
Prediction of Disease-related microRNAs through Integrating Attributes of microRNA Nodes and Multiple Kinds of Connecting Edges

Abstract
:1. Introduction
2. Results and Discussion
2.1. Evaluation Metrics
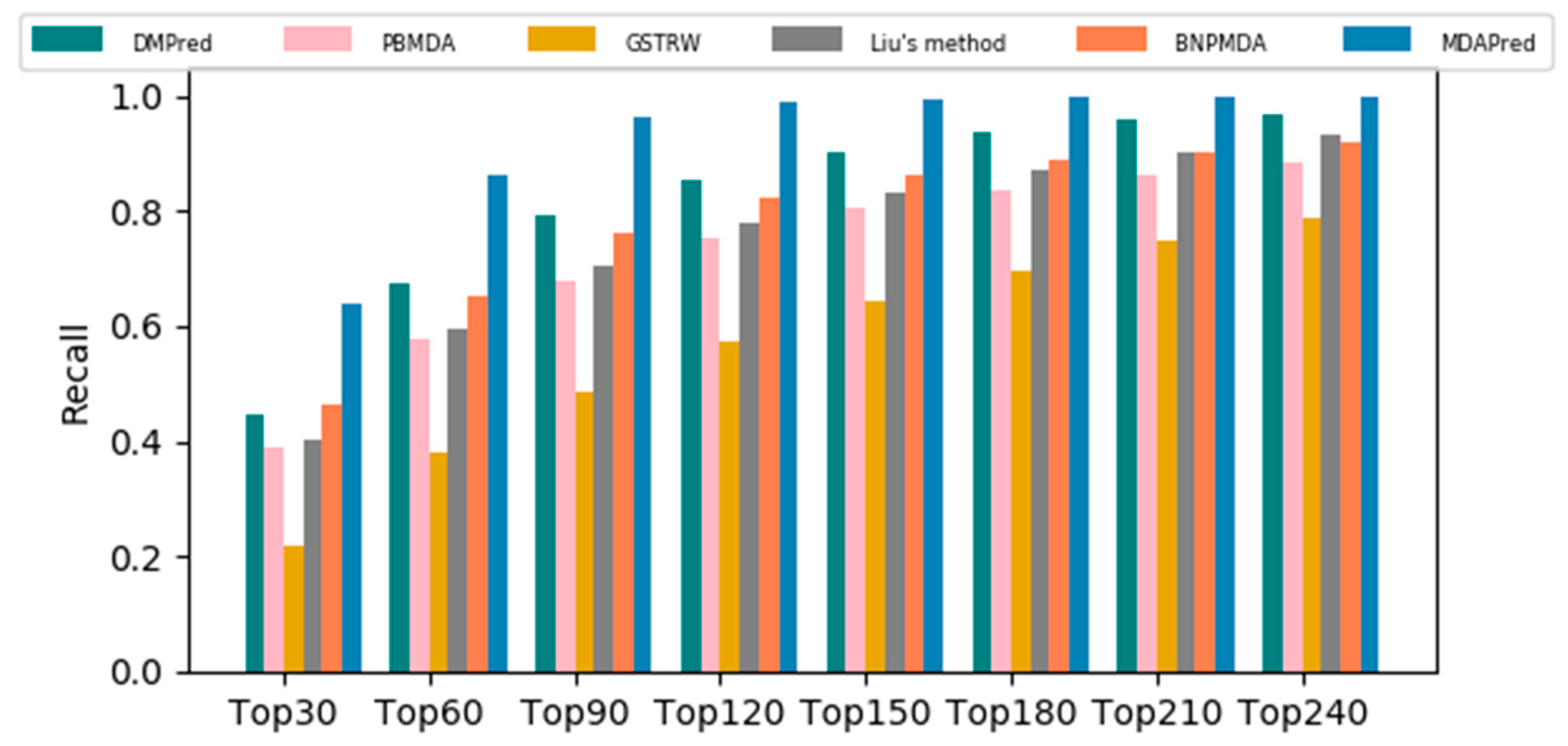
2.2. Comparison with Other Methods
2.3. Case Studies
3. Materials and Methods
3.1. Data Representation of miRNAs and Diseases
3.2. Prediction Models for Disease–miRNA Associations
3.3. Optimization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ambros, V.R. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.R. microRNAs: Tiny Regulators with Great Potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Guo, M.; Liu, X.; Wang, C.; Liu, Y.; Liu, G. Identify bilayer modules via pseudo-3D clustering: applications to miRNA-gene bilayer networks. Nucl. Acids Res. 2016, 44, e152. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA-Cancer Connection: The Beginning of a New Tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004, 20, 617–624. [Google Scholar] [CrossRef]
- Cheng, A.M.; Byrom, M.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucl. Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Fernando, T.R.; Rodriguezmalave, N.I.; Rao, D.S. MicroRNAs in B cell development and malignancy. J. Hematol. Oncol. 2012, 5, 7. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.; Jonesrhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Q.; Zheng, Y.; Lv, S.; Ning, S.; Sun, J.; Huang, T.; Zheng, Q.; Ren, H.; Xu, J. Prioritizing human cancer microRNAs based on genes’ functional consistency between microRNA and cancer. Nucl. Acids Res. 2011, 39, e153. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L. LRSSLMDA: Laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005912. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z.-H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2019, 20, 515–539. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and between score for MiRNA-disease association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef]
- Li, J.-Q.; Rong, Z.-H.; Chen, X.; Yan, G.-Y.; You, Z.-H. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.-N.; Li, J.-Q. Predicting miRNA–disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.; Yan, G. RWRMDA: Predicting novel human microRNA–disease associations. Mol. BioSyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, Y.; Li, J.; Li, X.; Zhong, Y.; Zhang, Z.; Ding, J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 2015, 31, 1805–1815. [Google Scholar] [CrossRef]
- Chen, M.; Liao, B.; Li, Z. Global Similarity Method Based on a Two-tier Random Walk for the Prediction of microRNA–Disease Association. Sci. Rep. 2018, 8, 6481. [Google Scholar] [CrossRef]
- Zhong, Y.; Xuan, P.; Wang, X.; Zhang, T.; Li, J.; Liu, Y.; Zhang, W. A non-negative matrix factorization based method for predicting disease-associated miRNAs in miRNA-disease bilayer network. Bioinformatics 2018, 34, 267–277. [Google Scholar] [CrossRef]
- Tan, V.Y.F.; Fevotte, C. Automatic Relevance Determination in Nonnegative Matrix Factorization with the $(\beta)$-Divergence. IEEE Trans. Pattern Anal. Machine Intellig. 2013, 35, 1592–1605. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L.; Xie, D.; Zhao, Q. EGBMMDA: Extreme gradient boosting machine for MiRNA-disease association prediction. Cell Death Disease 2018, 9, 3. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Yin, J. Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics 2019, 1, 9. [Google Scholar]
- Wang, L.; You, Z.-H.; Chen, X.; Li, Y.-M.; Dong, Y.-N.; Li, L.-P.; Zheng, K. LMTRDA: Using logistic model tree to predict MiRNA-disease associations by fusing multi-source information of sequences and similarities. PLoS Comput. Biol. 2019, 15, e1006865. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, Z.; Zhao, Y. ELLPMDA: Ensemble learning and link prediction for miRNA-disease association prediction. RNA Biol. 2018, 15, 807–818. [Google Scholar] [CrossRef]
- You, Z.; Huang, Z.; Zhu, Z.; Yan, G.; Li, Z.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. [Google Scholar] [CrossRef]
- Xuan, P.; Shen, T.; Wang, X.; Zhang, T.; Zhang, W. Inferring disease-associated microRNAs in heterogeneous networks with node attributes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 20, 1. [Google Scholar] [CrossRef]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffithsjones, S.; Eddy, S.R. Rfam: Updates to the RNA families database. Nucl. Acids Res. 2009, 37, 136–140. [Google Scholar] [CrossRef]
- Gu, C.; Liao, B.; Li, X.; Li, K. Network consistency projection for human miRNA-disease associations inference. Sci. Rep. 2016, 6, 36054. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef]
- Saito, T.; Rehmsmeier, M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS ONE 2015, 10, e0118432. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.; Liu, H. BNPMDA: Bipartite Network Projection for MiRNA–Disease Association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, X.; He, Z.; Zou, Q. Inferring MicroRNA-Disease Associations by Random Walk on a Heterogeneous Network with Multiple Data Sources. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 905–915. [Google Scholar] [CrossRef]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA–cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11, 1–8. [Google Scholar] [CrossRef]
- Nygren, M.K.; Tekle, C.; Ingebrigtsen, V.; Mäkelä, R.; Krohn, M.; Aure, M.; Nunes-Xavier, C.; Perälä, M.; Tramm, T.; Alsner, J. Identifying microRNAs regulating B7-H3 in breast cancer: The clinical impact of microRNA-29c. Br. J. Cancer 2014, 110, 2072. [Google Scholar] [CrossRef]
- Cilek, E.E.; Ozturk, H.; Dedeoglu, B.G. Construction of miRNA-miRNA networks revealing the complexity of miRNA-mediated mechanisms in trastuzumab treated breast cancer cell lines. PLoS ONE 2017, 12, e0185558. [Google Scholar] [CrossRef]
- Chaluvally-Raghavan, P.; Zhang, F.; Pradeep, S.; Hamilton, M.P.; Zhao, X.; Rupaimoole, R.; Moss, T.; Lu, Y.; Yu, S.; Pecot, C.V. Copy number gain of hsa-miR-569 at 3q26. 2 leads to loss of TP53INP1 and aggressiveness of epithelial cancers. Cancer Cell 2014, 26, 863–879. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, D.; Xu, C.; Zhu, G.; Wu, Z.; Wu, Q. Differential expression profile analysis of miRNAs with HER-2 overexpression and intervention in breast cancer cells. Int. J. Clin. Exp. Pathol. 2017, 10, 5039–5062. [Google Scholar]
- Chu, J.; Zhu, Y.; Liu, Y.; Sun, L.; Lv, X.; Wu, Y.; Hu, P.; Su, F.; Gong, C.; Song, E. E2F7 overexpression leads to tamoxifen resistance in breast cancer cells by competing with E2F1 at miR-15a/16 promoter. Oncotarget 2015, 6, 31944. [Google Scholar] [CrossRef]
- Ruepp, A.; Kowarsch, A.; Schmidl, D.; Buggenthin, F.; Brauner, B.; Dunger, I.; Fobo, G.; Frishman, G.; Montrone, C.; Theis, F.J. PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010, 11, 1–11. [Google Scholar] [CrossRef]
- Lee, T.; Shim, S.; Yu, U.; Park, H.O. Pharmaceutical Composition for Treating Cancer Comprising Microrna as Active Ingredient. WO 2016/137235, 1 September 2016. [Google Scholar]
- Park, J.; Jeong, S.; Park, K.; Yang, K.; Shin, S. Expression profile of microRNAs following bone marrow-derived mesenchymal stem cell treatment in lipopolysaccharide-induced acute lung injury. Exp. Therap. Med. 2018, 15, 5495–5502. [Google Scholar] [CrossRef]
- Sun, C.; Li, S.; Zhang, F.; Xi, Y.; Wang, L.; Bi, Y.; Li, D. Long non-coding RNA NEAT1 promotes non-small cell lung cancer progression through regulation of miR-377-3p-E2F3 pathway. Oncotarget 2016, 7, 51784. [Google Scholar] [CrossRef]
- Koyama, N.; Ishikawa, Y.; Iwai, Y.; Aoshiba, K.; Nakamura, H.; Hagiwara, K. Mutual Negative Regulation of EZH2 and miR-4448 for Tumor Progression via Epithelial Mesenchymal Transition in Small Cell Lung Cancer; AACR: Atlanta, GA, USA, 2019. [Google Scholar]
- Guo, H.; Chen, J.; Meng, F. Identification of novel diagnosis biomarkers for lung adenocarcinoma from the cancer genome atlas. Orig. Artic 2016, 9, 7908–7918. [Google Scholar]
- Zhou, R.; Zhou, X.; Yin, Z.; Guo, J.; Hu, T.; Jiang, S.; Liu, L.; Dong, X.; Zhang, S.; Wu, G. Tumor invasion and metastasis regulated by microRNA-184 and microRNA-574-5p in small-cell lung cancer. Oncotarget 2015, 6, 44609. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucl. Acids Res. 2014, 42, 1070–1074. [Google Scholar] [CrossRef]
- Hoehndorf, R.; Schofield, P.N.; Gkoutos, G.V. Analysis of the human diseasome using phenotype similarity between common, genetic, and infectious diseases. Sci. Rep. 2015, 5, 10888. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucl. Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name | AUC | |||||
|---|---|---|---|---|---|---|
| MDAPred | DMPred | PBMDA | GSTRW | Liu’s Method | BNPMDA | |
| Breast neoplasms | 0.986 | 0.974 | 0.906 | 0.837 | 0.920 | 0.902 |
| Hepatocellular carcinoma | 0.982 | 0.931 | 0.910 | 0.791 | 0.929 | 0.900 |
| Glioma | 0.957 | 0.855 | 0.882 | 0.786 | 0.914 | 0.843 |
| Acute myeloid leukemia | 0.979 | 0.963 | 0.885 | 0.796 | 0.910 | 0.865 |
| Lung neoplasms | 0.964 | 0.944 | 0.862 | 0.813 | 0.906 | 0.855 |
| Melanoma | 0.978 | 0.910 | 0.849 | 0.758 | 0.893 | 0.839 |
| Osteosarcoma | 0.968 | 0.985 | 0.860 | 0.771 | 0.897 | 0.859 |
| Ovarian neoplasms | 0.970 | 0.967 | 0.888 | 0.844 | 0.918 | 0.877 |
| Pancreatic neoplasms | 0.956 | 0.821 | 0.879 | 0.833 | 0.902 | 0.870 |
| Alzheimer Disease | 0.968 | 0.958 | 0.833 | 0.816 | 0.875 | 0.830 |
| Carcinoma, Renal Cell | 0.964 | 0.894 | 0.856 | 0.784 | 0.900 | 0.854 |
| Diabetes Mellitus, Type 2 | 0.964 | 0.936 | 0.870 | 0.870 | 0.905 | 0.869 |
| Glioblastoma | 0.938 | 0.951 | 0.849 | 0.759 | 0.889 | 0.843 |
| Heart failure | 0.962 | 0.959 | 0.884 | 0.814 | 0.909 | 0.882 |
| Atherosclerosis | 0.962 | 0.955 | 0.891 | 0.822 | 0.910 | 0.876 |
| Average AUC | 0.964 | 0.933 | 0.873 | 0.806 | 0.904 | 0.839 |
| Disease Name | AUPR | |||||
|---|---|---|---|---|---|---|
| MDAPred | DMPred | PBMDA | GSTRW | Liu’s Method | BNPMDA | |
| Breast neoplasms | 0.818 | 0.800 | 0.718 | 0.389 | 0.725 | 0.566 |
| Hepatocellular carcinoma | 0.816 | 0.715 | 0.767 | 0.483 | 0.749 | 0.676 |
| Glioma | 0.613 | 0.175 | 0.390 | 0.224 | 0.436 | 0.386 |
| Acute myeloid leukemia | 0.544 | 0.466 | 0.386 | 0.122 | 0.408 | 0.324 |
| Lung neoplasms | 0.686 | 0.620 | 0.561 | 0.370 | 0.596 | 0.542 |
| Melanoma | 0.689 | 0.366 | 0.482 | 0.205 | 0.524 | 0.491 |
| Osteosarcoma | 0.601 | 0.620 | 0.356 | 0.181 | 0.373 | 0.327 |
| Ovarian neoplasms | 0.714 | 0.366 | 0.529 | 0. 400 | 0.236 | 0.496 |
| Pancreatic neoplasms | 0.692 | 0.569 | 0.457 | 0.333 | 0.556 | 0.478 |
| Alzheimer Disease | 0.522 | 0.351 | 0.136 | 0.086 | 0.485 | 0.220 |
| Carcinoma, Renal Cell | 0.481 | 0.206 | 0.314 | 0.135 | 0.143 | 0.299 |
| Diabetes Mellitus, Type 2 | 0.549 | 0.398 | 0.259 | 0.132 | 0.356 | 0.268 |
| Glioblastoma | 0.533 | 0.284 | 0.346 | 0.161 | 0.303 | 0.336 |
| Heart failure | 0.599 | 0.393 | 0.301 | 0.134 | 0.348 | 0.300 |
| Atherosclerosis | 0.315 | 0.309 | 0.304 | 0.084 | 0.297 | 0.218 |
| Average PR | 0.603 | 0.500 | 0.436 | 0.233 | 0.463 | 0.359 |
| p-Value between MDAPred and Other Methods | DMPred | PBMDA | GSTRW | Liu’s Method | BNPMDA |
|---|---|---|---|---|---|
| p-values of ROC curves | 2.4983 × 10−41 | 3.2311 × 10−5 | 6.3212 × 10−16 | 6.9812 × 10−8 | 2.9742 × 10−6 |
| p-values of PR curves | 2.2341 × 10−35 | 1.8643 × 10−6 | 1.6542 × 10−6 | 3.4521 × 10−5 | 8.8432 × 10−4 |
| Rank | MiRNA name | Evidence | Rank | MiRNA name | Description |
|---|---|---|---|---|---|
| 1 | hsa-mir-186 | dbDEMC, PhenomiR | 26 | hsa-mir-885 | literature [40] |
| 2 | hsa-mir-99b | dbDEMC, PhenomiR | 27 | hsa-mir-6838 | Unconfirmed |
| 3 | hsa-mir-483 | PhenomiR | 28 | hsa-mir-323a | dbDEMC, PhenomiR |
| 4 | hsa-mir-4480 | literature [41] | 29 | hsa-mir-1244 | dbDEMC |
| 5 | hsa-mir-181d | dbDEMC, PhenomiR, miRCancer | 30 | hsa-mir-361 | PhenomiR, miRCancer |
| 6 | hsa-mir-28 | dbDEMC, PhenomiR | 31 | hsa-mir-216a | dbDEMC, PhenomiR, miRCancer |
| 7 | hsa-mir-455 | PhenomiR, miRCancer | 32 | hsa-mir-136 | dbDEMC, PhenomiR |
| 8 | hsa-mir-154 | dbDEMC, PhenomiR, miRCancer | 33 | hsa-mir-569 | literature [42] |
| 9 | hsa-mir-330 | dbDEMC, PhenomiR, miRCancer | 34 | hsa-mir-336 | dbDEMC |
| 10 | hsa-mir-454 | dbDEMC, PhenomiR | 35 | hsa-mir-325 | dbDEMC, PhenomiR |
| 11 | hsa-mir-181 | dbDEMC, PhenomiR, miRCancer | 36 | hsa-mir-571 | dbDEMC |
| 12 | hsa-mir-208b | dbDEMC, PhenomiR | 37 | hsa-mir-95 | dbDEMC, PhenomiR |
| 13 | hsa-mir-663 | dbDEMC, PhenomiR | 38 | hsa-mir-517b | dbDEMC, PhenomiR, miRCancer |
| 14 | hsa-mir-133 | dbDEMC, PhenomiR, miRCancer | 39 | hsa-mir-323 | dbDEMC, PhenpmiR |
| 15 | hsa-mir-30 | dbDEMC, PhenomiR, miRCancer | 40 | hsa-mir-633 | dbDEMC |
| 16 | hsa-mir-504 | dbDEMC | 41 | hsa-mir-1183 | dbDEMC |
| 17 | hsa-mir-543 | dbDEMC | 42 | hsa-mir-4454 | literature [43] |
| 18 | hsa-mir-217 | dbDEMC, PhenomiR, miRCancer | 43 | hsa-mir-705 | dbDEMC |
| 19 | hsa-mir-33 | dbDEMC, PhenomiR, miRCancer | 44 | hsa-mir-532 | dbDEMC, PhenomiR |
| 20 | hsa-mir-211 | dbDEMC, PhenomiR, miRCancer | 45 | hsa-mir-126a | dbDEMC, miRCancer |
| 21 | hsa-mir-449b | dbDEMC, PhenomiR, miRCancer | 46 | hsa-mir-1909 | dbDEMC |
| 22 | hsa-mir-362 | miRCancer | 47 | hsa-mir-539 | dbDEMC, PhenomiR, miRCancer |
| 23 | hsa-mir-208 | dbDEMC, PhenomiR | 48 | hsa-mir-520f | PhenomiR, miRCancer |
| 24 | hsa-mir-433 | dbDEMC, PhenomiR, miRCancer | 49 | hsa-mir-498 | miRCancer |
| 25 | hsa-mir-520e | dbDEMC, PhenomiR, miRCancer | 50 | hsa-mir-3135b | literature [44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Li, L.; Zhang, T.; Zhang, Y.; Song, Y. Prediction of Disease-related microRNAs through Integrating Attributes of microRNA Nodes and Multiple Kinds of Connecting Edges. Molecules 2019, 24, 3099. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173099
Xuan P, Li L, Zhang T, Zhang Y, Song Y. Prediction of Disease-related microRNAs through Integrating Attributes of microRNA Nodes and Multiple Kinds of Connecting Edges. Molecules. 2019; 24(17):3099. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173099
Chicago/Turabian StyleXuan, Ping, Lingling Li, Tiangang Zhang, Yan Zhang, and Yingying Song. 2019. "Prediction of Disease-related microRNAs through Integrating Attributes of microRNA Nodes and Multiple Kinds of Connecting Edges" Molecules 24, no. 17: 3099. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173099