAT3 [205], FGFR2 [206], etc.). For instance, GSA11129, which can interact with a G4 in the promoter of the gene for PDGFR-β to shift the equilibrium to a G4 species, was demonstrated to reduce the transcription level and to inhibit PDGF-β-driven cell proliferation and migration [203]. The G-rich element of the proximal promoter in the gene for PDGF-A also forms a stable G4 structure, even in the duplex context, and TMPyP4 reduced the basal promoter activity of PDGF-A, suggesting that targeting the PDGF-A G4 by the ligand specific for this G4 may be feasible as a cancer therapy for gliomas, sarcomas, and astrocytomas [204,207,208,209,210,211].
4. Alternative Nucleic Acid Form as a Biomedical Target—G-triplex
The G-triplex was initially regarded as a transient DNA form and a possible intermediate in the G4 folding process [212]. A growing body of literature suggests that such a structure forms stably under physiological conditions [75,213,214,215,216]. Along with their potential biological significance, small molecules targeting G-triplexes increasingly command considerable attention. Acridone–PNA conjugates highlight dual-site targeting by a planar acridone moiety appended to a Gly-GGG-Lys PNA sequence (Figure 10a) [217]. The PNA moiety associates with one guanine of three G-tetrads, to form a hybrid PNA+DNA G4. This ligand is thought to prefer a G-rich sequence in a single-stranded context over a pre-folded G4; thus, it might be especially useful for targeting G-triplex structures in such cellular dynamics. A dihydropyrimidin-4-one derivative is identified as a G-triplex and G4 ligand from the Mcule chemical database, by using simple docking programs (Figure 10b) [218].
Our group has devised a nanoplatform constructed by DNA origami for studying such intermediates of G4, such as G-triplex and G-hairpin, and has found that pyridostatin (PDC), a well-known ligand interacting with G4, unexpectedly recognized the G-triplex and G-hairpin structures (Figure 10c) [219]. Considering this, the ability to recognize the intermediates of G4 might be an essential component for the high binding affinity, selectivity, or inducing ability of the G4 structures from the stable duplex or single-stranded DNA. The platform manifests the power to assess unprecedented G4-binding properties of a ligand.
5. Spatially Indirect G4 Targeting by Py-Im Polyamide Molecules
By taking advantage of the sequence-selectivity of PIPs, as mentioned earlier, a head-to-head-type polyamide dimer was developed for the selective targeting of a designated G4, which displayed an inducible effect toward G4 formation (Figure 11) [220]. This strategy relies on a totally new ligand design based on the targeting of the duplex region on either side of an intended G4 in the genome. Although the results presented are still preliminary, such simultaneous dual-duplex binding across the targeted G4 has the potential advantages of being (1) independent of G4 topology, (2) targetable to other higher-order DNA structures such as i-motifs, and (3) programmable for a large variety of targets at different genome locations.
6. Summary and Outlook
Although a number of biomedical targets in the approach toward tumor therapy are recognized and investigators are pursuing the development of drugs, results have been limited thus far. The G-quadruplex (G4) has been recently considered to be a potential biomedical target, particularly for tumor therapy, and a considerable body of evidence has been accumulating that G4-interacting drugs exhibit good antitumor activities. As protein-targeting drugs face similar situations, G4-interacting drugs displayed low selectivity to the targeted G4 structure, mainly because of the similar skeletons among different G4 forms observed in the genome. In this review, we addressed the ligands interacting with G4 that were devised to gain selectivity for a particular G4 structure and to exhibit selective bioactivity for tumor cells. The selectivity issues remain incompletely solved, but if accomplished, would substantially impact cancer therapy.
The G4-driven oncogenes introduced here are known to be usually well-correlated and concertedly influence tumorigenesis, tumor growth, and malignant transition [164,167,168,190,221,222]. Although this relationship is not fully elucidated, combinatorial approaches may be a good option for further therapeutic advancements [223].
Importantly, G4 has an influence, not only on telomere or tumor-related genes, but also on differentiation- and neuron-related genes. As mentioned earlier, OCT4 expression may be governed to some degree by G4 formation at the proximal promoter in human embryonic stem cells (CCTL14), as shown by a reporter assay system [49], while the excessive occurrence of poly G4 structures on the expandable repeats accounts for some neurogenetic disorders [50]. On the contary, G4 structures at the CpG island located in xl3b are recognized by ATRX, contributing to appropriate synaptic function [51]. These data clearly imply an even more global impact of G4s on overall ontogenesis. In addition to the research direction of specific G4 ligands toward cancer therapy, studies on specific G4-interacting ligands may also focus on these issues and depict an entire map of G4 functions.
Author Contributions
S.A., S.O., and H.S. wrote the manuscript, and Z.Y., T.B., and H.S. gave suggestions for the content.
Funding
S.A. would like to thank a JSPS research fellowship to young scientists for financial support. This work was supported by NIH grant (1R01CA236350 to H.S.) and the Basic Science and Platform Technology Program for Innovative Biological Medicine by Japan Agency for Medical Research (18am0301005h0005 to H.S.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix Formation by Guanylic Acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of Parallel Four-Stranded Complexes by Guanine-Rich Motifs in DNA and its Implications for Meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.M.; Lyons, S.M.; Ivanov, P. RNA G-Quadruplexes in Biology: Principles and Molecular Mechanisms. J. Mol. Biol. 2017, 429, 2127–2147. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Perreault, J.; Topisirovic, I.; Richard, S. RNA G-Quadruplexes and their Potential Regulatory Roles in Translation. Translation 2016, 4, e1244031. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, P.; Pandey, S.; Maiti, S. The Tale of RNA G-Quadruplex. Org. Biomol. Chem. 2015, 13, 5570–5585. [Google Scholar] [CrossRef] [PubMed]
- Grand, C.L.; Powell, T.J.; Nagle, R.B.; Bearss, D.J.; Tye, D.; Gleason-Guzman, M.; Hurley, L.H. Mutations in the G-Quadruplex Silencer Element and their Relationship to c-MYC Overexpression, NM23 Repression, and Therapeutic Rescue. Proc. Natl. Acad. Sci. USA 2004, 101, 6140–6145. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct Evidence for a G-Quadruplex in a Promoter Region and its Targeting with a Small Molecule to Repress c-MYC Transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Li, Y.; Syed, J.; Suzuki, Y.; Asamitsu, S.; Shioda, N.; Wada, T.; Sugiyama, H. Effect of ATRX and G-Quadruplex Formation by the VNTR Sequence on α-Globin Gene Expression. ChemBioChem 2016, 17, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 Family Helicases Suppress Genome Instability at G-Quadruplex Motifs. Nature 2013, 497, 458–462. [Google Scholar] [CrossRef]
- Paeschke, K.; Capra, J.A.; Zakian, V.A. DNA Replication through G-Quadruplex Motifs is Promoted by the Saccharomyces Cerevisiae Pif1 DNA Helicase. Cell 2011, 145, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.; Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.P.; Foiani, M.; Nicolas, A. G-Quadruplex-Induced Instability during Leading-Strand Replication. EMBO J. 2011, 30, 4033–4046. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule–induced DNA Damage Identifies Alternative DNA Structures in Human Genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, I.; Owen-Hughes, T. ATRX: Put Me on Repeat. Cell 2010, 143, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valton, A.L.; Hassan-Zadeh, V.; Lema, I.; Boggetto, N.; Alberti, P.; Saintome, C.; Riou, J.F.; Prioleau, M.N. G4 Motifs Affect Origin Positioning and Efficiency in Two Vertebrate Replicators. EMBO J. 2014, 33, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P.; Reams, C.; Simpson, L.J.; Sale, J.E. Epigenetic Instability due to Defective Replication of Structured DNA. Mol. Cell 2010, 40, 703–713. [Google Scholar] [CrossRef]
- Schiavone, D.; Jozwiakowski, S.K.; Romanello, M.; Guilbaud, G.; Guilliam, T.A.; Bailey, L.J.; Sale, J.E.; Doherty, A.J. PrimPol is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol. Cell 2016, 61, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. Prevalence of Quadruplexes in the Human Genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. G-Quadruplexes in Promoters Throughout the Human Genome. Nucleic Acids Res. 2006, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.J.E.; Lim, K.W.; Phan, A.T. Formation of (3+1) G-Quadruplexes with a Long Loop by Human Telomeric DNA Spanning Five Or More Repeats. J. Am. Chem. Soc. 2011, 133, 11462–11465. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, V.T.; Phan, A.T. Bulges in G-Quadruplexes: Broadening the Definition of G-Quadruplex-Forming Sequences. J. Am. Chem. Soc. 2013, 135, 5017–5028. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.; Fox, K.R. Stability of Intramolecular DNA Quadruplexes: Comparison with DNA Duplexes. Biochemistry 2003, 42, 6507–6513. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.W.; Phan, A.T. Structural Basis of DNA quadruplex–duplex Junction Formation. Angew. Chem. 2013, 125, 8728–8731. [Google Scholar] [CrossRef]
- Lam, E.Y.N.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-Quadruplex Structures are Stable and Detectable in Human Genomic DNA. Nat. Commun. 2013, 4, 1796. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, A.; Balasubramanian, S. A Sequence-Independent Study of the Influence of Short Loop Lengths on the Stability and Topology of Intramolecular DNA G-Quadruplexes. Biochemistry 2008, 47, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Laughlan, G.; Murchie, A.I.; Norman, D.G.; Moore, M.H.; Moody, P.C.; Lilley, D.M.; Luisi, B. The High-Resolution Crystal Structure of a Parallel-Stranded Guanine Tetraplex. Science 1994, 265, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Zhang, X.; Ratliff, R.; Moyzis, R.; Rich, A. Crystal Structure of Four-Stranded Oxytricha Telomeric DNA. Nature 1992, 356, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human Telomere, Oncogenic Promoter and 5′-UTR G-Quadruplexes: Diverse Higher Order DNA and RNA Targets for Cancer Therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Neidle, S. The Structures of Quadruplex Nucleic Acids and their Drug Complexes. Curr. Opin. Struct. Biol. 2009, 19, 239–250. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal Structure of Parallel Quadruplexes from Human Telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Solution Structure of the Human Telomeric Repeat d [AG3(T2AG3)3] G-Tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef]
- Luu, K.N.; Phan, A.T.; Kuryavyi, V.; Lacroix, L.; Patel, D.J. Structure of the Human Telomere in K Solution: An Intramolecular (3+1) G-Quadruplex Scaffold. J. Am. Chem. Soc. 2006, 128, 9963–9970. [Google Scholar] [CrossRef]
- Adrian, M.; Heddi, B.; Phan, A.T. NMR Spectroscopy of G-Quadruplexes. Methods 2012, 57, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Patel, D.J. DNA Architecture: From G to Z. Curr. Opin. Struct. Biol. 2006, 16, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Wilner, O.I.; Willner, B.; Willner, I. DNA nanotechnology. Adv. Exp. Med. Biol. 2012, 733, 97–114. [Google Scholar] [PubMed]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of Human Telomerase by a G-Quadruplex-Interactive Compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef] [PubMed]
- Wheelhouse, R.T.; Sun, D.; Han, H.; Han, F.X.; Hurley, L.H. Cationic Porphyrins as Telomerase Inhibitors: The Interaction of Tetra-(N-Methyl-4-Pyridyl) Porphine with Quadruplex DNA. J. Am. Chem. Soc. 1998, 120, 3261–3262. [Google Scholar] [CrossRef]
- Lipps, H.J.; Rhodes, D. G-Quadruplex Structures: In Vivo Evidence and Function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a Novel Telomerase Inhibitor from Streptomyces Anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef]
- Kim, M.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a Potent Telomerase Inhibitor that Interacts quite Specifically with the Human Telomeric Intramolecular G-Quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer Activity of CX-3543: A Direct Inhibitor of rRNA Biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.K.; Brooks, T.A. Targeting Promoter G-Quadruplexes for Transcriptional Control. In Small-Molecule Transcription Factor Inhibitors in Oncology; Royal Society of Chemistry: Cambridge, UK, 2016; Chapter 7. [Google Scholar]
- Palumbo, S.L.; Ebbinghaus, S.W.; Hurley, L.H. Formation of a Unique End-to-End Stacked Pair of G-Quadruplexes in the hTERT Core Promoter with Implications for Inhibition of Telomerase by G-Quadruplex-Interactive Ligands. J. Am. Chem. Soc. 2009, 131, 10878–10891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, S.; Reszka, A.P.; Huppert, J.; Zloh, M.; Parkinson, G.N.; Todd, A.K.; Ladame, S.; Balasubramanian, S.; Neidle, S. Putative DNA Quadruplex Formation within the Human c-Kit Oncogene. J. Am. Chem. Soc. 2005, 127, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Quadrifoglio, F.; Xodo, L.E. G-Rich Oligonucleotide Inhibits the Binding of a Nuclear Protein to the Ki-Ras Promoter and Strongly Reduces Cell Growth in Human Carcinoma Pancreatic Cells. Biochemistry 2004, 43, 2512–2523. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.K.; Batra, H.; Gaerig, V.C.; Hockings, J.; Brooks, T.A. Identification and Characterization of a New G-Quadruplex Forming Region within the kRAS Promoter as a Transcriptional Regulator. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T.S.; Sun, D.; Hurley, L.H. Deconvoluting the Structural and Drug-Recognition Complexity of the G-Quadruplex-Forming Region Upstream of the Bcl-2 P1 Promoter. J. Am. Chem. Soc. 2006, 128, 5404–5415. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Guo, K.; Rusche, J.J.; Hurley, L.H. Facilitation of a Structural Transition in the polypurine/polypyrimidine Tract within the Proximal Promoter Region of the Human VEGF Gene by the Presence of Potassium and G-Quadruplex-Interactive Agents. Nucleic Acids Res. 2005, 33, 6070–6080. [Google Scholar] [CrossRef] [PubMed]
- Renčiuk, D.; Ryneš, J.; Kejnovská, I.; Foldynová-Trantírková, S.; Andäng, M.; Trantírek, L.; Vorlíčková, M. G-Quadruplex Formation in the Oct4 Promoter Positively Regulates Oct4 Expression. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2017, 1860, 175–183. [Google Scholar] [CrossRef]
- Simone, R.; Fratta, P.; Neidle, S.; Parkinson, G.N.; Isaacs, A.M. G-Quadruplexes: Emerging Roles in Neurodegenerative Diseases and the Non-Coding Transcriptome. FEBS Lett. 2015, 589, 1653–1668. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Yamaguchi, K.; Onozato, M.; Li, Y.; Kurosawa, K.; Tanabe, H.; Okamoto, N.; Era, T.; Sugiyama, H.; et al. Targeting G-Quadruplex DNA as Cognitive Function Therapy for ATR-X Syndrome. Nat. Med. 2018, 24, 802–813. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative Visualization of DNA G-Quadruplex Structures in Human Cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Wang, Y.; Huang, Z.; Tan, J.; Huang, Z.; Chen, S. Natural Alkaloids and Heterocycles as G-Quadruplex Ligands and Potential Anticancer Agents. Molecules 2018, 23, 493. [Google Scholar] [Green Version]
- Tian, T.; Chen, Y.; Wang, S.; Zhou, X. G-Quadruplex: A Regulator of Gene Expression and its Chemical Targeting. Chem 2018, 4, 1314–1344. [Google Scholar] [CrossRef]
- Brooks, T.A.; Kendrick, S.; Hurley, L. Making Sense of G-quadruplex and i-motif Functions in Oncogene Promoters. FEBS J. 2010, 277, 3459–3469. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Bando, T.; Sugiyama, H. Ligand Design to Acquire Specificity to Intended G-quadruplex Structures. Chem. Eur. J. 2019, 25, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-Quadruplex Stabilizer with Selective Lethality in BRCA1/2 Deficient Tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Nakamura, T.; Okabe, S.; Yoshida, H.; Iida, K.; Ma, Y.; Sasaki, S.; Yamori, T.; Shin-ya, K.; Nakano, I.; Nagasawa, K.; et al. Targeting Glioma Stem Cells in Vivo by a G-Quadruplex-Stabilizing Synthetic Macrocyclic Hexaoxazole. Sci. Rep. 2017, 7, 3605. [Google Scholar] [CrossRef]
- Marchetti, C.; Zyner, K.G.; Ohnmacht, S.A.; Robson, M.; Haider, S.M.; Morton, J.P.; Marsico, G.; Vo, T.; Laughlin-Toth, S.; Ahmed, A.A.; et al. Targeting Multiple Effector Pathways in Pancreatic Ductal Adenocarcinoma with a G-Quadruplex-Binding Small Molecule. J. Med. Chem. 2018, 61, 2500–2517. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.; Berardinelli, F.; Leone, S.; Coluzzi, E.; di Masi, A.; Doria, F.; Freccero, M.; Sgura, A.; Folini, M.; Antoccia, A. Naphthalene diimide-derivatives G-quadruplex Ligands Induce Cell Proliferation Inhibition, Mild Telomeric Dysfunction and Cell Cycle Perturbation in U251MG Glioma Cells. FEBS J. 2018, 285, 3769–3785. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-Throughput Sequencing of DNA G-Quadruplex Structures in the Human Genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Monchaud, D.; Teulade-Fichou, M. A Hitchhiker’s Guide to G-Quadruplex Ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, N.W. Targeting G-Quadruplex DNA with Small Molecules. CHIMIA Int. J. Chem. 2009, 63, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xiang, J.; Yang, Q.; Sun, H.; Guan, A.; Tang, Y. G4LDB: A Database for Discovering and Studying G-Quadruplex Ligands. Nucleic Acids Res. 2012, 41, D1115–D1123. [Google Scholar] [CrossRef] [PubMed]
- Anantha, N.V.; Azam, M.; Sheardy, R.D. Porphyrin Binding to Quadruplexed T4G4. Biochemistry 1998, 37, 2709–2714. [Google Scholar] [CrossRef]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-Quadruplex-Interactive Molecule BRACO-19 Inhibits Tumor Growth, Consistent with Telomere Targeting and Interference with Telomerase Function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.; Müller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.; Balasubramanian, S. A Novel Small Molecule that Alters Shelterin Integrity and Triggers a DNA-Damage Response at Telomeres. J. Am. Chem. Soc. 2008, 130, 15758–15759. [Google Scholar] [CrossRef]
- De Cian, A.; DeLemos, E.; Mergny, J.; Teulade-Fichou, M.; Monchaud, D. Highly Efficient G-Quadruplex Recognition by Bisquinolinium Compounds. J. Am. Chem. Soc. 2007, 129, 1856–1857. [Google Scholar] [CrossRef]
- Tera, M.; Ishizuka, H.; Takagi, M.; Suganuma, M.; Shin-ya, K.; Nagasawa, K. Macrocyclic Hexaoxazoles as sequence-and mode-selective G-quadruplex Binders. Angew. Chem. 2008, 120, 5639–5642. [Google Scholar] [CrossRef]
- Tera, M.; Iida, K.; Ishizuka, H.; Takagi, M.; Suganuma, M.; Doi, T.; Shin-ya, K.; Nagasawa, K. Synthesis of a Potent G-quadruplex-binding Macrocyclic Heptaoxazole. ChemBioChem 2009, 10, 431–435. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The Protein Complex that Shapes and Safeguards Human Telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Human Telomeric G-quadruplex: The Current Status of Telomeric G-quadruplexes as Therapeutic Targets in Human Cancer. FEBS J. 2010, 277, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Abraham Punnoose, J.; Cui, Y.; Koirala, D.; Yangyuoru, P.M.; Ghimire, C.; Shrestha, P.; Mao, H. Interaction of G-Quadruplexes in the Full-Length 3′ Human Telomeric Overhang. J. Am. Chem. Soc. 2014, 136, 18062–18069. [Google Scholar] [CrossRef] [PubMed]
- Russo Krauss, I.; Ramaswamy, S.; Neidle, S.; Haider, S.; Parkinson, G.N. Structural Insights into the Quadruplex–Duplex 3′ Interface Formed from a Telomeric Repeat: A Potential Molecular Target. J. Am. Chem. Soc. 2016, 138, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Liao, T.; Li, Z.; Gonzalez-Garcia, J.; Reynolds, M.; Zou, M.; Vilar, R. Dinickel–Salphen Complexes as Binders of Human Telomeric Dimeric G-Quadruplexes. Chem. Eur. J. 2017, 23, 4713–4722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yang, J.; Dong, C.; Wang, Y.; Sun, B.; Chen, J.; Xu, Y.; Chen, W. Highly Selective, Sensitive and Fluorescent Sensing of Dimeric G-Quadruplexes by a Dimeric Berberine. Organ. Biomol. Chem. 2016, 14, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Abraham Punnoose, J.; Ma, Y.; Li, Y.; Sakuma, M.; Mandal, S.; Nagasawa, K.; Mao, H. Adaptive and Specific Recognition of Telomeric G-Quadruplexes Via Polyvalency Induced Unstacking of Binding Units. J. Am. Chem. Soc. 2017, 139, 7476–7484. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, L.; Wu, B.; Huo, Y.; Duan, N.; Kong, D. Two Cationic Porphyrin Isomers Showing Different Multimeric G-Quadruplex Recognition Specificity Against Monomeric G-Quadruplexes. Nucleic Acids Res. 2014, 42, 8719–8731. [Google Scholar] [CrossRef]
- Zhao, C.; Wu, L.; Ren, J.; Xu, Y.; Qu, X. Targeting Human Telomeric Higher-Order DNA: Dimeric G-Quadruplex Units Serve as Preferred Binding Site. J. Am. Chem. Soc. 2013, 135, 18786–18789. [Google Scholar] [CrossRef]
- Yu, H.; Wang, X.; Fu, M.; Ren, J.; Qu, X. Chiral Metallo-Supramolecular Complexes Selectively Recognize Human Telomeric G-Quadruplex DNA. Nucleic Acids Res. 2008, 36, 5695–5703. [Google Scholar] [CrossRef]
- Qin, H.; Zhao, C.; Sun, Y.; Ren, J.; Qu, X. Metallo-Supramolecular Complexes Enantioselectively Eradicate Cancer Stem Cells in Vivo. J. Am. Chem. Soc. 2017, 139, 16201–16209. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, K.; Sannohe, Y.; Kaieda, S.; Tanaka, K.; Osuga, H.; Tahara, H.; Xu, Y.; Kawase, T.; Bando, T.; Sugiyama, H. A Chiral Wedge Molecule Inhibits Telomerase Activity. J. Am. Chem. Soc. 2010, 132, 3778–3782. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Chen, S.; Wang, B.; Ou, T.; Gu, L.; Tan, J.; Huang, Z. Specific Targeting of Telomeric Multimeric G-Quadruplexes by a New Triaryl-Substituted Imidazole. Nucleic Acids Res. 2016, 45, 1606–1618. [Google Scholar] [CrossRef]
- Lin, C.; Wu, G.; Wang, K.; Onel, B.; Sakai, S.; Shao, Y.; Yang, D. Molecular Recognition of the Hybrid-2 Human Telomeric G-quadruplex by Epiberberine: Insights into Conversion of Telomeric G-quadruplex Structures. Angew. Chem. Int. Ed. 2018, 57, 10888–10893. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Eilers, M. Transcriptional Regulation and Transformation by Myc Proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. C-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Brooks, T.A.; Hurley, L.H. The Role of Supercoiling in Transcriptional Control of MYC and its Importance in Molecular Therapeutics. Nat. Rev. Cancer 2009, 9, 849–861. [Google Scholar] [CrossRef]
- González, V.; Hurley, L.H. The c-MYC NHE III1: Function and Regulation. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that Drug-Targeted Down-Regulation of MYC in Non-Hodgkins Lymphoma is Directly Mediated through the Promoter G-Quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef] [PubMed]
- Boddupally, P.V.; Hahn, S.; Beman, C.; De, B.; Brooks, T.A.; Gokhale, V.; Hurley, L.H. Anticancer Activity and Cellular Repression of c-MYC by the G-Quadruplex-Stabilizing 11-Piperazinylquindoline is Not Dependent on Direct Targeting of the G-Quadruplex in the c-MYC Promoter. J. Med. Chem. 2012, 55, 6076–6086. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Obata, S.; Phan, A.T.; Hashiya, K.; Bando, T.; Sugiyama, H. Simultaneous Binding of Hybrid Molecules Constructed with Dual DNA-Binding Components to a G-Quadruplex and its Proximal Duplex. Chem. Eur. J. 2018, 24, 4428–4435. [Google Scholar] [CrossRef] [PubMed]
- Dervan, P.B.; Edelson, B.S. Recognition of the DNA Minor Groove by Pyrrole-Imidazole Polyamides. Curr. Opin. Struct. Biol. 2003, 13, 284–299. [Google Scholar] [CrossRef]
- Dutta, D.; Debnath, M.; Müller, D.; Paul, R.; Das, T.; Bessi, I.; Schwalbe, H.; Dash, J. Cell Penetrating Thiazole Peptides Inhibit c-MYC Expression Via Site-Specific Targeting of c-MYC G-Quadruplex. Nucleic Acids Res. 2018, 46, 5355–5365. [Google Scholar] [CrossRef]
- Hu, M.; Wang, Y.; Yu, Z.; Hu, L.; Ou, T.; Chen, S.; Huang, Z.; Tan, J. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef]
- Chen, S.-B.; Tan, J.-H.; Ou, T.-M.; Huang, S.-L.; An, L.-K.; Luo, H.-B.; Li, D.; Gu, L.-Q.; Huang, Z.-S. Pharmacophore-based discovery of triaryl-substituted imidazole as new telomeric G-quadruplex ligand. Bioorg. Med. Chem. Lett. 2011, 21, 1004–1009. [Google Scholar] [CrossRef]
- Hu, M.-H.; Chen, S.-B.; Wang, Y.-Q.; Zeng, Y.-M.; Ou, T.-M.; Li, D.; Gu, L.-Q.; Huang, Z.-S.; Tan, J.-H. Accurate high-throughput identification of parallel G-quadruplex topology by a new tetraarylsubstituted imidazole. Biosens. Bioelectron. 2016, 83, 77–84. [Google Scholar] [CrossRef]
- Chen, S.-B.; Wu, W.-B.; Hu, M.-H.; Ou, T.-M.; Gu, L.-Q.; Tan, J.-H.; Huang, Z.-S. Discovery of a new fluorescent light-up probe specific to parallel G-quadruplexes. Chem. Commun. 2014, 50, 12173–12176. [Google Scholar] [CrossRef]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S., Jr. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2015, 11, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Saha, P.; Das, T.; Dash, J. Target Guided Synthesis using DNA Nano-Templates for Selectively Assembling a G-Quadruplex Binding c-MYC Inhibitor. Nat. Commun. 2017, 8, 16103. [Google Scholar] [CrossRef] [PubMed]
- Martiny-Baron, G.; Marmé, D. VEGF-Mediated Tumour Angiogenesis: A New Target for Cancer Therapy. Curr. Opin. Biotechnol. 1995, 6, 675–680. [Google Scholar] [CrossRef]
- Bikfalvi, A.; Bicknell, R. Recent Advances in Angiogenesis, Anti-Angiogenesis and Vascular Targeting. Trends Pharmacol. Sci. 2002, 23, 576–582. [Google Scholar] [CrossRef]
- Finkenzeller, G.; Sparacio, A.; Technau, A.; Marmé, D.; Siemeister, G. Sp1 Recognition Sites in the Proximal Promoter of the Human Vascular Endothelial Growth Factor Gene are Essential for Platelet-Derived Growth Factor-Induced Gene Expression. Oncogene 1997, 15, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Le, X.; Abbruzzese, J.L.; Peng, Z.; Qian, C.N.; Tang, H.; Xiong, Q.; Wang, B.; Li, X.C.; Xie, K. Constitutive Sp1 Activity is Essential for Differential Constitutive Expression of Vascular Endothelial Growth Factor in Human Pancreatic Adenocarcinoma. Cancer Res. 2001, 61, 4143–4154. [Google Scholar] [PubMed]
- Willett, C.G.; Boucher, Y.; Di Tomaso, E.; Duda, D.G.; Munn, L.L.; Tong, R.T.; Chung, D.C.; Sahani, D.V.; Kalva, S.P.; Kozin, S.V.; et al. Direct Evidence that the VEGF-Specific Antibody Bevacizumab has Antivascular Effects in Human Rectal Cancer. Nat. Med. 2004, 10, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Gerber, H.; Novotny, W. Discovery and Development of Bevacizumab, an Anti-VEGF Antibody for Treating Cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Jacobs, C.S.; Dervan, P.B. Modifications at the C-Terminus to Improve Pyrrole–Imidazole Polyamide Activity in Cell Culture. J. Med. Chem. 2009, 52, 7380–7388. [Google Scholar]
- Sun, D.; Liu, W.J.; Guo, K.; Rusche, J.J.; Ebbinghaus, S.; Gokhale, V.; Hurley, L.H. The Proximal Promoter Region of the Human Vascular Endothelial Growth Factor Gene has a G-Quadruplex Structure that can be Targeted by G-Quadruplex-Interactive Agents. Mol. Cancer Ther. 2008, 7, 880–889. [Google Scholar] [CrossRef]
- Taka, T.; Joonlasak, K.; Huang, L.; Lee, T.R.; Chang, S.T.; Tuntiwechapikul, W. Down-Regulation of the Human VEGF Gene Expression by Perylene Monoimide Derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zan, L.; Wang, X.; Lu, Y.; Ou, T.; Lin, J.; Huang, Z.; Gu, L. Stabilization of VEGF G-Quadruplex and Inhibition of Angiogenesis by Quindoline Derivatives. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 2970–2977. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Chakraborty, S.; Chorell, E.; Sengupta, P.K.; Bhowmik, S. Importance of the Hydroxyl Substituents in the B–ring of Plant Flavonols on their Preferential Binding Interactions with VEGF G–quadruplex DNA: Multi-Spectroscopic and Molecular Modeling Studies. Int. J. Biol. Macromol. 2018, 118, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Sengupta, P.K.; Bhowmik, S. Exploring the Preferential Interaction of Quercetin with VEGF Promoter G-Quadruplex DNA and Construction of a pH-Dependent DNA-Based Logic Gate. RSC Adv. 2017, 7, 37230–37240. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 Protein Family: Arbiters of Cell Survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Radha, G.; Raghavan, S.C. BCL2: A Promising Cancer Therapeutic Target. Biochim. Biophys. Acta (BBA) Rev. Cancer 2017, 1868, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vidler, L.R.; Filippakopoulos, P.; Fedorov, O.; Picaud, S.; Martin, S.; Tomsett, M.; Woodward, H.; Brown, N.; Knapp, S.; Hoelder, S. Discovery of Novel Small-Molecule Inhibitors of BRD4 using Structure-Based Virtual Screening. J. Med. Chem. 2013, 56, 8073–8088. [Google Scholar] [CrossRef]
- Klasa, R.J.; Gillum, A.M.; Klem, R.E.; Frankel, S.R. Oblimersen Bcl-2 Antisense: Facilitating Apoptosis in Anticancer Treatment. Antisense Nucleic Acid Drug Dev. 2002, 12, 193–213. [Google Scholar] [CrossRef]
- Tzung, S.; Kim, K.M.; Basañez, G.; Giedt, C.D.; Simon, J.; Zimmerberg, J.; Zhang, K.Y.; Hockenbery, D.M. Antimycin A Mimics a Cell-Death-Inducing Bcl-2 Homology Domain 3. Nat. Cell Biol. 2001, 3, 183–191. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Rosenthal, C.T.; Lowe, S.W. Genetic Analysis of Chemoresistance in Primary Murine Lymphomas. Nat. Med. 2000, 6, 1029–1035. [Google Scholar] [CrossRef]
- Amundson, S.A.; Myers, T.G.; Scudiero, D.; Kitada, S.; Reed, J.C.; Fornace, A.J., Jr. An Informatics Approach Identifying Markers of Chemosensitivity in Human Cancer Cell Lines. Cancer Res. 2000, 60, 6101–6110. [Google Scholar] [PubMed]
- Douarre, C.; Gomez, D.; Morjani, H.; Zahm, J.; O’Donohue, M.; Eddabra, L.; Mailliet, P.; Riou, J.; Trentesaux, C. Overexpression of Bcl-2 is Associated with Apoptotic Resistance to the G-Quadruplex Ligand 12459 but is Not Sufficient to Confer Resistance to Long-Term Senescence. Nucleic Acids Res. 2005, 33, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, S.; Chen, W.; Spicer, E.K.; Courtenay-Luck, N.; Fernandes, D.J. The Nucleolin Targeting Aptamer AS1411 Destabilizes Bcl-2 Messenger RNA in Human Breast Cancer Cells. Cancer Res. 2008, 68, 2358–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rantanen, S.; Imonni, O.; Joensuu, H.; Franssila, K.; Knuutila, S. Causes and Consequences of BCL2 Overexpression in Diffuse Large B-Cell Lymphoma. Leuk. Lymphoma 2001, 42, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Jaeger, U.; Hockett, R.D.; Graninger, W.; Bennett, S.; Goldman, P.; Korsmeyer, S.J. Alternative Promoters and Exons, Somatic Mutation and Deregulation of the Bcl-2-Ig Fusion Gene in Lymphoma. EMBO J. 1988, 7, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Young, R.L.; Korsmeyer, S.J. A Negative Regulatory Element in the Bcl-2 5′-Untranslated Region Inhibits Expression from an Upstream Promoter. Mol. Cell. Biol. 1993, 13, 3686–3697. [Google Scholar] [CrossRef] [PubMed]
- Heckman, C.; Mochon, E.; Arcinas, M.; Boxer, L.M. The WT1 Protein is a Negative Regulator of the Normal Bcl-2 Allele in t(14;18) Lymphomas. J. Biol. Chem. 1997, 272, 19609–19614. [Google Scholar] [CrossRef]
- Gomez-Manzano, C.; Mitlianga, P.; Fueyo, J.; Lee, H.Y.; Hu, M.; Spurgers, K.B.; Glass, T.L.; Koul, D.; Liu, T.J.; McDonnell, T.J.; et al. Transfer of E2F-1 to Human Glioma Cells Results in Transcriptional Up-Regulation of Bcl-2. Cancer Res. 2001, 61, 6693–6697. [Google Scholar]
- Liu, Y.; Boxer, L.M.; Latchman, D.S. Activation of the Bcl-2 Promoter by Nerve Growth Factor is Mediated by the p42/p44 MAPK Cascade. Nucleic Acids Res. 1999, 27, 2086–2090. [Google Scholar] [CrossRef]
- Dai, J.; Chen, D.; Jones, R.A.; Hurley, L.H.; Yang, D. NMR Solution Structure of the Major G-Quadruplex Structure Formed in the Human BCL2 Promoter Region. Nucleic Acids Res. 2006, 34, 5133–5144. [Google Scholar] [CrossRef]
- Agrawal, P.; Lin, C.; Mathad, R.I.; Carver, M.; Yang, D. The Major G-Quadruplex Formed in the Human BCL-2 Proximal Promoter Adopts a Parallel Structure with a 13-Nt Loop in K Solution. J. Am. Chem. Soc. 2014, 136, 1750–1753. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ou, T.; Lu, Y.; Li, Z.; Xu, Z.; Xi, C.; Tan, J.; Huang, S.; An, L.; Li, D. Turning Off Transcription of the Bcl-2 Gene by Stabilizing the Bcl-2 Promoter Quadruplex with Quindoline Derivatives. J. Med. Chem. 2010, 53, 4390–4398. [Google Scholar] [CrossRef] [PubMed]
- Onel, B.; Carver, M.; Wu, G.; Timonina, D.; Kalarn, S.; Larriva, M.; Yang, D. A New G-Quadruplex with Hairpin Loop Immediately Upstream of the Human BCL2 P1 Promoter Modulates Transcription. J. Am. Chem. Soc. 2016, 138, 2563–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, J.; Pagano, A.; Capasso, D.; Di Gaetano, S.; Giustiniano, M.; Novellino, E.; Randazzo, A.; Pagano, B. Targeting the BCL2 Gene Promoter G-Quadruplex with a New Class of Furopyridazinone-Based Molecules. ChemMedChem 2018, 13, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lin, D.; Tang, Y.; Fei, X.; Wang, C.; Zhang, B.; Zhou, J. A Light-Up Probe Targeting for Bcl-2 2345 G-Quadruplex DNA with Carbazole TO. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 191, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Onyshchenko, M.I.; Gaynutdinov, T.I.; Englund, E.A.; Appella, D.H.; Neumann, R.D.; Panyutin, I.G. Stabilization of G-Quadruplex in the BCL2 Promoter Region in Double-Stranded DNA by Invading Short PNAs. Nucleic Acids Res. 2009, 37, 7570–7580. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kendrick, S.; Hecht, S.M.; Hurley, L.H. The Transcriptional Complex between the BCL2 i-Motif and hnRNP LL is a Molecular Switch for Control of Gene Expression that can be Modulated by Small Molecules. J. Am. Chem. Soc. 2014, 136, 4172–4185. [Google Scholar] [CrossRef]
- Kendrick, S.; Kang, H.; Alam, M.P.; Madathil, M.M.; Agrawal, P.; Gokhale, V.; Yang, D.; Hecht, S.M.; Hurley, L.H. The Dynamic Character of the BCL2 Promoter i-Motif Provides a Mechanism for Modulation of Gene Expression by Compounds that Bind Selectively to the Alternative DNA Hairpin Structure. J. Am. Chem. Soc. 2014, 136, 4161–4171. [Google Scholar] [CrossRef]
- Shaw, T.J.; Keszthelyi, E.J.; Tonary, A.M.; Cada, M.; Vanderhyden, B.C. Cyclic AMP in Ovarian Cancer Cells both Inhibits Proliferation and Increases c-KIT Expression. Exp. Cell Res. 2002, 273, 95–106. [Google Scholar] [CrossRef]
- Edling, C.E.; Hallberg, B. C-Kit—A Hematopoietic Cell Essential Receptor Tyrosine Kinase. Int. J. Biochem. Cell Biol. 2007, 39, 1995–1998. [Google Scholar] [CrossRef]
- Ashman, L.K. The Biology of Stem Cell Factor and its Receptor C-Kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Robertson, S.C.; Tynan, J.A.; Donoghue, D.J. RTK Mutations and Human Syndromes: When Good Receptors Turn Bad. Trends Genet. 2000, 16, 265–271. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic Kinase Signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Fletcher, J.A.; Heinrich, M.C. Biology of Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2004, 22, 3813–3825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardelmann, E.; Merkelbach-Bruse, S.; Pauls, K.; Thomas, N.; Schildhaus, H.U.; Heinicke, T.; Speidel, N.; Pietsch, T.; Buettner, R.; Pink, D.; et al. Polyclonal Evolution of Multiple Secondary KIT Mutations in Gastrointestinal Stromal Tumors Under Treatment with Imatinib Mesylate. Clin. Cancer Res. 2006, 12, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Spitaleri, G.; Biffi, R.; Barberis, M.; Fumagalli, C.; Toffalorio, F.; Catania, C.; Noberasco, C.; Lazzari, C.; de Marinis, F.; De Pas, T. Inactivity of Imatinib in Gastrointestinal Stromal Tumors (GISTs) Harboring a KIT Activation-Loop Domain Mutation (Exon 17 Mutation pN822K). Oncol. Targets Ther. 2015, 8, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tojo, A.; Aoki, N.; Shibuya, M. Characterization of the Promoter Region of the Human c-kit proto-oncogene. Jpn. J. Cancer Res. 1993, 84, 1136–1144. [Google Scholar] [CrossRef]
- Yasuda, H.; Galli, S.J.; Geissler, E. Cloning and Functional Analysis of the Mouse c-Kit Promoter. Biochem. Biophys. Res. Commun. 1993, 191, 893–901. [Google Scholar] [CrossRef]
- Park, G.H.; Plummer, H.K., 3rd; Krystal, G.W. Selective Sp1 Binding is Critical for Maximal Activity of the Human c-Kit Promoter. Blood 1998, 92, 4138–4149. [Google Scholar]
- GSE31477: ENCODE Transcription Factor Binding Sites by ChIP-Seq from Stanford/Yale/USC/Harvard. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/sra/?term=SRX186618 (accessed on 10 December 2018).
- Hsu, S.D.; Varnai, P.; Bugaut, A.; Reszka, A.P.; Neidle, S.; Balasubramanian, S. A G-Rich Sequence within the c-Kit Oncogene Promoter Forms a Parallel G-Quadruplex having Asymmetric G-Tetrad Dynamics. J. Am. Chem. Soc. 2009, 131, 13399–13409. [Google Scholar] [CrossRef]
- Dhamodharan, V.; Harikrishna, S.; Bhasikuttan, A.C.; Pradeepkumar, P. Topology Specific Stabilization of Promoter Over Telomeric G-Quadruplex DNAs by Bisbenzimidazole Carboxamide Derivatives. ACS Chem. Biol. 2014, 10, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Diveshkumar, K.V.; Sakrikar, S.; Rosu, F.; Harikrishna, S.; Gabelica, V.; Pradeepkumar, P. Specific Stabilization of c-MYC and c-KIT G-Quadruplex DNA Structures by Indolylmethyleneindanone Scaffolds. Biochemistry 2016, 55, 3571–3585. [Google Scholar] [CrossRef] [PubMed]
- Zorzan, E.; Da Ros, S.; Musetti, C.; Shahidian, L.Z.; Coelho, N.F.R.; Bonsembiante, F.; Létard, S.; Gelain, M.E.; Palumbo, M.; Dubreuil, P. Screening of Candidate G-Quadruplex Ligands for the Human c-KIT Promotorial Region and their Effects in Multiple in-Vitro Models. Oncotarget 2016, 7, 21658–21675. [Google Scholar] [CrossRef] [PubMed]
- Głuszyńska, A.; Juskowiak, B.; Kuta-Siejkowska, M.; Hoffmann, M.; Haider, S. Carbazole Derivatives’ Binding to c-KIT G-Quadruplex DNA. Molecules 2018, 23, 1134. [Google Scholar] [CrossRef] [PubMed]
- Bejugam, M.; Sewitz, S.; Shirude, P.S.; Rodriguez, R.; Shahid, R.; Balasubramanian, S. Trisubstituted Isoalloxazines as a New Class of G-Quadruplex Binding Ligands: Small Molecule Regulation of c-Kit Oncogene Expression. J. Am. Chem. Soc. 2007, 129, 12926–12927. [Google Scholar] [CrossRef]
- Gunaratnam, M.; Swank, S.; Haider, S.M.; Galesa, K.; Reszka, A.P.; Beltran, M.; Cuenca, F.; Fletcher, J.A.; Neidle, S. Targeting Human Gastrointestinal Stromal Tumor Cells with a Quadruplex-Binding Small Molecule. J. Med. Chem. 2009, 52, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- McLuckie, K.I.; Waller, Z.A.; Sanders, D.A.; Alves, D.; Rodriguez, R.; Dash, J.; McKenzie, G.J.; Venkitaraman, A.R.; Balasubramanian, S. G-Quadruplex-Binding Benzo [a] Phenoxazines Down-Regulate c-KIT Expression in Human Gastric Carcinoma Cells. J. Am. Chem. Soc. 2011, 133, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, C.; Yan, J.; Hou, J.; Chen, S.; Ou, T.; Gu, L.; Huang, Z.; Tan, J. Synthesis and Evaluation of Quinazolone Derivatives as a New Class of c-KIT G-Quadruplex Binding Ligands. ACS Med. Chem. Lett. 2013, 4, 909–914. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R.A. Telomeres and Telomerase in Cancer. Carcinogenesis 2009, 31, 9–18. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of Telomeres and Telomerase in Cancer, and Advances in Telomerase-Targeted Therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomeres and Human Disease: Ageing, Cancer and Beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, D.; Rizzo, A.; Trisciuoglio, D.; Cardinali, G.; Torrisi, M.R.; Zangemeister-Wittke, U.; Zupi, G.; Biroccio, A. Involvement of hTERT in Apoptosis Induced by Interference with Bcl-2 Expression and Function. Cell Death Differ. 2005, 12, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The Telomerase Reverse Transcriptase Regulates Chromatin State and DNA Damage Responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Kang, M.K.; Dicterow, E.; Kameta, A.; Baluda, M.A.; Park, N.H. Introduction of Human Telomerase Reverse Transcriptase to Normal Human Fibroblasts Enhances DNA Repair Capacity. Clin. Cancer Res. 2004, 10, 2551–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N. TERT Promotes Epithelial Proliferation through Transcriptional Control of a Myc-and Wnt-Related Developmental Program. PLoS Genet. 2008, 4, e10. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E. Telomerase Regulates MYC-Driven Oncogenesis Independent of its Reverse Transcriptase Activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT Promoter Mutations Occur Frequently in Gliomas and a Subset of Tumors Derived from Cells with Low Rates of Self-Renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Kang, H.; Cui, Y.; Yin, H.; Scheid, A.; Hendricks, W.P.; Schmidt, J.; Sekulic, A.; Kong, D.; Trent, J.M.; Gokhale, V. A Pharmacological Chaperone Molecule Induces Cancer Cell Death by Restoring Tertiary DNA Structures in Mutant hTERT Promoters. J. Am. Chem. Soc. 2016, 138, 13673–13692. [Google Scholar] [CrossRef] [Green Version]
- Krens, L.L.; Baas, J.M.; Gelderblom, H.; Guchelaar, H. Therapeutic Modulation of k-Ras Signaling in Colorectal Cancer. Drug Discov. Today 2010, 15, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Inoue, T.; Taylor, R.D.; Watanabe, T.; Koshikawa, N.; Yoda, H.; Shinohara, K.; Bando, T.; Sugiyama, H.; Nagase, H.; et al. Inhibition of KRAS Codon 12 Mutants using a Novel DNA-Alkylating pyrrole–imidazole Polyamide Conjugate. Nat. Commun. 2015, 6, 6706. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Paramasivam, M.; Membrino, A.; Yokoyama, K.K.; Xodo, L.E. The KRAS Promoter Responds to Myc-Associated Zinc Finger and Poly(ADP-Ribose) Polymerase 1 Proteins, which Recognize a Critical Quadruplex-Forming GA-Element. J. Biol. Chem. 2010, 285, 22003–22016. [Google Scholar] [CrossRef] [PubMed]
- Amato, J.; Madanayake, T.W.; Iaccarino, N.; Novellino, E.; Randazzo, A.; Hurley, L.H.; Pagano, B. HMGB1 Binds to the KRAS Promoter G-Quadruplex: A New Player in Oncogene Transcriptional Regulation? Chem. Commun. 2018, 54, 9442–9445. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Paramasivam, M.; Spolaore, B.; Xodo, L.E. Structural Polymorphism within a Regulatory Element of the Human KRAS Promoter: Formation of G4-DNA Recognized by Nuclear Proteins. Nucleic Acids Res. 2008, 36, 3765–3780. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G-Quadruplex Formation within the Promoter of the KRAS Proto-Oncogene and its Effect on Transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef]
- Kaiser, C.E.; Van Ert, N.A.; Agrawal, P.; Chawla, R.; Yang, D.; Hurley, L.H. Insight into the Complexity of the i-Motif and G-Quadruplex DNA Structures Formed in the KRAS Promoter and Subsequent Drug-Induced Gene Repression. J. Am. Chem. Soc. 2017, 139, 8522–8536. [Google Scholar] [CrossRef]
- Cogoi, S.; Paramasivam, M.; Filichev, V.; Géci, I.; Pedersen, E.B.; Xodo, L.E. Identification of a New G-Quadruplex Motif in the KRAS Promoter and Design of Pyrene-Modified G4-Decoys with Antiproliferative Activity in Pancreatic Cancer Cells. J. Med. Chem. 2008, 52, 564–568. [Google Scholar] [CrossRef]
- Lavrado, J.; Brito, H.; Borralho, P.M.; Ohnmacht, S.A.; Kim, N.; Leitão, C.; Pisco, S.; Gunaratnam, M.; Rodrigues, C.M.; Moreira, R.; et al. KRAS Oncogene Repression in Colon Cancer Cell Lines by G-Quadruplex Binding Indolo [3, 2-c] Quinolines. Sci. Rep. 2015, 5, 9696. [Google Scholar] [CrossRef]
- Ramsay, R.G.; Gonda, T.J. MYB Function in Normal and Cancer Cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Klempnauer, K.; Gonda, T.J.; Bishop, J.M. Nucleotide Sequence of the Retroviral Leukemia Gene v-Myb and its Cellular Progenitor c-Myb: The Architecture of a Transduced Oncogene. Cell 1982, 31, 453–463. [Google Scholar] [CrossRef]
- Greig, K.T.; Carotta, S.; Nutt, S.L. Critical Roles for c-Myb in Hematopoietic Progenitor Cells. Semin. Immunol. 2008, 20, 247–256. [Google Scholar] [CrossRef]
- Zuber, J.; Rappaport, A.R.; Luo, W.; Wang, E.; Chen, C.; Vaseva, A.V.; Shi, J.; Weissmueller, S.; Fellmann, C.; Taylor, M.J.; et al. An Integrated Approach to Dissecting Oncogene Addiction Implicates a Myb-Coordinated Self-Renewal Program as Essential for Leukemia Maintenance. Genes Dev. 2011, 25, 1628–1640. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Benassi, B.; D’Agnano, I.; D’Angelo, C.; Buglioni, S.; Mottolese, M.; Ricciotti, A.; Citro, G.; Cosimelli, M.; Ramsay, R.G. C-Myb and Bcl-x Overexpression Predicts Poor Prognosis in Colorectal Cancer: Clinical and Experimental Findings. Am. J. Pathol. 2001, 158, 1289–1299. [Google Scholar] [CrossRef]
- Miao, R.Y.; Drabsch, Y.; Cross, R.S.; Cheasley, D.; Carpinteri, S.; Pereira, L.; Malaterre, J.; Gonda, T.J.; Anderson, R.L.; Ramsay, R.G. MYB is Essential for Mammary Tumorigenesis. Cancer Res. 2011, 71, 7029–7037. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Andrén, Y.; Moskaluk, C.A.; Frierson, H.F., Jr.; Cooke, S.L.; Futreal, P.A.; Kling, T.; Nelander, S.; Nordkvist, A.; Persson, F. Clinically Significant Copy Number Alterations and Complex Rearrangements of MYB and NFIB in Head and Neck Adenoid Cystic Carcinoma. Genes Chromosomes Cancer 2012, 51, 805–817. [Google Scholar] [CrossRef]
- Guerra, J.; Withers, D.A.; Boxer, L.M. Myb Binding Sites Mediate Negative Regulation of c-Myb Expression in T-Cell Lines. Blood 1995, 86, 1873–1880. [Google Scholar]
- McCann, S.; Sullivan, J.; Guerra, J.; Arcinas, M.; Boxer, L.M. Repression of the c-Myb Gene by WT1 Protein in T and B Cell Lines. J. Biol. Chem. 1995, 270, 23785–23789. [Google Scholar] [CrossRef]
- Perrotti, D.; Melotti, P.; Skorski, T.; Casella, I.; Peschle, C.; Calabretta, B. Overexpression of the Zinc Finger Protein MZF1 Inhibits Hematopoietic Development from Embryonic Stem Cells: Correlation with Negative Regulation of CD34 and c-Myb Promoter Activity. Mol. Cell. Biol. 1995, 15, 6075–6087. [Google Scholar] [CrossRef]
- Bellon, T.; Perrotti, D.; Calabretta, B. Granulocytic Differentiation of Normal Hematopoietic Precursor Cells Induced by Transcription Factor PU.1 Correlates with Negative Regulation of the c-Myb Promoter. Blood 1997, 90, 1828–1839. [Google Scholar] [PubMed]
- Sullivan, J.; Feeley, B.; Guerra, J.; Boxer, L.M. Identification of the Major Positive Regulators of c-Myb Expression in Hematopoietic Cells of Different Lineages. J. Biol. Chem. 1997, 272, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Correa, I.; Casadevall, C.; Travali, S.; Soprano, K.J.; Calabretta, B. The Jun Family Members, c-Jun and JunD, Transactivate the Human c-Myb Promoter Via an Ap1-Like Element. J. Biol. Chem. 1992, 267, 19665–19672. [Google Scholar] [PubMed]
- Palumbo, S.L.; Memmott, R.M.; Uribe, D.J.; Krotova-Khan, Y.; Hurley, L.H.; Ebbinghaus, S.W. A Novel G-Quadruplex-Forming GGA Repeat Region in the c-Myb Promoter is a Critical Regulator of Promoter Activity. Nucleic Acids Res. 2008, 36, 1755–1769. [Google Scholar] [CrossRef] [PubMed]
- Matsugami, A.; Ouhashi, K.; Kanagawa, M.; Liu, H.; Kanagawa, S.; Uesugi, S.; Katahira, M. An Intramolecular Quadruplex of (GGA) 4 Triplet Repeat DNA with a G: G: G: G Tetrad and a G (: A): G (: A): G (: A): G Heptad, and its Dimeric Interaction 1. J. Mol. Biol. 2001, 313, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Matsugami, A.; Okuizumi, T.; Uesugi, S.; Katahira, M. Intramolecular Higher Order Packing of Parallel Quadruplexes Comprising a G:G:G:G Tetrad and a G(:A):G(:A):G(:A):G Heptad of GGA Triplet Repeat DNA. J. Biol. Chem. 2003, 278, 28147–28153. [Google Scholar] [CrossRef]
- University of Washington Human Reference Epigenome Mapping Project. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/sra/?term=SRX213515 (accessed on 10 December 2018).
- GSE59395: Genome-Wide Map of Regulatory Interactions in the Human Genome. Available online: https://www.ncbi.nlm.nih.gov/sra/?term=SRX2370636 (accessed on 10 December 2018).
- Li, F.; Zhou, J.; Xu, M.; Yuan, G. Exploration of G-Quadruplex Function in c-Myb Gene and its Transcriptional Regulation by Topotecan. Int. J. Biol. Macromol. 2018, 107, 1474–1479. [Google Scholar] [CrossRef]
- Miyazaki, T.; Pan, Y.; Joshi, K.; Purohit, D.; Hu, B.; Demir, H.; Mazumder, S.; Okabe, S.; Yamori, T.; Viapiano, M.; et al. Telomestatin Impairs Glioma Stem Cell Survival and Growth through the Disruption of Telomeric G-Quadruplex and Inhibition of the Proto-Oncogene, c-Myb. Clin. Cancer Res. 2012, 18, 1268–1280. [Google Scholar] [CrossRef]
- Brown, R.V.; Wang, T.; Chappeta, V.R.; Wu, G.; Onel, B.; Chawla, R.; Quijada, H.; Camp, S.M.; Chiang, E.T.; Lassiter, Q.R.; et al. The Consequences of Overlapping G-Quadruplexes and i-Motifs in the Platelet-Derived Growth Factor Receptor β Core Promoter Nuclease Hypersensitive Element Can Explain the Unexpected Effects of Mutations and Provide Opportunities for Selective Targeting of both Structures by Small Molecules to Downregulate Gene Expression. J. Am. Chem. Soc. 2017, 139, 7456–7475. [Google Scholar]
- Qin, Y.; Rezler, E.M.; Gokhale, V.; Sun, D.; Hurley, L.H. Characterization of the G-Quadruplexes in the Duplex Nuclease Hypersensitive Element of the PDGF-A Promoter and Modulation of PDGF-A Promoter Activity by TMPyP4. Nucleic Acids Res. 2007, 35, 7698–7713. [Google Scholar] [CrossRef]
- Lin, S.; Li, S.; Chen, Z.; He, X.; Zhang, Y.; Xu, X.; Xu, M.; Yuan, G. Formation, Recognition and Bioactivities of a Novel G-Quadruplex in the STAT3 Gene. Bioorg. Med. Chem. Lett. 2011, 21, 5987–5991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, W.; Zhou, J.; Xu, M.; Yuan, G. Investigation of G-Quadruplex Formation in the FGFR2 Promoter Region and its Transcriptional Regulation by Liensinine. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ustach, C.; Kim, H.C. Platelet-Derived Growth Factor Signaling and Human Cancer. J. Biochem. Mol. Biol. 2003, 36, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Westermark, B.; Heldin, C.; Nistér, M. Platelet-derived Growth Factor in Human Glioma. Glia 1995, 15, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sulzbacher, I.; Birner, P.; Trieb, K.; Träxler, M.; Lang, S.; Chott, A. Expression of Platelet-Derived Growth Factor-AA is Associated with Tumor Progression in Osteosarcoma. Mod. Pathol. 2003, 16, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afrakhte, M.; Mistér, M.; Östman, A.; Westermark, B.; Paulsson, Y. Production of cell-associated PDGF-AA by a Human Sarcoma Cell Line: Evidence for a Latent Autocrine Effect. Int. J. Cancer 1996, 68, 802–809. [Google Scholar] [CrossRef]
- Guha, A.; Dashner, K.; Mc Black, P.L.; Wagner, J.A.; Stiles, C.D. Expression of PDGF and PDGF Receptors in Human Astrocytoma Operation Specimens Supports the Existence of an Autocrine Loop. Int. J. Cancer 1995, 60, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Yagi, H.; Sannohe, Y.; Rajendran, A.; Sugiyama, H. Folding Pathways of Human Telomeric Type-1 and Type-2 G-Quadruplex Structures. J. Am. Chem. Soc. 2010, 132, 14910–14918. [Google Scholar] [CrossRef]
- Limongelli, V.; De Tito, S.; Cerofolini, L.; Fragai, M.; Pagano, B.; Trotta, R.; Cosconati, S.; Marinelli, L.; Novellino, E.; Bertini, I. The G-Triplex DNA. Angew. Chem. 2013, 125, 2325–2329. [Google Scholar] [CrossRef]
- Rajendran, A.; Endo, M.; Hidaka, K.; Sugiyama, H. Direct and Single-Molecule Visualization of the Solution-State Structures of G-Hairpin and G-Triplex Intermediates. Angew. Chem. Int. Ed. 2014, 53, 4107–4112. [Google Scholar] [CrossRef]
- Cerofolini, L.; Amato, J.; Giachetti, A.; Limongelli, V.; Novellino, E.; Parrinello, M.; Fragai, M.; Randazzo, A.; Luchinat, C. G-Triplex Structure and Formation Propensity. Nucleic Acids Res. 2014, 42, 13393–13404. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Cui, Y.; Zhao, T.; Fu, H.; Koirala, D.; Punnoose, J.A.; Kong, D.; Mao, H. Divalent Cations and Molecular Crowding Buffers Stabilize G-Triplex at Physiologically Relevant Temperatures. Sci. Rep. 2015, 5, 9255. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Sengupta, P.; Krishnan, Y.; Ladame, S. Combining G-Quadruplex Targeting Motifs on a Single Peptide Nucleic Acid Scaffold: A Hybrid (3+1) PNA–DNA Bimolecular Quadruplex. Chem. Eur. J. 2008, 14, 8682–8689. [Google Scholar] [CrossRef] [PubMed]
- Amato, J.; Pagano, A.; Cosconati, S.; Amendola, G.; Fotticchia, I.; Iaccarino, N.; Marinello, J.; De Magis, A.; Capranico, G.; Novellino, E.; et al. Discovery of the First Dual G-triplex/G-Quadruplex Stabilizing Compound: A New Opportunity in the Targeting of G-Rich DNA Structures? Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, A.; Endo, M.; Hidaka, K.; Teulade-Fichou, M.; Mergny, J.; Sugiyama, H. Small Molecule Binding to a G-Hairpin and a G-Triplex: A New Insight into Anticancer Drug Design Targeting G-Rich Regions. Chem. Commun. 2015, 51, 9181–9184. [Google Scholar] [CrossRef] [PubMed]
- Obata, S.; Asamitsu, S.; Hashiya, K.; Bando, T.; Sugiyama, H. G-Quadruplex Induction by the Hairpin Pyrrole–Imidazole Polyamide Dimer. Biochemistry 2017, 57, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct Activation of TERT Transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Zaanan, A.; Okamoto, K.; Kawakami, H.; Khazaie, K.; Huang, S.; Sinicrope, F.A. The Mutant KRAS Gene Up-Regulates BCL-XL Protein Via STAT3 to Confer Apoptosis Resistance that is Reversed by BIM Protein Induction and BCL-XL Antagonism. J. Biol. Chem. 2015, 290, 23838–23849. [Google Scholar] [CrossRef]
- Kendrick, S.; Muranyi, A.; Gokhale, V.; Hurley, L.H.; Rimsza, L.M. Simultaneous Drug Targeting of the Promoter MYC G-Quadruplex and BCL2 i-Motif in Diffuse Large B-Cell Lymphoma Delays Tumor Growth. J. Med. Chem. 2017, 60, 6587–6597. [Google Scholar] [CrossRef]
Figure 1.
(a) Structure and schematic illustration of a G-tetrad. (b) Schematic illustrations of typical intramolecular G-quadruplex (G4) structures: (left) crystal structure of parallel type telomere G4 (PDB code: 1kf1); (center) solution structure of antiparallel type telomere G4 (PDB code: 143d); (right) solution structure of hybrid type telomere G4 (PDB code: 2gku). The figures were adapted with permission from Reference 56. PDB; Protein Data Bank.
Figure 1.
(a) Structure and schematic illustration of a G-tetrad. (b) Schematic illustrations of typical intramolecular G-quadruplex (G4) structures: (left) crystal structure of parallel type telomere G4 (PDB code: 1kf1); (center) solution structure of antiparallel type telomere G4 (PDB code: 143d); (right) solution structure of hybrid type telomere G4 (PDB code: 2gku). The figures were adapted with permission from Reference 56. PDB; Protein Data Bank.

Figure 2.
Selective telomere G4 targeting by ligands. (a) Telomere G-stretch sequences potentially adopt non-canonical G4s that offer specific binding motifs. (b) Several telomere G4-preferred binders based on the specific-motif recognition. It is worth noting that the by Ni-M and IZNP1 exhibit differential antitumor activities, likely based on the specific-motif recognition of telomere G4s. (c) EPI and the solution structure of the EPI-wtTel26 complex (PDB code: 6ccw). The bases that do not participate in the interaction event with EPI are omitted for clarity. Figure 2a,c were adapted with permission from References 56 and 86, respectively.
Figure 2.
Selective telomere G4 targeting by ligands. (a) Telomere G-stretch sequences potentially adopt non-canonical G4s that offer specific binding motifs. (b) Several telomere G4-preferred binders based on the specific-motif recognition. It is worth noting that the by Ni-M and IZNP1 exhibit differential antitumor activities, likely based on the specific-motif recognition of telomere G4s. (c) EPI and the solution structure of the EPI-wtTel26 complex (PDB code: 6ccw). The bases that do not participate in the interaction event with EPI are omitted for clarity. Figure 2a,c were adapted with permission from References 56 and 86, respectively.

Figure 3.
(a) The c-MYC promoter has one putative G4-forming sequence (PQS). (b) Molecules preferentially interacting with its G4 DNAs over other ones. N-Methylpyrrole is highlighted in blue and N-methylimidazole is highlighted in red. Antitumor activities of TH3, IZCZ-3, benzofuran derivative, and Tz 1 are summarized in Table 1.
Figure 3.
(a) The c-MYC promoter has one putative G4-forming sequence (PQS). (b) Molecules preferentially interacting with its G4 DNAs over other ones. N-Methylpyrrole is highlighted in blue and N-methylimidazole is highlighted in red. Antitumor activities of TH3, IZCZ-3, benzofuran derivative, and Tz 1 are summarized in Table 1.

Figure 4.
(a) The VEGF promoter has one putative G4-forming sequence (PQS) located close to the transcription start site (TSS) and hormone response element (HRE) that regulate the transcription. (b) Molecules interacting with its G4 DNAs and efficiently suppressing VEGF protein expression. It is worth noting that SYUIQ-FM05 has potential to reduce VEGF-stimulated tumor angiogenesis.
Figure 4.
(a) The VEGF promoter has one putative G4-forming sequence (PQS) located close to the transcription start site (TSS) and hormone response element (HRE) that regulate the transcription. (b) Molecules interacting with its G4 DNAs and efficiently suppressing VEGF protein expression. It is worth noting that SYUIQ-FM05 has potential to reduce VEGF-stimulated tumor angiogenesis.

Figure 5.
(a) The BCL2 promoter has two G4-forming elements that were shown to attenuate the BCL2 promoter activity. (b) Molecules preferentially interacting with the G4 (distal one) over other G4 structures. An antitumor activity of furopyridazinone derivative is summarized in Table 1.
Figure 5.
(a) The BCL2 promoter has two G4-forming elements that were shown to attenuate the BCL2 promoter activity. (b) Molecules preferentially interacting with the G4 (distal one) over other G4 structures. An antitumor activity of furopyridazinone derivative is summarized in Table 1.

Figure 6.
(a) The c-kit promoter has two PQSs, where several transcription factors are likely involved. (b) Molecules interacting with its G4 DNA and showing the downregulation of the c-kit gene transcription.
Figure 6.
(a) The c-kit promoter has two PQSs, where several transcription factors are likely involved. (b) Molecules interacting with its G4 DNA and showing the downregulation of the c-kit gene transcription.

Figure 7.
(a) The hTERT promoter has several PQSs, where two tandemly aligned G4s are suggested, and a molecule interacting with the specific motif of its G4 DNA and showing the downregulation of the hTERT gene transcription. The arrows with asterisks in the right illustration of the (b) represent bases that are protected in the presence of GTC365 from methylation by dimethyl sulfate (DMS) in the experiment performed in Reference 102. The protection suggests the occupied site of GTC365. The numbers represent the order of the runs of poly G observed in the hTERT promoter, which are numbered in Reference 102. The antitumor activity of GTC365 is summarized in Table 1. The right illustration of the Figure 7 (b) was adapted with permission from Reference. 172.
Figure 7.
(a) The hTERT promoter has several PQSs, where two tandemly aligned G4s are suggested, and a molecule interacting with the specific motif of its G4 DNA and showing the downregulation of the hTERT gene transcription. The arrows with asterisks in the right illustration of the (b) represent bases that are protected in the presence of GTC365 from methylation by dimethyl sulfate (DMS) in the experiment performed in Reference 102. The protection suggests the occupied site of GTC365. The numbers represent the order of the runs of poly G observed in the hTERT promoter, which are numbered in Reference 102. The antitumor activity of GTC365 is summarized in Table 1. The right illustration of the Figure 7 (b) was adapted with permission from Reference. 172.

Figure 8.
(a) The human KRAS promoter has three PQSs. In particular, the G4 formation at the most proximal PQS (PQS1) is shown to act as a stronger transcriptional repressor. (b) Molecules interacting with its G4 DNA and causing antitumor activities.
Figure 8.
(a) The human KRAS promoter has three PQSs. In particular, the G4 formation at the most proximal PQS (PQS1) is shown to act as a stronger transcriptional repressor. (b) Molecules interacting with its G4 DNA and causing antitumor activities.

Figure 9.
(a) The c-myb promoter has multiple PQSs. (b) A molecule interacting with its G4 DNA and efficiently repressing MYB protein expression.
Figure 9.
(a) The c-myb promoter has multiple PQSs. (b) A molecule interacting with its G4 DNA and efficiently repressing MYB protein expression.

Figure 10.
(a–c) G-triplex-targeting ligands. (c) A platform for their evaluation constructed by DNA origami.
Figure 10.
(a–c) G-triplex-targeting ligands. (c) A platform for their evaluation constructed by DNA origami.

Figure 11.
Head-to-head polyamide dimer that can target a certain G4 by simultaneously binding to dual-duplex sites across the G4. N-Methylpyrrole is highlighted in blue, and N-methylimidazole is highlighted in red.
Figure 11.
Head-to-head polyamide dimer that can target a certain G4 by simultaneously binding to dual-duplex sites across the G4. N-Methylpyrrole is highlighted in blue, and N-methylimidazole is highlighted in red.


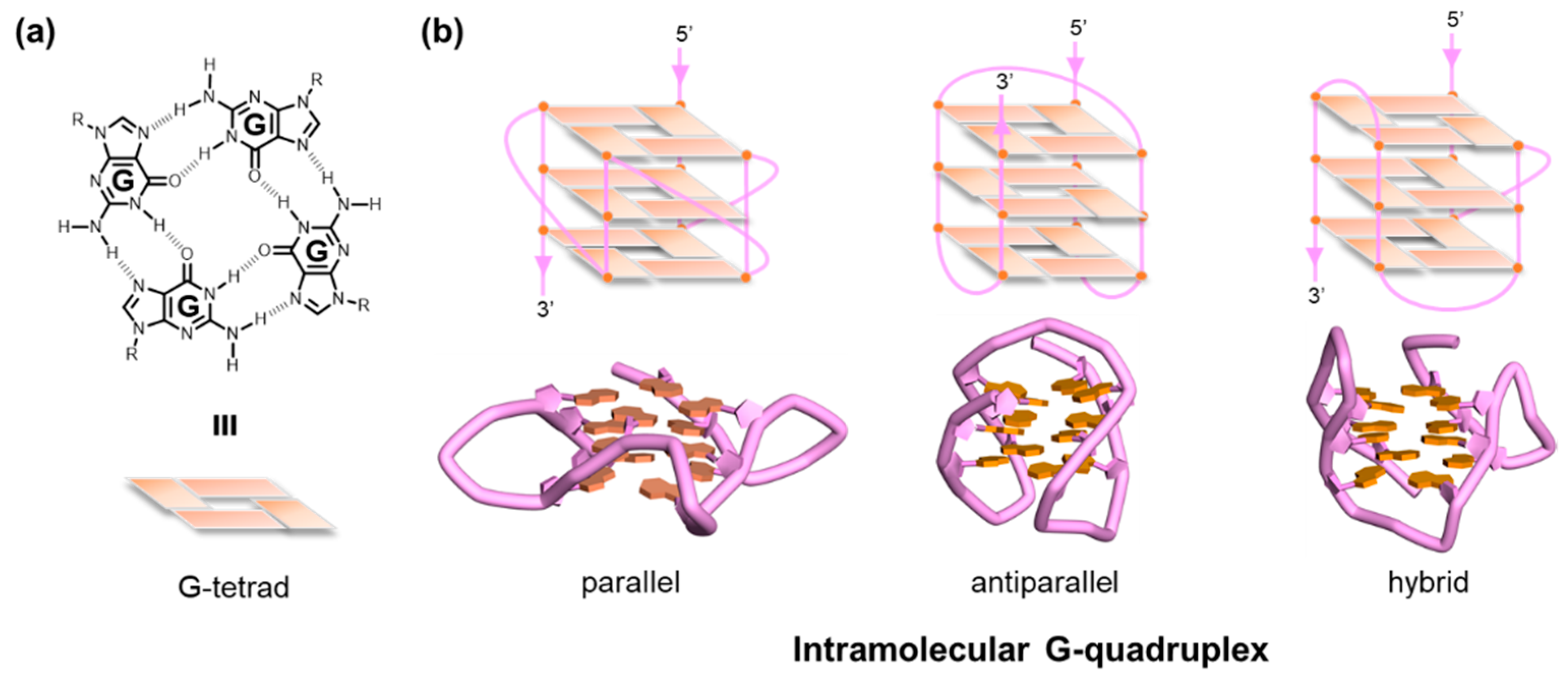{kind=link}
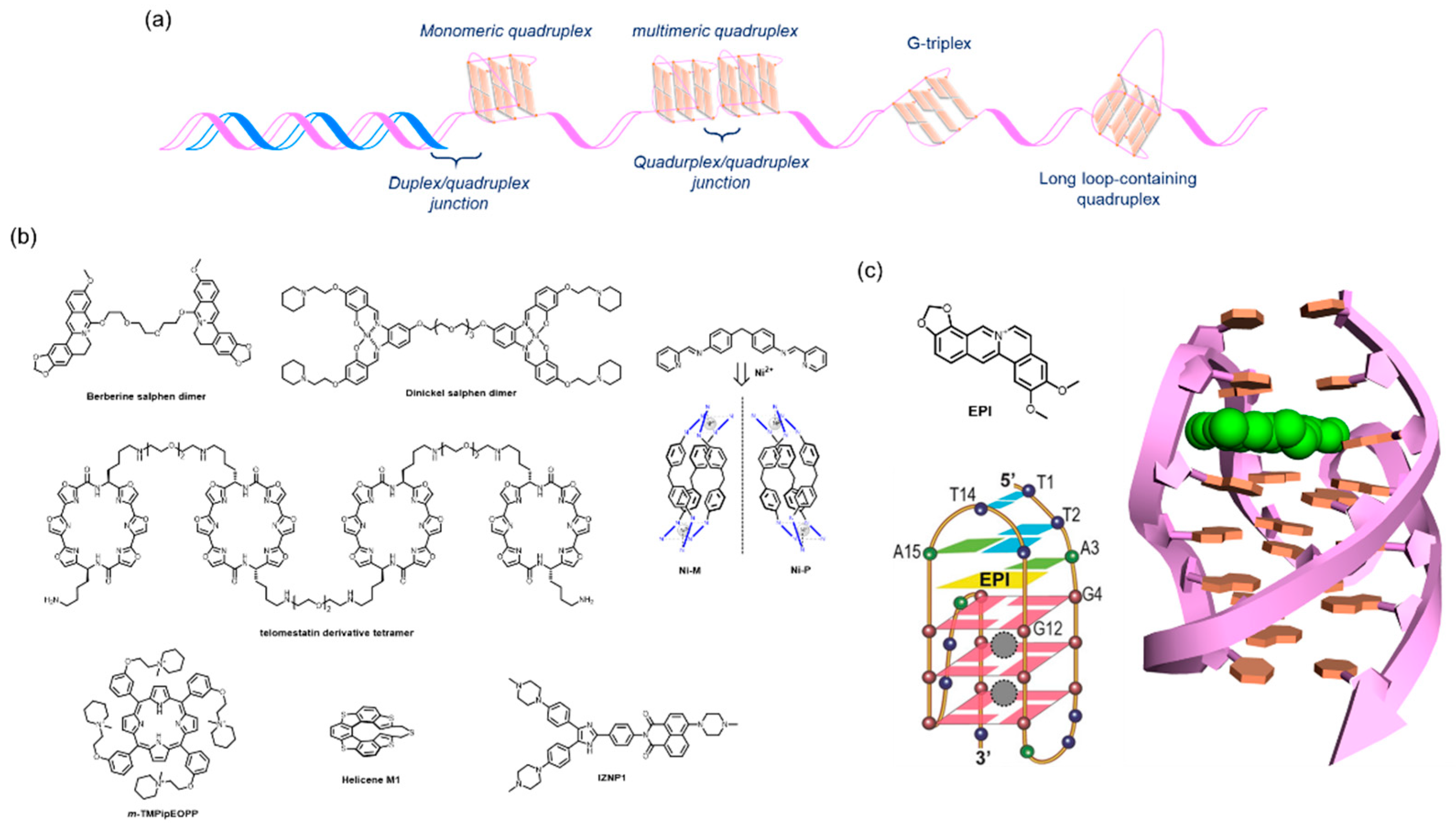{kind=link}
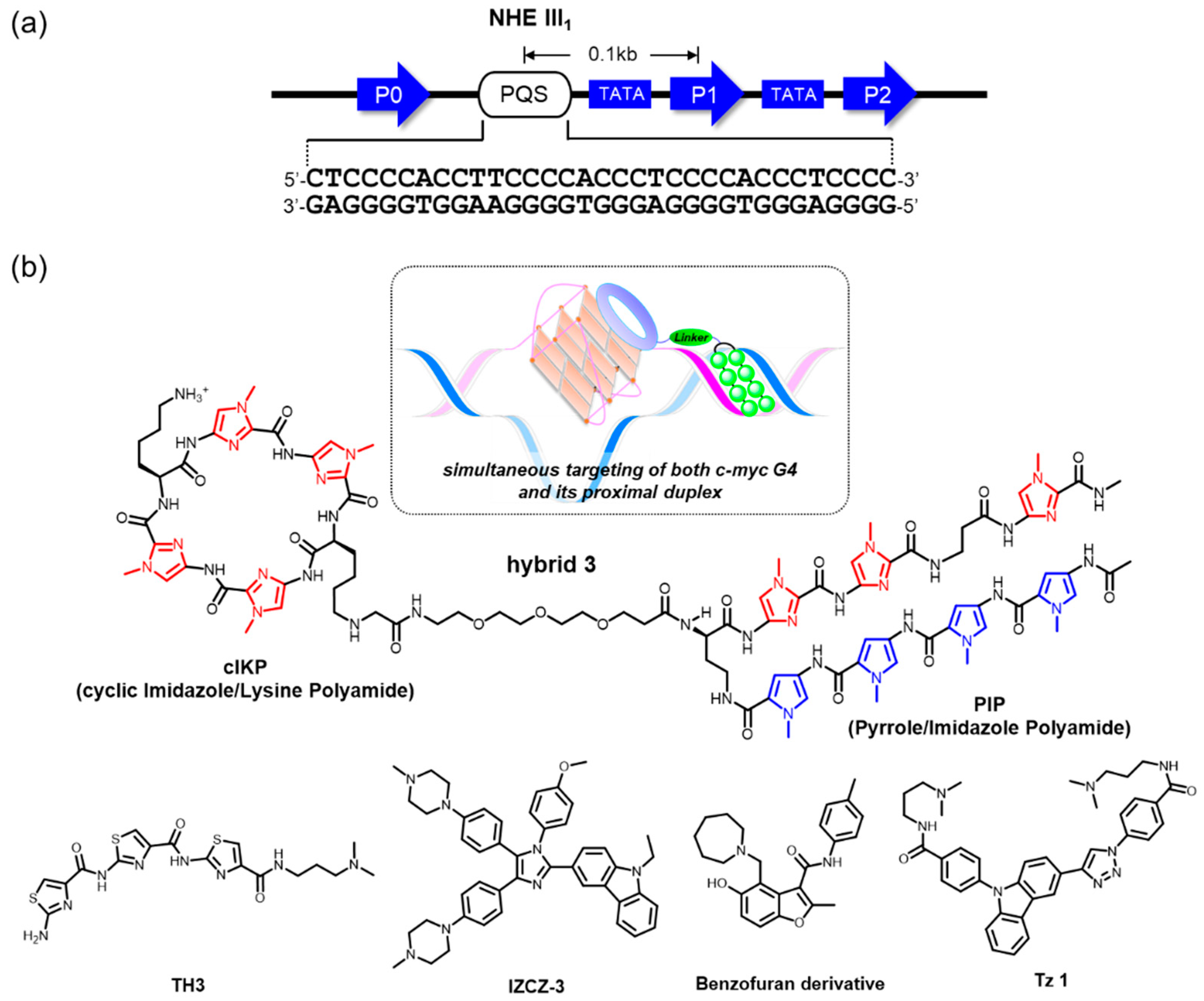{kind=link}
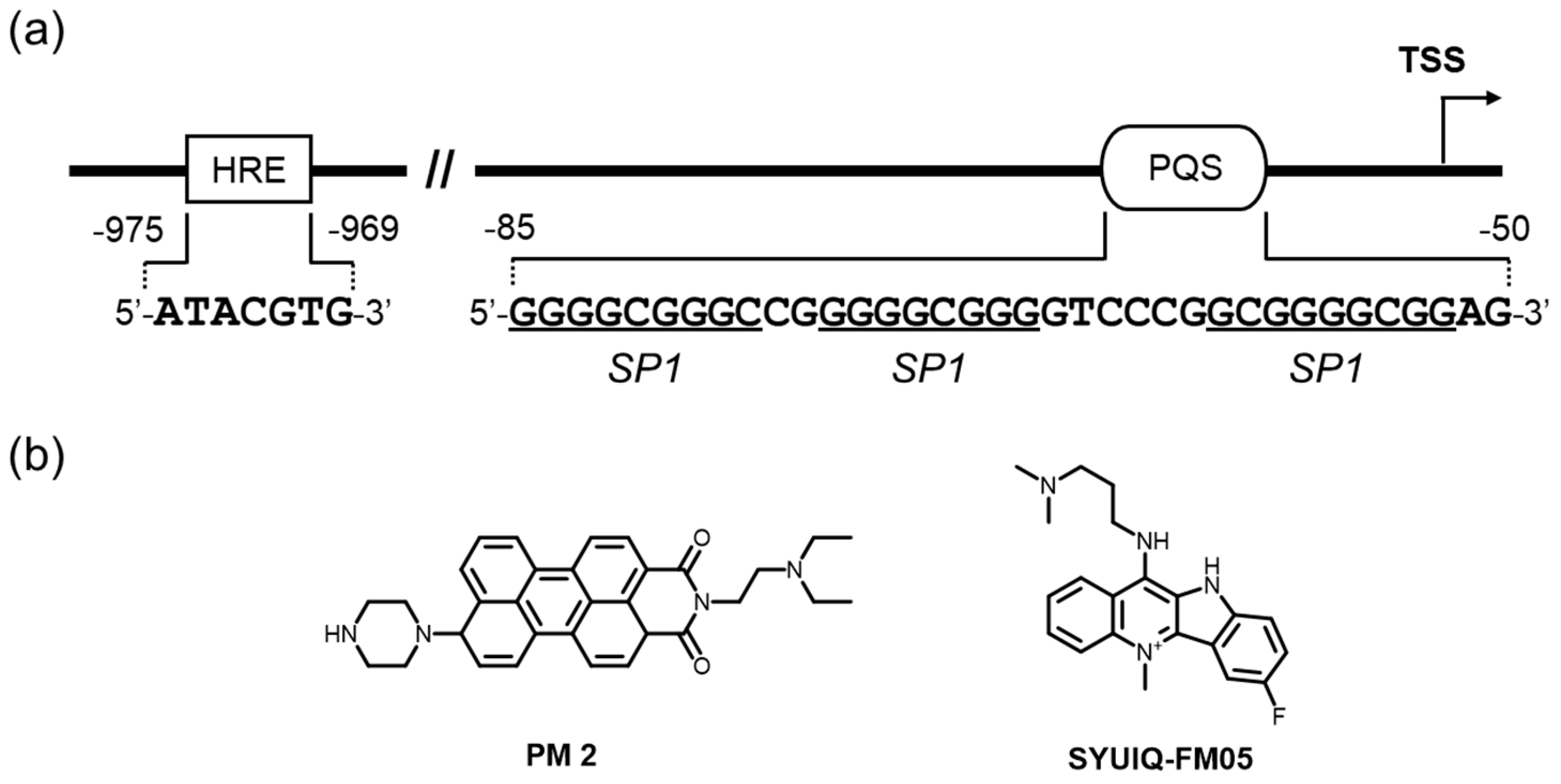{kind=link}
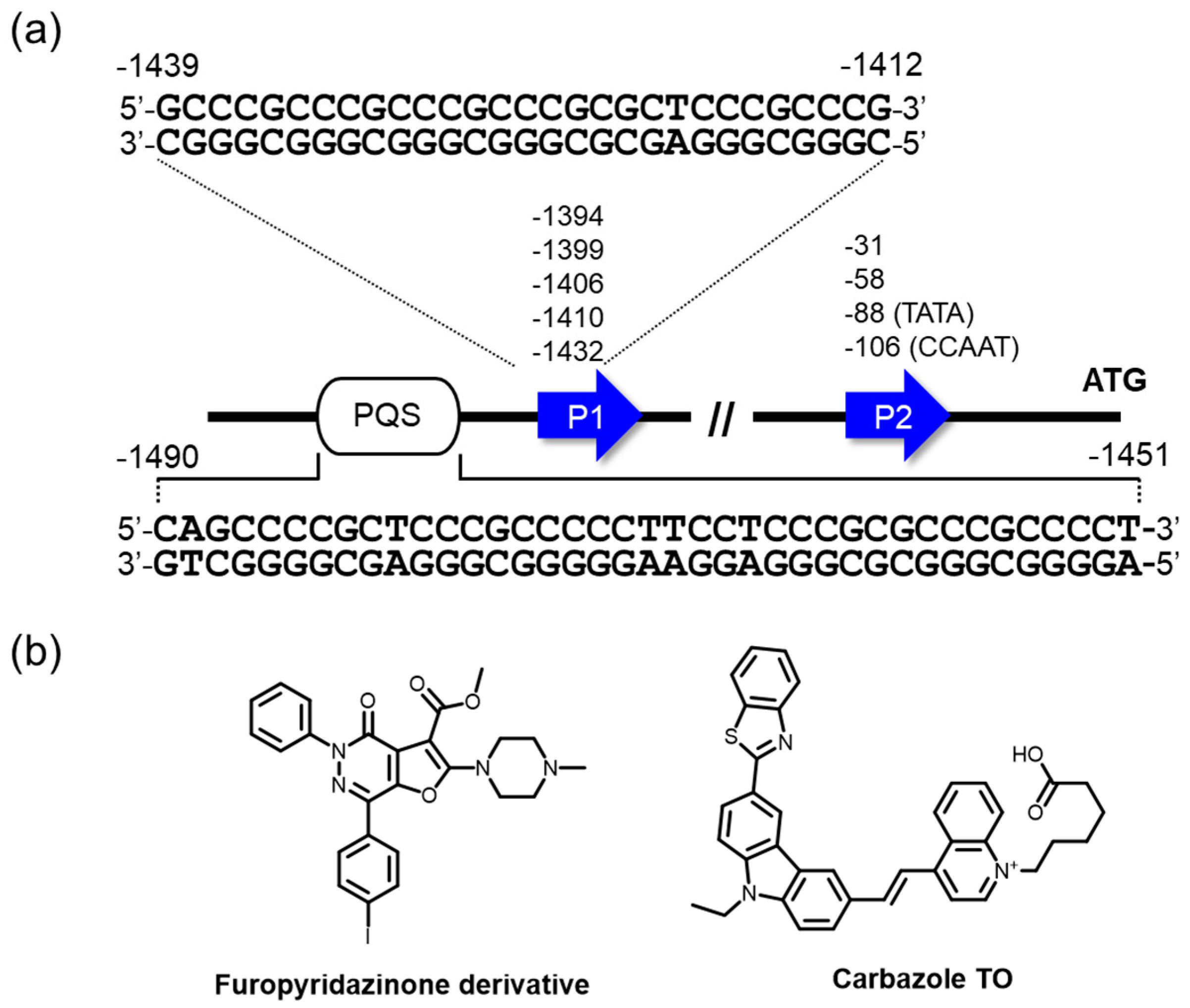{kind=link}
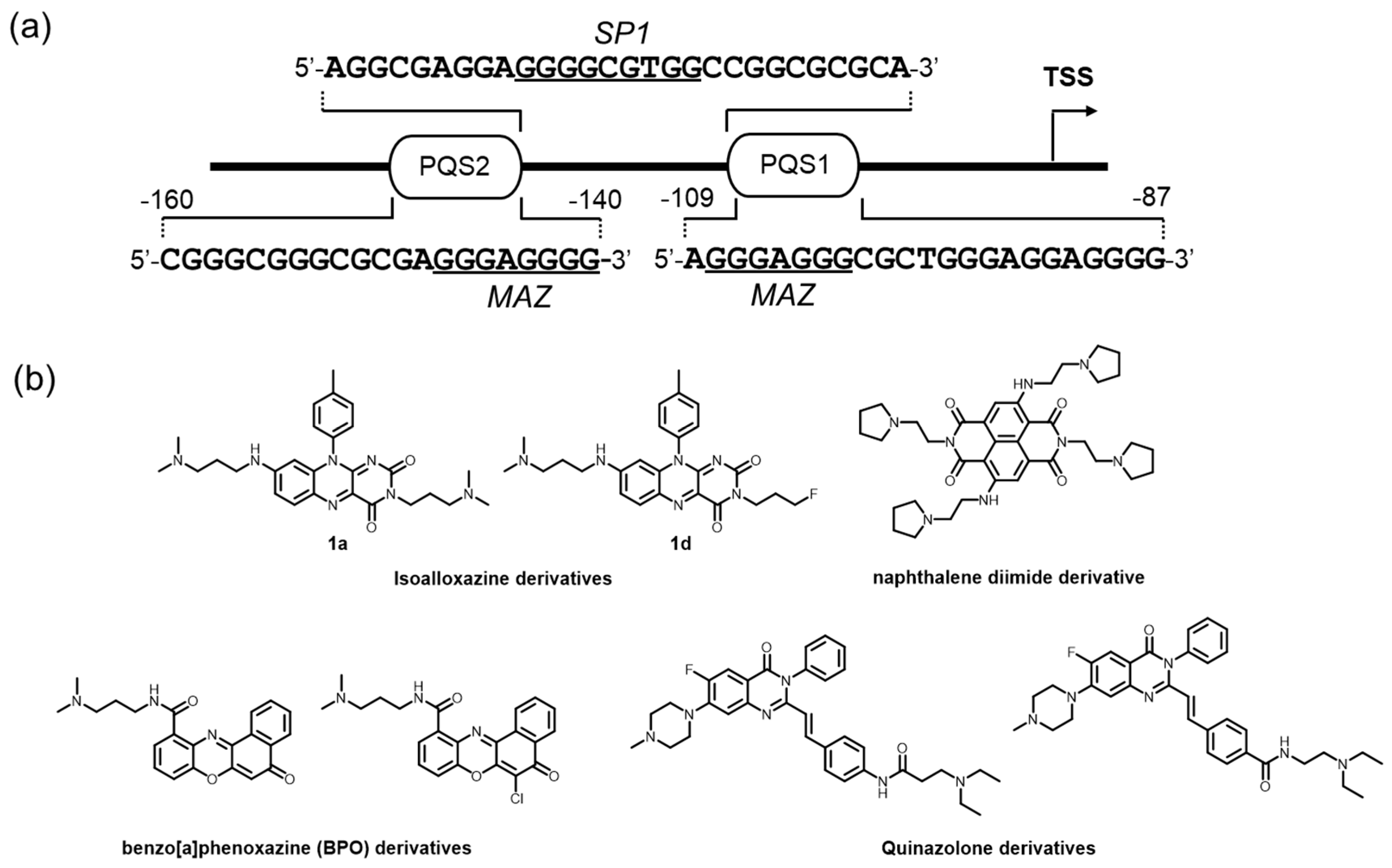{kind=link}
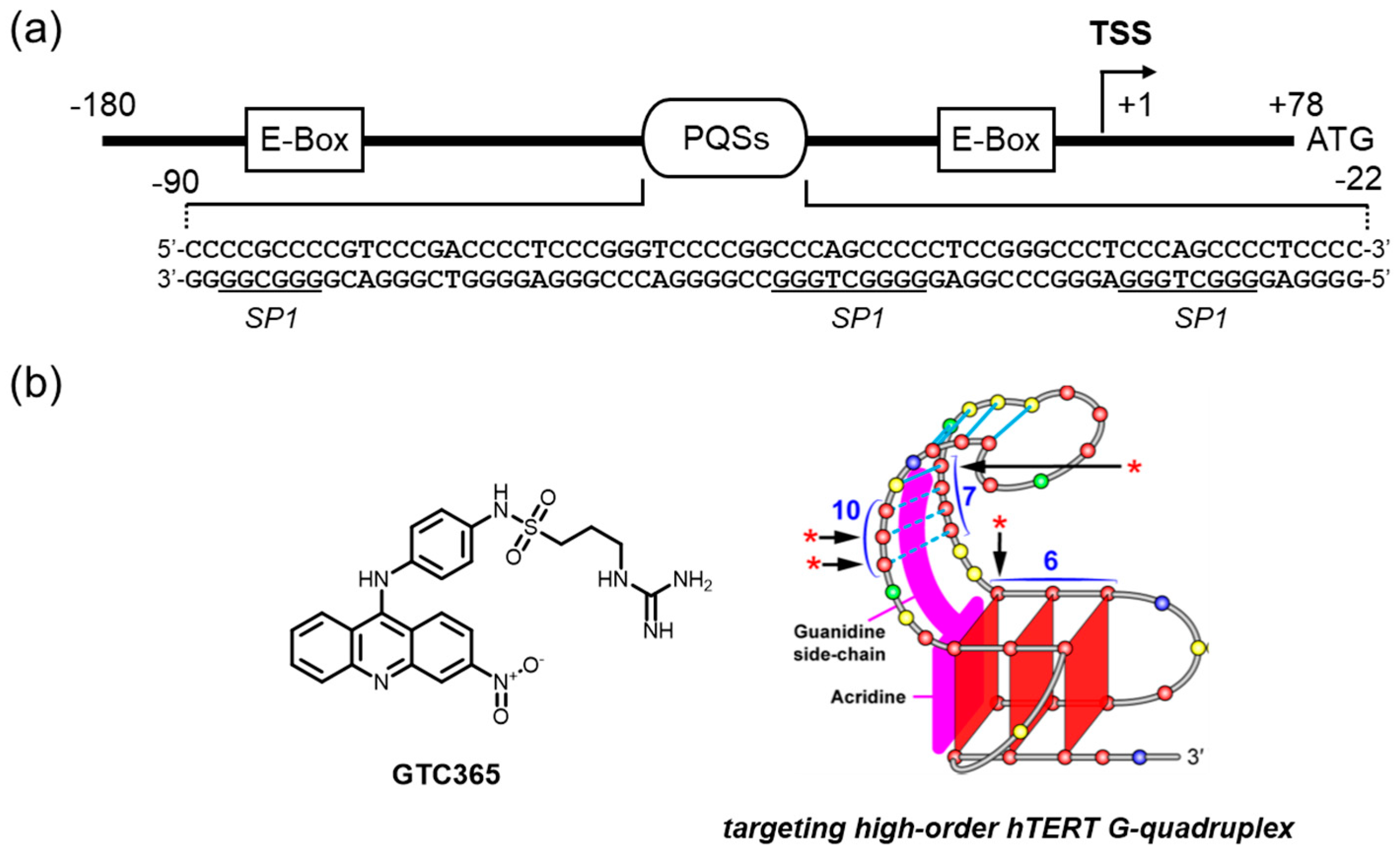{kind=link}
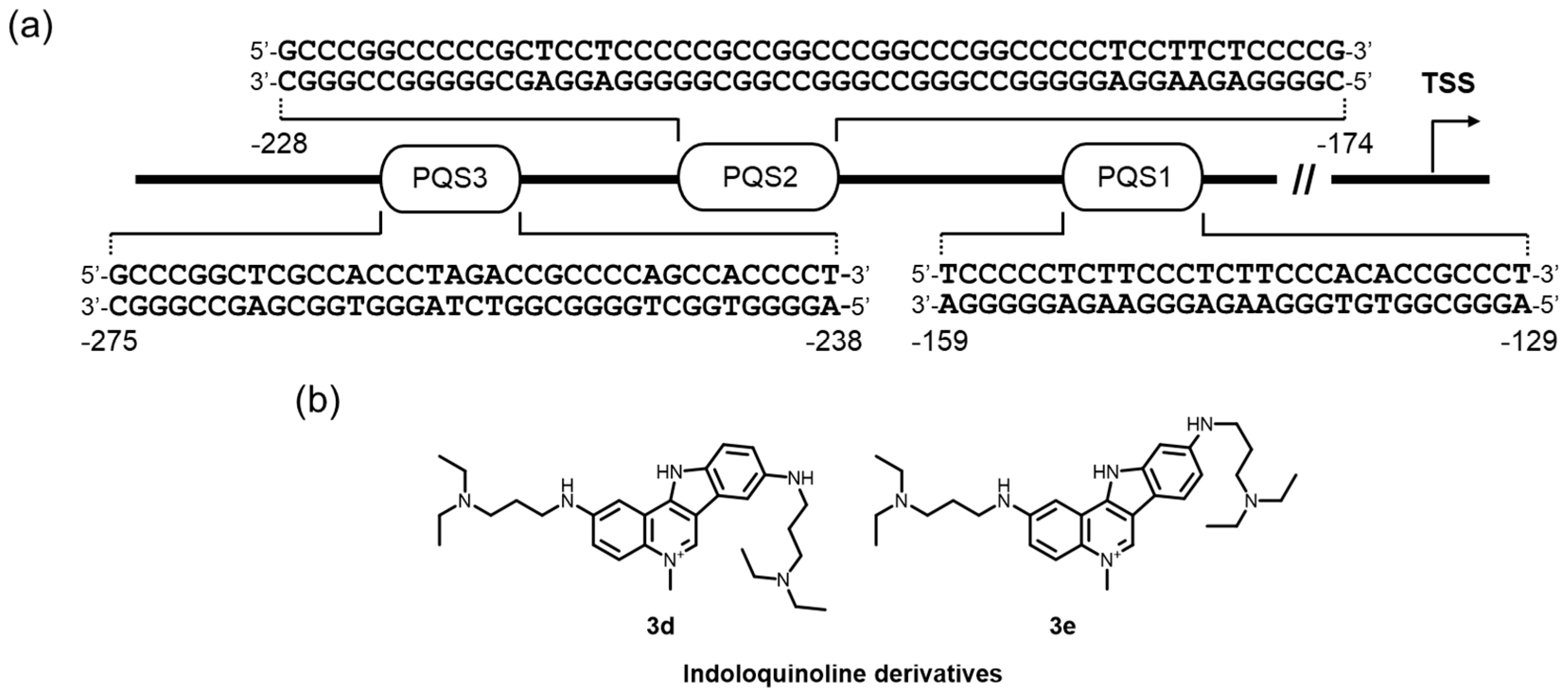{kind=link}
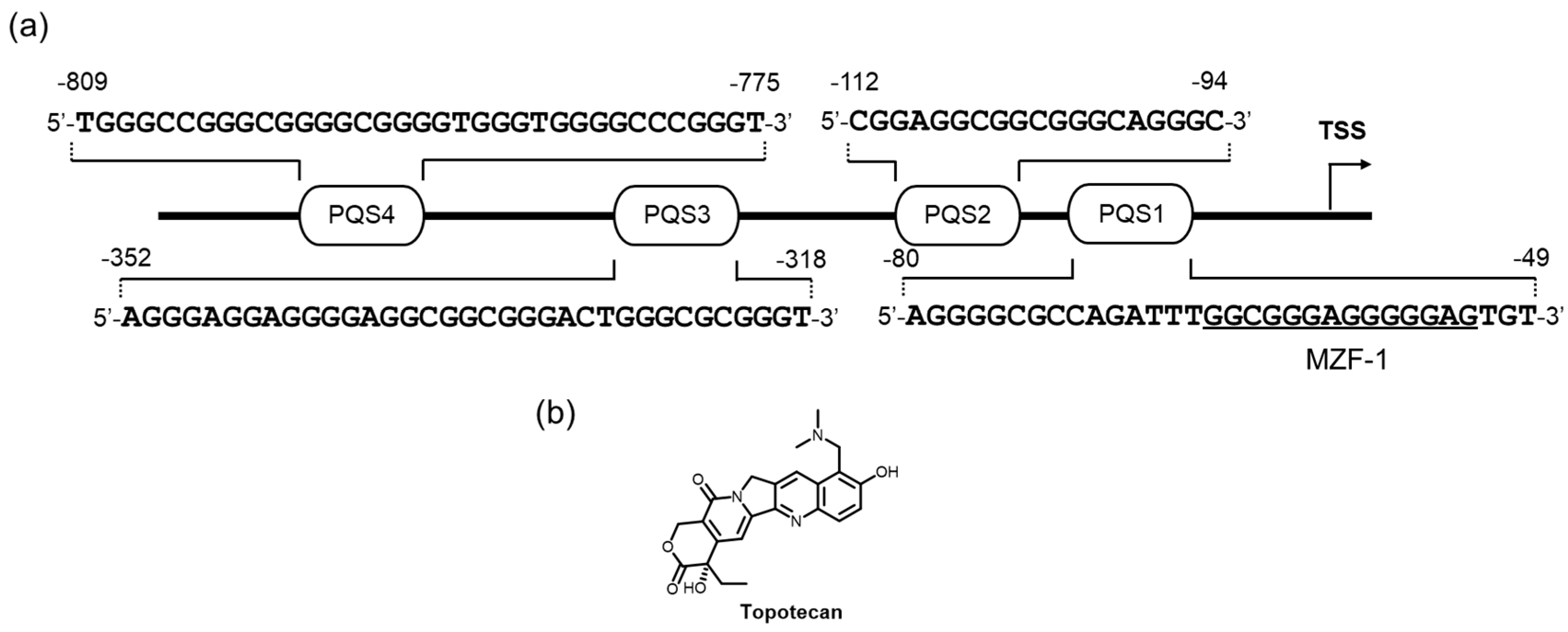{kind=link}
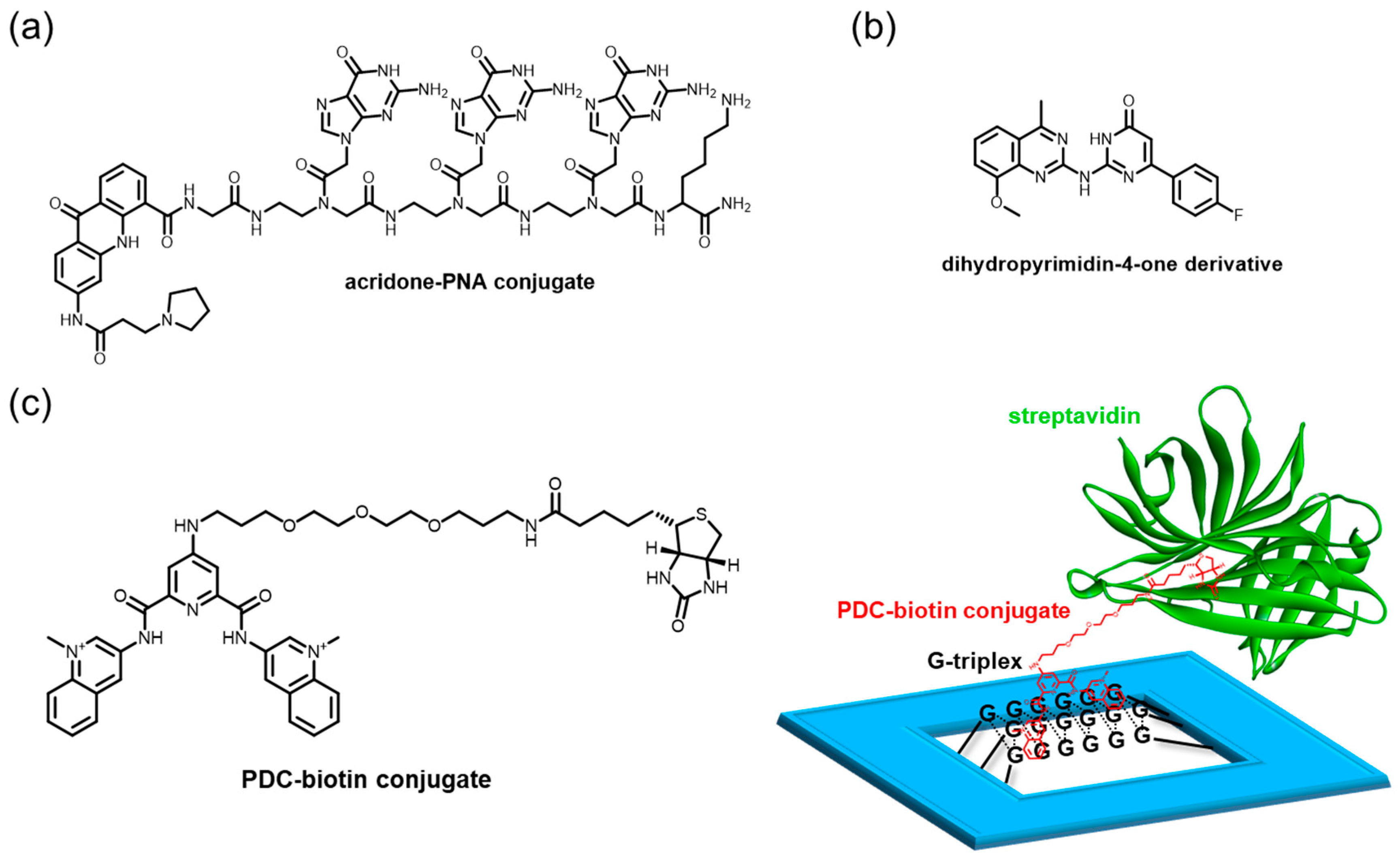{kind=link}
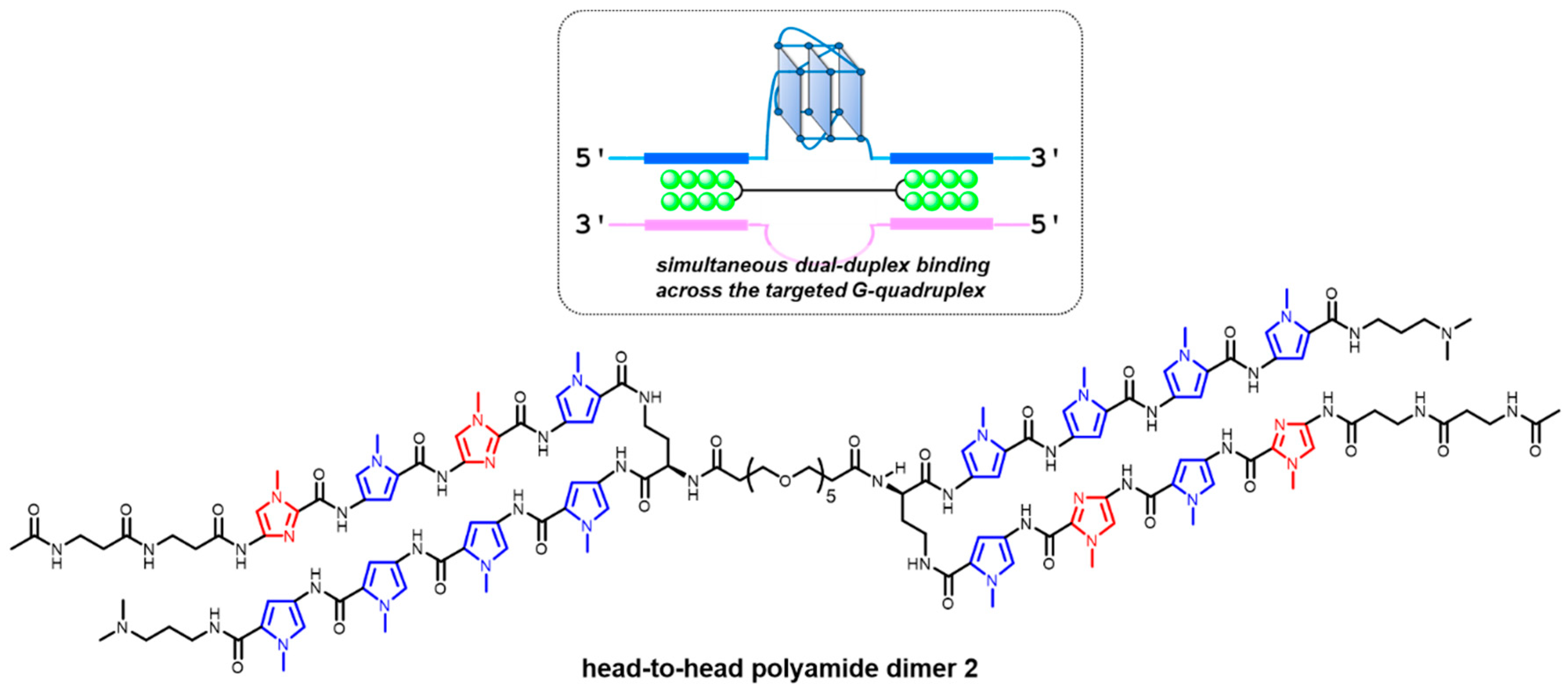{kind=link}
Table 1.
Summary of targeted G-quadruplex (G4)-preferred ligands that exhibit antitumor activities.
| Ligand a | Target G4 or Gene b | Preferred Target Structure c | Cell Line d | Tumor Type | Effect | Notes | References |
|---|---|---|---|---|---|---|---|
| Ni-P | telomere | hybrid | MDA-MB-231 | breast cancer (adenocarcinoma) | · Caner stem cell-specific apoptosis · Bulk cancer-specific apoptosis and senescence · Negligible cytotoxicity to normal somatic cells | · Tumor growth suppression in a MDA-MB-231 xenograft model vivo | [82,83] |
| MCF-7 | breast cancer (adenocarcinoma) | ||||||
| IZNP1 | telomere | dimeric G4s | SiHa | squamous cell carcinoma | · Apoptosis · Senescence | ·Telomere dysfunction (DNA damage and telomere uncapping) | [85] |
| TH3 | c-MYC | c-MYC (parallel) | A549 | lung cancer | · Antiproliferative effect (apoptosis) · Negligible cytotoxicity to normal somatic cells | · Validation of the minimal effects for a G4-driven gene (BCL2) | [97] |
| Hela | cervical cancer | ||||||
| IZCZ-3 | c-MYC | c-MYC (parallel) | SiHa | squamous cell carcinoma | · Antiproliferative effect (apoptosis) · Negligible cytotoxicity to normal somatic cells | · Validation of c-MYC G4-dependent gene suppression · Tumor growth suppression in a SiHa xenograft model in vivo | [98] |
| Hela | cervical cancer | ||||||
| Huh7 | liver cancer | ||||||
| A375 | malignant melanoma | ||||||
| Benzofuran derivative | c-MYC | c-MYC (parallel) | L363 MM1S MM1R etc. | myeloma | · Antiproliferative effect (apoptosis) · Negligible cytotoxicity to normal cells | · Validation of the minimal effects for other G4-driven genes | [102] |
| Tz 1 | c-MYC | c-MYC (parallel) | HCT116 | colorectal carcinoma | · Apoptosis | · Validation of c-MYC G4-dependent gene suppression | [103] |
| Furopyridazinone derivative | BCL2 | BCL2 (hybrid) | Jurkat | human acute T cell leukemia | · Antiproliferative effect (apoptosis) · Negligible cytotoxicity to normal cells | - | [135] |
| GTC365 | hTERT | hTERT (stem-loop-containing hybrid) | MCF-7 | breast cancer (adenocarcinoma) | · Apoptosis · Senescence | · Validation of decreased telomerase activity and telomere length | [172] |
a This column describes G-quadruplex (G4)-interacting ligands that were reported to exhibit antitumor activities likely based on the clear selectivity to target G4s. b This column a G4-related genomic structure (telomere) or G4-driven genes the were intended to be targeted by each ligand. c This column describes G4 structures or topologies that were preferred by each ligand among other G4s examined in the respective papers. d This column describes cell lines in which antitumor activities of ligands were examined.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 2019, 24, 429. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030429
AMA Style
Asamitsu S, Obata S, Yu Z, Bando T, Sugiyama H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules. 2019; 24(3):429. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030429
Chicago/Turabian StyleAsamitsu, Sefan, Shunsuke Obata, Zutao Yu, Toshikazu Bando, and Hiroshi Sugiyama. 2019. "Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy" Molecules 24, no. 3: 429. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24030429