
Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs


Abstract
:1. Introduction
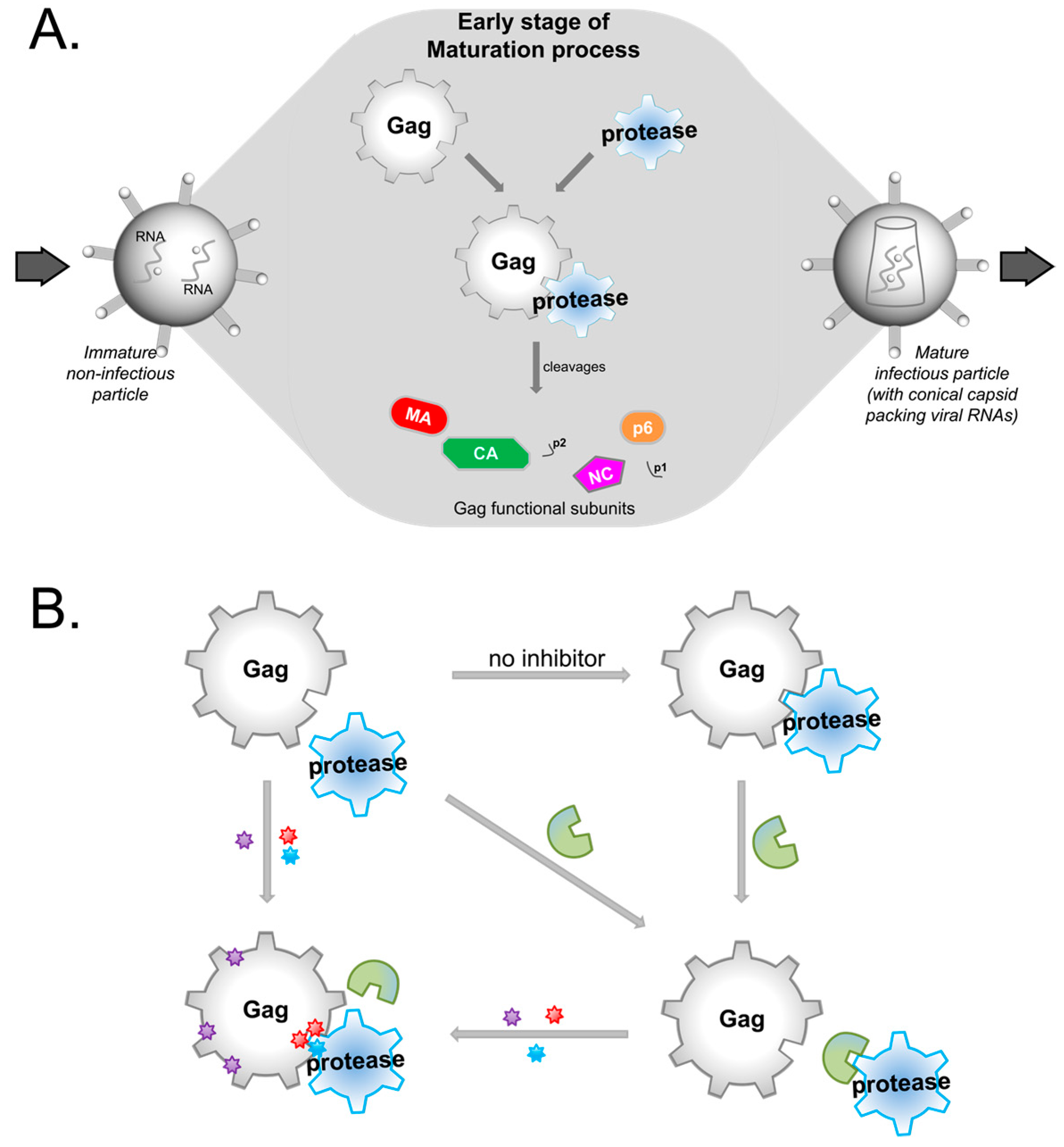
2. Possible Targets in Gag
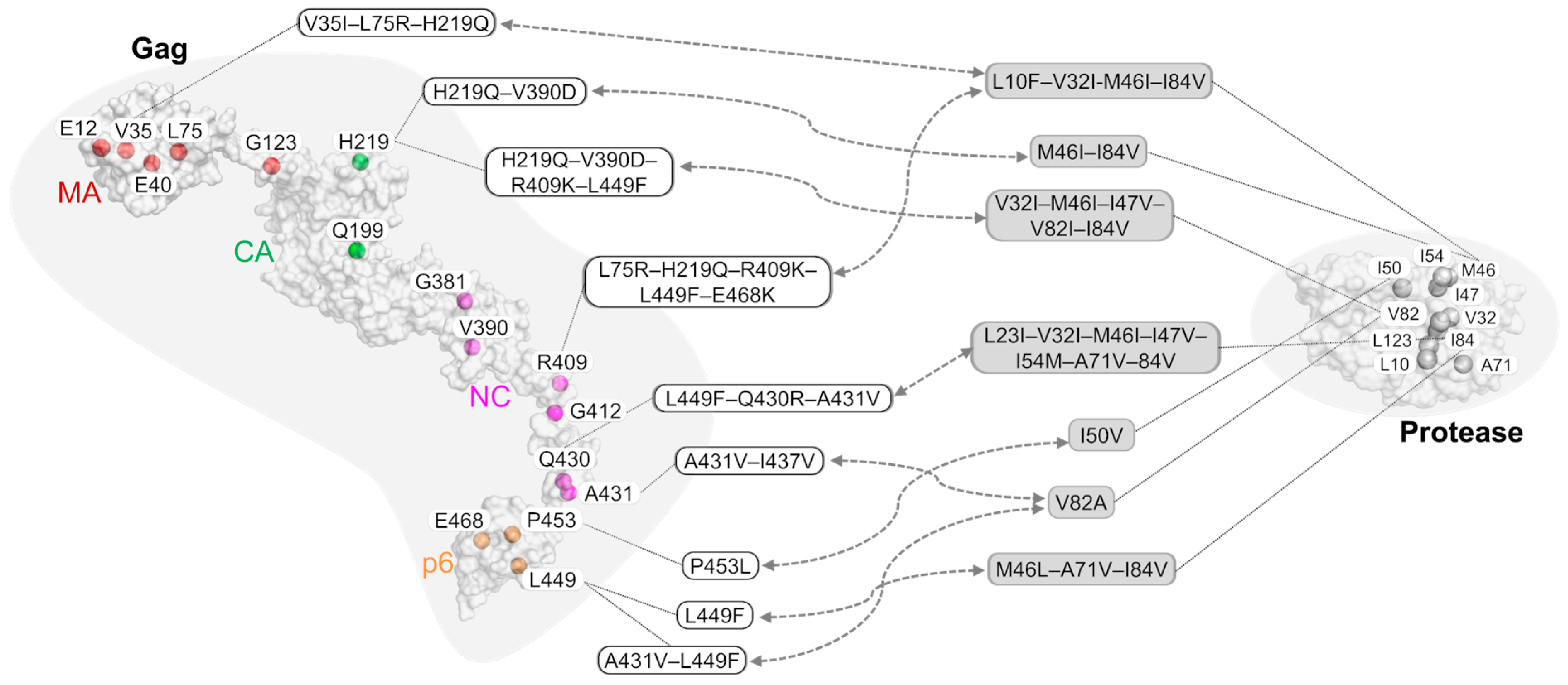
3. The Role of Gag Mutations in Restoring Gag–Protease Synergy in PI Resistance
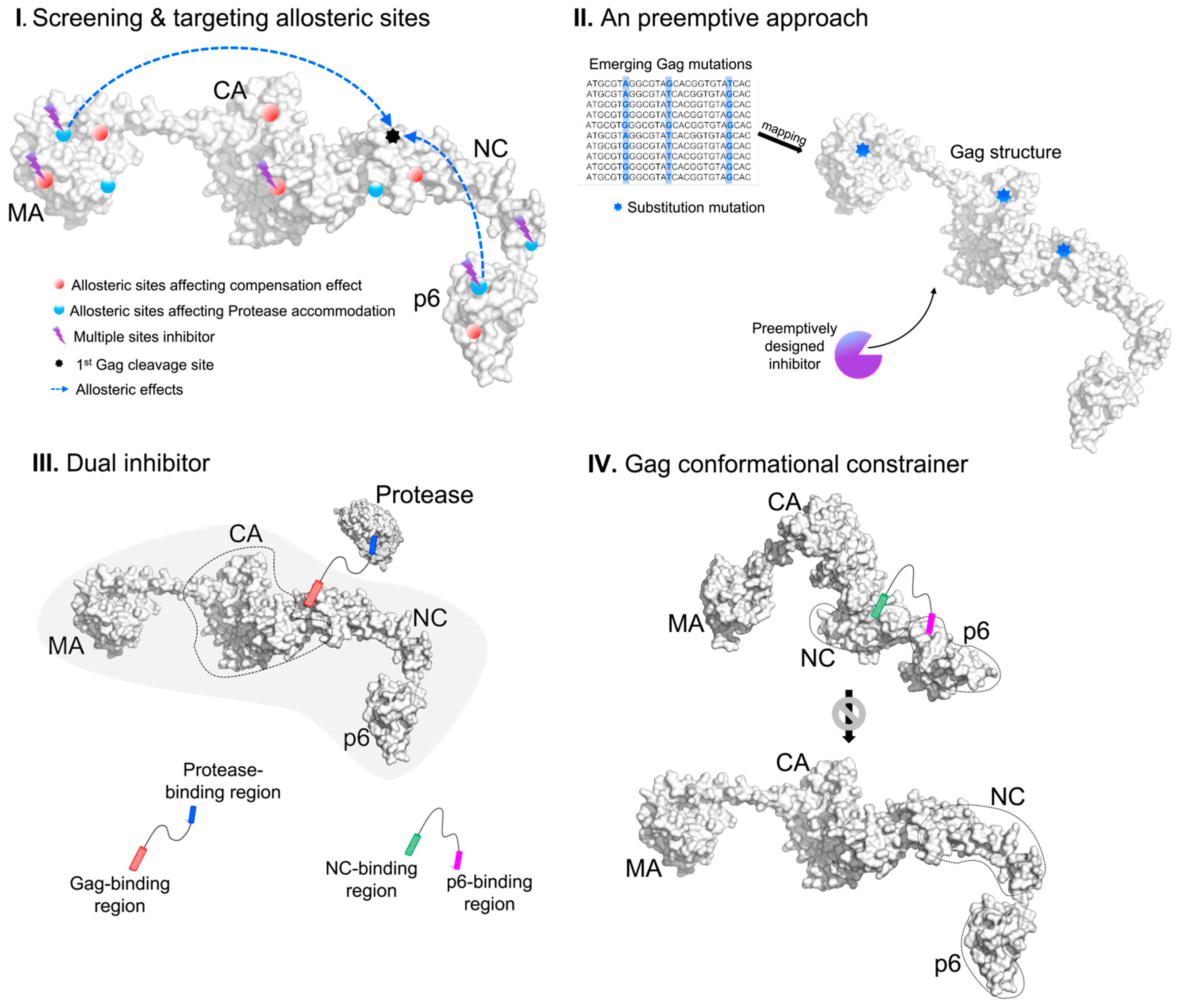
4. Conceptual Novel Designs of Gag Inhibitors
5. Conclusion and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Bhatti, A.B.; Usman, M.; Kandi, V. Current scenario of HIV/AIDS, treatment options, and major challenges with compliance to antiretroviral therapy. Cureus 2016, 8, e515. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Pannek, S.; Fichtenbaum, C. Emerging reverse transcriptase inhibitors for HIV-1 infrection. Expert Opin. Emerg. Drugs 2018, 23, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Orkin, C.; Llibre, J.; Gallien, S.; Antinori, A.; Behrens, G.; Carr, A. Nucleoside reverse transcriptase inhibitor-reducing strategies in HIV treatment: Assessing the evidence. HIV Med. 2017, 19, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Debyser, Z.; Vansant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV integration site selection provides a block-and-lock strategy for a functional cure of HIV infection. Viruses 2019, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, C.; Descamps, D. Resistance to HIV integrase inhibitors: About R263K and E157Q mutations. Viruses 2018, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Favarato, G.; Townsend, C.L.; Bailey, H.; Peters, H.; Tookey, P.A.; Taylor, G.P.; Thorne, C. Protease inhibitors and preterm delivery: Another piece in the puzzle. AIDS 2018, 32, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl.) 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Castain, L.; Perrier, M.; Charpentier, C.; Palich, R.; Desire, N.; Wirden, M.; Descamps, D.; Sayon, S.; Landman, R.; Valantin, M.-A.; et al. New machanism of resistance in virological failure to protease inhibitors: Selection of non-described protease, Gag and Gp41 mutations. J. Antimicrob. Chemother. 2019, 74, 2019–2023. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; Calvez, V.; Günthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2017 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2017, 24, 132. [Google Scholar] [PubMed]
- Park, J.; Sayer, J.; Aniana, A.; Yu, X.; Weber, I.; Harrison, R.; Louis, J. Binding of clinical inhibitors to a model precursor of a rationally selected multidrug resistant HIV-1 protease is significantly weaker than that to the released mature enzyme. Biochemistry 2016, 55, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, J.; Shao, Q.; Shi, J.; Zhu, W. Effects of drug-resistant mutations on the dynamic properties of HIV-1 protease and inhibition by amprenavir and darunavir. Sci. Rep. 2015, 5, 10517. [Google Scholar] [CrossRef] [PubMed]
- Kletenkov, K.; Hoffman, D.; Böni, J.; Yerly, S.; Aubert, V.; Schöni-Affolter, F.; Struck, D.; Verheyen, J.; Klimkait, T. Role of Gag mutations in PI resistance in the Swiss HIV cohort study: Bystanders or contributors? J. Antimicrob. Chemother. 2016, 72, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Flynn, W.F.; Chang, M.W.; Tan, Z.; Oliveira, G.; Yuan, J.; Okulicz, J.F.; Torbett, B.E.; Levy, R.M. Deep sequencing of Protease Inhibitor resistant HIV patient isolates reveals patterns of correlated mutations in Gag and protease. PLoS Comput. Biol. 2015, 11, e1004249. [Google Scholar] [CrossRef] [PubMed]
- Ghosn, J.; Delaugerre, C.; Flandre, P.; Galimand, J.; Cohen-Codar, I.; Raffi, F.; Delfraissy, J.-F.; Rouzioux, C.; Chaix, M. Polymorphism in Gag gene cleavage sites of HIV-1 non-B subtype and virological outcome of a first-line lopinavir/ritonavir single drug regimen. PLoS ONE 2011, 6, e24798. [Google Scholar] [CrossRef] [PubMed]
- Dam, E.; Quercia, R.; Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Krausslich, H.-G.; Hance, A.J.; Clavel, F.; Group, A.S. Gag mutations strongly contribute to HIV-1 resistance to Protease Inhibitors in highly drug-experienced patients besides compensating for fitness loss. PLoS Pathog. 2009, 5, e1000345. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Venzon, D.J.; Koh, Y.; Aoki-Ogata, H.; Miyakawa, T.; Yoshimura, K.; Maeda, K.; Mitsuya, H. Non-cleavage site Gag mutations in Amprenavir-resisitant Human Immunodeficiency Virus Type 1 (HIV-1) predispose HIV-1 to rapid acquisition of Amprenavir resistance by delay development of resistance to other Protease Inhibitors. J. Virol. 2009, 83, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Parry, C.M.; Kohli, A.; Boinett, C.J.; Towers, G.J.; McCormick, A.L.; Pillay, D. Gag determinants of fitness and drug susceptibility in protease inhibitor-resistant human immunodeficiency virus type 1. J. Virol. 2009, 83, 9094–9101. [Google Scholar] [CrossRef]
- Tamiya, S.; Mardy, S.; Kavlick, M.F.; Yoshimura, K.; Mitsuya, H. Amino acid insertions near Gag cleavage sites restore the otherwise compromised replication of human immunodeficiency virus type 1 variants resistant to Protease Inhibitors. J. Virol. 2004, 78, 12030–12040. [Google Scholar] [CrossRef]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.; Wrin, T.; et al. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Gatanaga, H.; Suzuki, Y.; Tsang, H.; Yoshimura, K.; Kavlick, M.K.; Nagashima, K.; Gorelick, R.J.; Mardy, S.; Tang, C.; Summers, M.F.; et al. Amino acid substitutions in Gag protein at non-cleavage sites are indispensable for the development of a high multitude of HIV-1 resistance against protease inhibitors. J. Biol. Chem. 2002, 277, 5952–5961. [Google Scholar] [CrossRef] [PubMed]
- Codoner, F.M.; Pena, R.; Blanch-Lombarte, O.; Jimenez-Moyano, E.; Pino, M.; Vollbretch, T.; Clotet, B.; Martinez-Picado, J.; Draenert, R.; Prado, J.G. Gag-protease coevolution analyses define novel structural surfaces in the HIV-1 matrix and capsid involved in resistance to Protease Inhibitors. Sci. Rep. 2017, 7, 3717. [Google Scholar] [CrossRef] [PubMed]
- Clavel, F.; Mammano, F. Review: Role of Gag in HIV resistance to protease inhibitors. Viruses 2010, 2, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Ozen, A.; Lin, K.-H.; Yilmaz, N.K.; Schiffer, C.A. Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. USA 2014, 111, 15993–15998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, L.; Matsuda, M.; Matsuda, Z.; Yokomaku, Y.; Chiba, T.; Okano, A.; Yamada, K.; Sugiura, W. Gag non-cleavage site mutations contribute to full recovery of viral fitness in protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2004, 48, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. Mini review: HIV-1 Gag proteins: Diverse functions in the virus life cycle. Virology 1998, 251, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pettit, S.C.; Moody, M.D.; Wehbie, R.S.; Kaplan, A.H.; Nantermet, P.V.; Klein, C.A.; Swanstrom, R. The p2 domain of Human Immunodeficiency Virus Type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 1994, 68, 8017–8027. [Google Scholar]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Fun, A.; Wensing, A.M.; Verheyen, J.; Nijhuis, M. Human Immunodeficiency Virus gag and protease: Partners in resistance. Retrovirology 2012, 9, 63. [Google Scholar] [CrossRef]
- Pettit, S.C.; Lindquist, J.N.; Kaplan, A.H.; Swanstrom, R. Processing sites in the human immunodeficiency virus type 1 (HIV-1) Gag-Pro-Pol precursor are cleaved by the viral protease at different rates. Retrovirology 2005, 2, 66. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.; Hon, G.; Chandonia, J.; Brenner, S. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Stephens, R. Sequence Logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.W.; Shafer, R.W. HIV-1 Antiretroviral Resistance: Scientific principles and clinical applications. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.J.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Fumero, E.; Podzamczer, D. New patterns of HIV-1 resistance during HAART. Clin. Microbiol. Infect. 2003, 9, 1077–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loulergue, P.; Delaugerre, C.; Jullien, V.; Viard, J.-P. HIV Drug resistance on HAART despite an undetectable viral load. Curr. HIV Res. 2011, 9, 623–624. [Google Scholar] [CrossRef] [PubMed]
- Fun, A.; van Maarseveen, N.M.; Pokorna, J.; Maas, R.E.; Schipper, P.J.; Konvalinka, J.; Nijhuis, M. HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology 2011, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.F.; Pond, S.L.K.; Richman, D.D.; Frost, S.D. Mapping Protease Inhibitor Resistance to HIV Type 1 sequence polymorphisma within patients. J. Virol. 2007, 81, 13598–13607. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.-L.; Varghese, V.; Rhee, S.-Y.; Gatell, J.M.; Shafer, R.W. HIV-1 integrase inhibitor resistance and its clinical implications. J. Infect. Dis. 2011, 203, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, H.; Maeda, K.; Das, D.; Ghosh, A. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv. Pharmacol. 2008, 56, 169–197. [Google Scholar] [PubMed]
- Gallant, J.E. Initial therapy of HIV Infection. J. Clin. Virol. 2002, 25, 317–333. [Google Scholar] [CrossRef]
- Barrie, K.; Perez, E.; Lamers, S.; Farmerie, W.; Dunn, B.; Sleasman, J.; Goodenow, M. Natural variation in HIV-1 protease, Gag p7 and p6, and protease cleavage sites within gag/pol polyproteins: Amino acid substitutions in the absence of protease inhibitors in mothers and children infected by human immunodeficiency virus type 1. J. Virol. 1996, 219, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Ode, H.; Suzuki, K.; Fujino, M.; Maejima, M.; Kimura, Y.; Masaoka, T.; Hattori, J.; Matsuda, M.; Hachiya, A.; et al. Unique flap conformation in an HIV-1 protease with high-level darunavir resistance. Front. Microbiol. 2016, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Appadurai, R.; Senapati, S. Dynamical network of HIV-1 protease mutants reveals the mechanism of drug resistance and unhindered activity. Biochemistry 2016, 55, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Chang, Y.; Agniswamy, J.; Harrison, R.; Weber, I. Conformational variation of an extreme drug resistant mutant of HIV protease. J. Mol. Graph. Model. 2015, 62, 87–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, D.A.; Nalivaika, E.A.; Nalam, M.N.; Prachanronarong, K.L.; Cao, H.; Bandaranayake, R.M.; Cai, Y.; Kurt-Yilmaz, N.; Schiffer, C.A. Drug Resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc. 2014, 136, 11956–11963. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.D.; Gonzales, E.; Huang, X.; Smith, A.; de Vera, I.; D’Amore, P.; Rocca, J.; Goodenow, M.; Dunn, B.; Fanucci, G. Effects of PRE and POST therapy drug-pressure selected mutations on HIV-1 protease conformational sampling. FEBS Lett. 2014, 588, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Britto, M.; Kear-Scott, J.; Boone, C.; Rocca, J.; Simmerling, C.; McKenna, R.; Bieri, M.; Gooley, P.; Dunn, B.; et al. The role of select subtype polymorphisms on HIV-1 protease conformational sampling and dynamics. J. Biol. Chem. 2014, 289, 17203–17214. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Lazim, R.; Zhang, D. Understanding the basis of I50V-induced affinity decrease in HIV-1 protease via molecular dynamics simulations using polarized force field. J. Comput. Chem. 2015, 36, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.; Rigo, M.; Sinigaglia, M.; de Medeiros, R.; Junqueira, D.; SEM, A.; Vieira, G. New insights into the in silico prediction of HIV protease resistance to nelfinavir. PLoS ONE 2014, 9, e87520. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Jamal, S.; Gopal, S.; Jain, R.; Wahi, D.; Grover, A. Structural studies on molecular mechanisms of Nelfinavir resistance caused by non-active mutation V77I in HIV-1 protease. BMC Bioinform. 2015, 16, S10. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Robertson, D.L.; Carruthers, C.D.; Morrison, S.G.; Jian, B.; Chen, Y.; Shaw, G.M.; Sinoiussi, F.B.; Girard, M.; Srinivasan, A.; et al. A comprehensive panel of near-full length clones and references sequences for non-subtype B isolates of Human Immunodeficiency Virus Type 1. J. Virol. 1998, 72, 5680–5698. [Google Scholar] [PubMed]
- Martinez-Picado, J.; Savara, A.V.; Sutton, L.; D’Aquila, R.T. Replicative fitness of protease inhibitor-resistant mutants of human immunodefieciency virus type 1. J. Virol. 1999, 73, 3744–3752. [Google Scholar] [PubMed]
- Bally, F.; Martinez, R.; Peters, S.; Sudre, P.; Telenti, A. Polymorphism of HIV Type 1 Gag p7/p1 and p1/p6 cleavage sites: Clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retrovir. 2000, 16, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Verheyen, J.; Rhee, S.-Y.; Voet, A.; Vandamme, A.-M.; Theys, K. Functional conservation of HIV-1 Gag: Implications for rational drug design. Retrovirology 2013, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Yap, P.; Koh, D.W.S.; Su, C.T.T.; Chan, K.F.; Gan, S.K.E. Predicting mutations in HIV-1 Gag: Insight from in silico and an in vitro BSL2 platform on thermostability and allosteric effect. bioRXiv 2019. [Google Scholar] [CrossRef]
- Geller, R.; Domingo-Calap, P.; Cuevas, J.M.; Rossolillo, P.; Negroni, M.; Sanjuán, R. The external domains of the HIV-1 envelope are a mutational cold spot. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef]
- Su, C.T.T.; Kwoh, C.K.; Verma, C.S.; Gan, S.K.E. Modeling the full length HIV-1 Gag polyprotein reveals the role of its p6 subunit in viral maturation and the effect of non-cleavage site mutations in protease drug resistance. J. Biomol. Struct. Dyn. 2017. [Google Scholar] [CrossRef]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second locus involved in human immunodefiency virus type 1 resistance to protease inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [PubMed]
- Malet, I.; Roquebert, B.; Dalban, C.; Wirden, M.; Amellal, B.; Agher, R.; Simon, A.; Katlama, C.; Costagliola, D.; Calvez, V.; et al. Association of Gag cleavage sites to Protease mutations and to virological response in HIV-1 treated patients. J. Infect. 2007, 54, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Parry, C.M.; Kolli, M.; Myers, R.; Cane, P.A.; Schiffer, C.; Pillay, D. Three residues in HIV-1 matrix contribute to protease inhibitor susceptibility and replication capacity. Antimicrob. Agents Chemother. 2011, 55, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Su, C.T.T.; Lua, W.H.; Ling, W.L.; Gan, S.K.E. Structural analyses of 2015-updated drug-resistant mutations in HIV-1 protease: An implication of protease inhibitor cross-resistance. BMC Bioinform. 2016, 17, 500. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y.; Taylor, J.; Fessel, W.J.; Kaufman, D.; Towner, W.; Troia, P.; Ruane, P.; Hellinger, J.; Shirvani, V.; Zolopa, A.; et al. HIV-1 protease mutations and protease inhibitor cross-resistance. Antimicrob. Agents Chemother. 2010, 54, 4253–4261. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.; Nalivaika, E.A.; Ozen, A.; et al. Molecular basis for drug resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef] [PubMed]
- Voshavar, C. Protease inhibitors for the treatment of HIV/AIDS: Recent advances and future challenges. Curr. Top. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.S.; et al. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; Clercq, E.D. Twenty-six years of Anti-HIV Drug discovery: Where do we stand and where do we go? J. Med. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.; Azoulay, D. Anti-DFS70 autoantibodies in HIV-1 positive individuals. Curr. Opin. Rheumatol. 2018, 30, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.; Mascola, J.; Nabel, G.J. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: The end of the beginning. Nat. Rev. Immunol. 2013, 13, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.; Irimia, A.; Ofek, G.; Kwong, P.; Wilson, I.; Walensky, L. Stapled HIV-1 peptides recapitulate antigenic structures and engage broadly neutralizing antibodies. Nat. Struct. Mol. Biol. 2014, 21, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Pertea, M.; Rongvaux, A.; Wang, L.; Durand, C.M.; Ghiaur, G.; Lai, J.; McHugh, H.L.; Hao, H.; Zhang, H.; et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 2015, 517, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Salzwedel, K.; Allaway, G. Berivimat: A novel maturation inhibitor for the treatment of HIV-1 infection. Antivir. Chem. Chemother. 2008, 19, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Thenin-Houssier, S.; Valente, S.T. HIV-1 capsid inhibitors as antiretroviral agents. Curr. HIV Res. 2016, 14, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Feasley, C.L.; Jackson, K.W.; Nitz, T.J.; Salzwedel, K.; Air, G.M.; Sakalian, M. The prototype HIV-1 maturation inhibitor, berivimat, binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirology 2011, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.; Shi, D.; Chrustowicz, J.; Hattne, J.; Gonen, T.; Yeager, M. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor berivimat. Proc. Natl. Acad. Sci. USA 2018, 115, 13258–13263. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.W.; Adamson, C.S.; Heymann, J.B.; Freed, E.O.; Steven, A.C. HIV-1 maturation inhibitor Berivimat stabilizes the immature Gag lattice. J. Virol. 2011, 85, 1420–1428. [Google Scholar] [CrossRef]
- Dang, Z.; Qian, K.; Ho, P.; Zhu, L.; Lee, K.-H.; Huang, L.; Chen, C.-H. Synthesis of betulinic acid derivatives as entry inhibitors against HIV-1 and Berivimat-resistant HIV-1 variants. Bioorg. Med. Chem. Lett. 2012, 22, 5190–5194. [Google Scholar] [CrossRef]
- Seclen, E.; Gonzalez, M.M.; Corral, A.; de Mendoza, C.; Soriano, V.; Poveda, E. High prevalence of natural polymorphism in Gag (CA-SP1) associated with reduced response to Berivimat, an HIV-1 maturation inhibitor. AIDS 2010, 24, 467–469. [Google Scholar] [CrossRef]
- Margot, N.A.; Gibbs, C.S.; Miller, M.D. Phenotypic susceptibility to Berivimat in isolates from HIV-1 infected patients without prior exposure to Berivimat. Antimicrob. Agents Chemother. 2010, 54, 2345–2353. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, J.; Verhofstede, C.; Knops, E.; Vandekerckhove, L.; Fun, A.; Brunen, D.; Dauwe, K.; Wensing, A.M.; Pfister, H.; Kaiser, R.; et al. High prevalence of Berivimat resistance mutations in protease inhibitor-resistant HIV isolates. AIDS 2010, 24, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.; Li, T.; Lin, Z.; Protack, T.; van Ham, P.; Hwang, C.; Krystal, M.; Nijhuis, M.; Lataillade, M.; Dicker, I. The second-generation maturation inhibitor GSK3532795 maintains potent activity toward HIV Protease Inhibitor-resistant clinical isolates. J. Acquir. Immune Defic. Syndr. 2017, 75, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ramirez, J.; Bogner, J.R.; Molina, J.-M.; Lombaard, J.; Dicker, I.B.; Stock, D.A.; DeGrosky, M.; Gartland, M.; Dumitrescu, T.P.; Min, S.; et al. Safety, efficacy, and dose response of the maturation inhibitor GSK3532795 (formerly known as BMS-955176) plus tenofovir/emtricitabine once daily in treatment-naive HIV-1-infected adults: Week 24 primary analysis from a randomized Phase IIb trial. PLoS ONE 2018, 13, e0205368. [Google Scholar] [CrossRef] [PubMed]
- Phua, S.X.; Chan, K.F.; Su, C.T.T.; Poh, J.J.; Gan, S.K.E. Perspective: The promises of a holistic view of proteins—Impact on antibody engineering and drug discovery. Biosci. Rep. 2019, 39, BSR20181958. [Google Scholar] [CrossRef] [PubMed]
- Chiang, R.Z.H.; Gan, S.K.E.; Su, C.T.T. A computational study for rational HIV-1 non-nucleoside reverse transcriptase inhibitor selection and the discovery of novel allosteric pockets for inhibitor design. Biosci. Rep. 2018, 38, BSR20171113. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.F.; Phua, S.X.; Su, C.T.T.; Gan, S.K.E. Inhibiting HIV-1 and MMLV Reverse Transcriptase: The potential of an allosteric broad-spectrum inhibitor. bioRXiv 2019. [Google Scholar] [CrossRef]
- Richard, S.; Selle, F.; Lotz, J.; Khalil, A.; Gligorov, J.; Soares, D. Pertuzumab and trastuzumab: The rationale way to synergy. An. Acad. Bras. Cienc. 2016, 88, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Capelan, M.; Pugliano, L.; De Azambuja, E.; Bozovic, I.; Saini, K.; Sotiriou, C.; Loi, S.; Piccart-Gebhart, M. Pertuzumab: New hope for patients with HER2-positive breast cancer. Ann. Oncol. 2012, 24, 273–282. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, J.; Yan, Y.; Yao, X.; Fang, L.; Xiong, H.; Liu, Y.; Chu, Q.; Zhou, P.; Wu, K. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J. Exp. Clin. Cancer Res. 2019, 38, 355. [Google Scholar] [CrossRef]
- Maldonado, J.O.; Martin, J.L.; Mueller, J.D.; Zhang, W.; Mansky, L.M. New insight into retroviral Gag-Gag and Gag-membrane interactions. Front. Microbiol. 2014, 5, 302. [Google Scholar] [CrossRef] [PubMed]
- El Meshri, S.; Dujardin, D.; Godet, J.; Richert, L.; Boudier, C.; Darlix, J.; Didier, P.; Mely, Y.; de Rocquigny, H. Role of the nucleocapsid domain in HIV-1 Gag oligomerization and trafficking to the plasma membrane: A fluorescence lifetime imaging microscopy investigation. J. Mol. Biol 2015, 427, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Mendonca, L.M.; Angert, I.; Mueller, J.D.; Zhang, W.; Mansky, L.M. Disparate contributions of human retrovirus capsid subdomains to Gag-Gag oligomerization, virus morphology, and particle biogenesis. J. Virol. 2017, 91, e00298-17. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | ||
|---|---|---|---|
| Inhibitor | Strain or Lab Clone | Mutations on Gag | Mutations on Protease |
| Amprenavir | HIV-1 NL4-3 (pNL4-3) | V35I–L75R–H219Q | L10F–V32I–M46I–I84V |
| Amprenavir | HIV-1 NL4-3 (pNL4-3) | L75R–H219Q–R409K–L449F–E468K | L10F–V32I–M46I–I84V |
| Amprenavir | HIV-1 NL4-3 (pNL4-3) | E12K–V35I–L75R–H219Q–V390D–R409K–L449F–E468K | L10F–V32I–M46I–I54M–A71V–I84V |
| JE–2147 | HIV-1 NL4-3 (pNL4-3) | H219Q–V390D | M46I–I84V |
| JE–2147 | HIV-1 NL4-3 (pNL4-3) | H219Q–V390D–R409K–L449F | V32I–M46I–I47V–V82I–I84V |
| KNI–272 | HIV-1 NL4-3 (pNL4-3) | V35I–E40K–G123E–H219Q–G381S–R409K–A431V | V32I–M46I–A71V–V82I–I84V |
| UIC–94003 | HIV-1 NL4-3 (pNL4-3) | E12K–E40K–G123E–Q199H–H219Q–R409K–G412D–L449F–E468K | L10F–M46I–I50V–A71V |
| Amprenavir | HIV-1 HXB2 | P453L | I50V |
| BILA–1906BS | HIV-1 strain IIIB | L449F | M46L–A71V–I84V |
| BILA–2185BS | HIV-1 strain IIIB | L449F–Q430R–A431V | L23I–V32I–M46I–I47V–I54M–A71V–I84V |
| Indinavir | HIV-1 pNL4.3 | A431V–I437V | V82A |
| Ritonavir/Saquinavir | HIV-1 subtype B # | A431V–L449F | I84V |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.T.-T.; Koh, D.W.-S.; Gan, S.K.-E. Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs. Molecules 2019, 24, 3243. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24183243
Su CT-T, Koh DW-S, Gan SK-E. Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs. Molecules. 2019; 24(18):3243. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24183243
Chicago/Turabian StyleSu, Chinh Tran-To, Darius Wen-Shuo Koh, and Samuel Ken-En Gan. 2019. "Reviewing HIV-1 Gag Mutations in Protease Inhibitors Resistance: Insights for Possible Novel Gag Inhibitor Designs" Molecules 24, no. 18: 3243. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24183243