
Synthetic Access to Aromatic α-Haloketones


1
Department of Chemistry and Center for Sustainable Chemistry, Ghent University, 9000 Ghent, Belgium
2
VITO (Flemish Institute for Technological Research), Separation and Conversion Technology, Boeretang 200, 2400 Mol, Belgium
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(11), 3583; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113583
Submission received: 22 April 2022
/
Revised: 19 May 2022
/
Accepted: 20 May 2022
/
Published: 2 June 2022
(This article belongs to the Section Organic Chemistry)
Abstract
:α-Haloketones play an essential role in the synthesis of complex N-, S-, O-heterocycles; of which some exhibit a remarkable biological activity. Research further illustrated that α-bromo-, α-chloro-, and α-iodoketones are key precursors for blockbuster pharmacological compounds. Over the past twenty years, substantial advances have been made in the synthesis of these industrially relevant building blocks. Efforts have focused on rendering the synthetic protocols greener, more effective and versatile. In this survey, we summarised and thoroughly evaluated the progress of the field, established in the past two decades, in terms of generality, efficacy and sustainability.

| Table of Contents | |
| 1. Introduction............................................................................................................................................................................................................ | 2 |
| 2. Direct α-Halogenation of Aryl Ketones.............................................................................................................................................................. | 3 |
| 2.1. Electrophilic Halogenation............................................................................................................................................................................ | 3 |
| 2.1.1. Br2 as Halogenation Agent..................................................................................................................................................................... | 3 |
| 2.1.2. I2 as Halogenation Agent........................................................................................................................................................................ | 6 |
| 2.1.3. NXS as Halogenation Agents................................................................................................................................................................. | 11 |
| 2.1.4. Halides (X−) as X2 Sources...................................................................................................................................................................... | 15 |
| Metal Halides (MX)....................................................................................................................................................................................... | 15 |
| Quaternary Ammonium Halides (NH4X)................................................................................................................................................... | 16 |
| Hydrogen Halides (HX)................................................................................................................................................................................ | 17 |
| 2.1.5. Polymeric Halogenation Agents............................................................................................................................................................ | 20 |
| 2.1.6. Ionic Liquids as Halogenation Agent.................................................................................................................................................... | 22 |
| 2.1.7. Miscellaneous Halogenation Agents..................................................................................................................................................... | 24 |
| 2.2. Nucleophilic Halogenation........................................................................................................................................................................... | 26 |
| 3. Oxyhalogenation of Hydrocarbons..................................................................................................................................................................... | 28 |
| 3.1. Alkenes as Substrates..................................................................................................................................................................................... | 28 |
| 3.2. Alkynes as Substrates..................................................................................................................................................................................... | 32 |
| 4. Oxidative Halogenation of Secondary Alcohols................................................................................................................................................ | 34 |
| 5. Oxidation of Functionalised Hydrocarbons....................................................................................................................................................... | 35 |
| 5.1. Haloalkanes as Substrates............................................................................................................................................................................. | 35 |
| 5.2. Haloalkenes as Substrates............................................................................................................................................................................. | 36 |
| 5.3. Haloalcohols as Substrates............................................................................................................................................................................ | 37 |
| 6. Hydration of 1-Haloalkynes................................................................................................................................................................................ | 38 |
| 6.1. Metal-Catalysed Hydrations......................................................................................................................................................................... | 38 |
| 6.2. Non-Metal-Catalysed Hydrations................................................................................................................................................................ | 40 |
| 7. Miscellaneous Routes........................................................................................................................................................................................... | 41 |
| 7.1. α-Functionalised Ketones as Substrates...................................................................................................................................................... | 41 |
| 7.2. Alternative Substrates.................................................................................................................................................................................... | 43 |
| 8. Conclusions and Outlook..................................................................................................................................................................................... | 45 |
| References.................................................................................................................................................................................................................. | 46 |
1. Introduction
Over the past few decades, the versatility and synthetic value of α-haloketones in organic synthesis have been amply showcased in the literature. The existence of two adjacent electrophilic centres, namely the α-halocarbon and the carbonyl group, transforms these reactive carbonyl compounds into highly valuable building blocks for the construction of more complex structures. In that context, a wide variety of N, S, and O-heterocycles have been accessed using protocols involving α-haloketones [1,2,3,4,5,6,7,8,9,10,11]. These compounds have also proven to be important intermediates in the synthesis of various organometallic species. [12,13,14]. Other applications have demonstrated their utility as key synthetic intermediates for pharmaceutical blockbuster compounds (Figure 1) [15,16,17,18,19,20,21,22,23,24,25,26,27].
In recent years, a significant body of work has been dedicated to the design and development of new synthetic protocols to access α-haloketones. Even though these processes have been reviewed in the literature, a thorough and comprehensive survey is still lacking. For example, recent reviews have dealt with the α-bromination [28,29] and α-iodination [30] of carbonyl compounds; however, these were very specific and only involved pathways towards a very limited range of α-haloketones. In 2018, Verkariya and co-workers reviewed the synthetic access to α-iodocarbonyl compounds [31]. Although a broad range of iodinated products were considered, the work was restricted to direct iodination routes. In 2003, Erian and co-workers reported a survey involving the synthetic access to and application of α-haloketones [32]. Here, again, synthetic protocols yielding the desired ketones were only briefly discussed and the authors relied on the work published by De Kimpe and Verhé [33]. An up-to-date review of the synthetic access to α-haloketones is clearly needed. Therefore, the present contribution aims to describe and evaluate the diverse protocols leading to α-haloketones that have been published over the past two decades. Since the number of reports in the literature is quite significant, this review will be limited to methods generating carbonyl compounds carrying one halogen atom at their α-position, and will mainly focus on aromatic α-haloketones compounds, as these appear to have the highest synthetic value [34,35,36].
2. Direct α-Halogenation of Aryl Ketones
α-Haloketones are most commonly obtained from their corresponding carbonyl analogues through direct halogenation. Over the years, a variety of direct halogenation routes have been developed with apparent advantages and disadvantages. The readily available starting material for these reactions and the overall reaction efficacy have rendered these processes highly useful for synthetic chemists. At the same time, these direct halogenation reactions exhibited certain drawbacks, such as moderate conversions, long reaction times, cumbersome procedures and the use of hazardous and toxic reagents and solvents. In addition to the desired α-haloketone, some protocols also yielded the α,α-dihalogenated and/or aromatic ring halogenated by-products (Scheme 1). This moderate selectivity consequently complicates the purification process.
2.1. Electrophilic Halogenation
2.1.1. Br2 as Halogenation Agent
The most straightforward route to α-haloketones involves the reaction of enolizable aromatic ketones with electrophilic X2, under acidic (or basic) conditions; these conditions are typically needed to generate the nucleophilic enol (Scheme 2).
The procedure developed by Li, Xiang and co-workers in 2014 enables the bromination of aryl ketones under acidic conditions [37]. A small excess of Br2 (1.1 equiv.) was added to a solution of ketone in glacial HOAc and subsequently irradiated for 5 h in a microwave (MW). Figure 2 presents the bromination scope with respective isolated yields. The main drawback of this procedure is the use of HOAc as a solvent, which substantially limited the substrate scope; acid-sensitive functional groups (e.g., -OH, -NHx) cannot tolerate these reaction conditions.
Using a similar strategy, Nieuwland developed a continuous flow procedure for the α-bromination of acetophenone [38]. 2-Bromo-1-phenylethanone was isolated in 99% yield upon treatment of the substrate with HBr and bromine in 1,4-dioxane (Scheme 3). The authors claimed that ring brominated or dibrominated products could not be observed, which highlights the excellent selectivity of the process. This work showcased a highly effective α-bromination protocol which could be applicable on an industrial scale. However, the large-scale use of toxic and corrosive HBr and Br2 reagents would require important safety measures to avoid leakage or contact of the reagents with the environment/operators, which remains a significant drawback.
Attempts to substitute the Bronsted acid activation with a Lewis acid activation have shown promising results, via the in situ formation of ZnBr2 from Zn dust and bromine (Scheme 4) [39]. The aqueous procedure was well tolerated by substituents (i.e., ortho) close to the carbonyl centre. However, the use of toxic metallic Zn and Br2 counteracted the progress made in terms of a greener approach [40,41].
Another attempt to alleviate the acid sensitivity of substrates/products was developed by Ryu and co-workers, using a phase-vanishing protocol (Scheme 5) [42]. The reaction mixture consisted of four separate phases: (i) an aqueous phase, to trap HBr, (ii) an organic (CH3Cl) phase (Galden HT135), containing the substrate, (iii) a fluorous phase, and (iv) the bromine phase. The fluorous phase acted as a membrane to separate the Br2 and the substrate-containing phase. Upon stirring, the fluorous phase enables the former to mix with the latter phase. During the reaction, HBr migrates to the aqueous layer, and at the end of the reaction, the bromine phase vanishes. This procedure thus prevented possible product decomposition induced by HBr, as the substrate and the acid were contained in different phases. This quadruple phase method simplified the purification of the desired α-bromoketones. In total, five α-bromoketones were isolated in good to excellent yields in this manner. It should be noted that dibrominated products were observed in trace amounts, which could complicate purification.
Chen et al. improved the reaction by successfully performing the α-bromination of a very broad substrate scope (21) in good to excellent yields (84–99%) without any additional activator (Scheme 6) [43]. The presence of both electron-withdrawing (e.g., Cl, Br, CF3) and -donating (e.g., OMe, t-Bu, OH) groups was well tolerated. Moreover, this route’s efficacy was not significantly affected by the presence of steric bulk at the ortho-position.
In 2014, Krupadanam and co-workers published an acid-free, Br2-mediated halogenation [44]. One equivalent of bromine was added dropwise to an ice-cold solution of the substrate in diethyl ether, after which the solution was stirred for 2 h at room temperature. Eight 2-bromoacetophenones were isolated in moderate to good yields (Table 1). In addition, the procedure generated hetero-aromatic bromoketones, i.e., 2-bromo-1-(pyridin-3-yl)ethenone and 2-bromo-1-(thiophen-2-yl)ethanone, in moderate yields (70 and 76%, respectively). However, for the former, the bromination only proceeded upon addition of an acidic promotor (HBr or HOAc).
In 2018, Portilla described the bromination of four aromatic ketones mediated by bromine [45]. The corresponding α-bromoketones were isolated in good to excellent yields (Scheme 7). Krupadanam’s and Portilla’s routes stand out from other Br2-mediated halogenation reactions, as they demonstrated that the reaction can proceed without any promotor or catalyst. Additionally, the acid-free routes allowed a significant improvement in the halogenation yield.
All the above Br2-mediated methods are characterised by the same intrinsic drawbacks. These routes depict a moderate atom economy, since only one of the two available Br-atoms is incorporated into the final product. The “excess” bromine atom will interact with the α-H atom of the substrate, leading to corrosive and toxic HBr formation, inducing possible product decomposition. Furthermore, the toxic, irritating, and corrosive nature of bromine limits the applicability of a Br2-mediated route [46,47].
2.1.2. I2 as Halogenation Agent
In the direct iodination of ketones, Stavber and co-workers established an effective iodination route consisting of I2 (0.5 equiv.) and 30% aqueous H2O2 (0.6 equiv.) in methanol [48]. This system was only effective in the presence of an acid catalyst. The authors compared the efficacy of two catalytic systems; H2SO4 (cat A; 0.1 equiv.) and H4SiO4·12WO3·26H2O (POM) (cat B; 0.06 equiv.). Using both methods, three aromatic α-iodoketones were obtained in good to excellent yields (Scheme 8). The excellent iodine atom economy of this route should be stressed, as every I-atom is incorporated in the substrate. This method prevented the long-term persistence of harmful HI in the reaction mixture.
In 2007, Stavber updated the procedure by substituting the aqueous H2O2 with the solid urea–H2O2 (UHP) complex. This yielded a solvent-free direct iodination, though an increase in the I2 content from 0.5 to 1 equivalent was required [49]. The excellent iodine atom economy was thus not maintained. Furthermore, the iodination yields (22–58%) dropped significantly compared to those obtained from the previous route. Subsequently, it was demonstrated that both the I2/H2O2- and I2/UHP-mediated iodination could be performed in an ionic liquid (IL), thus easing the purification process [50]. The reaction was performed in both water-miscible and water-immiscible Ils, respectively, 1-butyl-3-methyl imidazolium tetrafluoroborate ([bmim]BF4) and 1-butyl-3-methyl imidazolium hexafluoro-phosphate ([bmim]PF6) (Scheme 9). Experimental data demonstrated that the substrates were less effectively iodinated in both Ils, compared to the solvent-free [49] and MeOH iodination protocols [48].
Additionally, Stavber reported a sodium nitrite-catalysed procedure (Scheme 10, method A) [51]. NaNO2 is activated via treatment with silica-supported sulfuric acid. After activation, HNO2 is obtained, which is then oxidised by air to NO2. Since only 0.5 equiv. of I2 is used, after depletion of all I2 molecules, the obtained HI will then react with NO2 to reform I2 and NO; the latter is then again oxidised to NO2 by air. In this manner, 100% of iodine atom economy was achieved as only 0.5 equivalent of iodine was required for the reaction to proceed to completion. The sustainability of this route was further demonstrated, as air was used as the stoichiometric oxidant. Compared to other iodination routes, the purification was more straightforward as it only required separation from the inorganic salts. Due to the inherent health hazards linked to SiO2 [52,53,54], this procedure was later modified (Scheme 10, method B) [55]. The iodination was carried out in an aqueous sodium dodecyl sulphate (SDS) solution and promoted by H2SO4. The SDS micelles acted as an ideal promotor of the α-iodination of aryl ketones. This route displayed excellent selectivity as side chain functionalisation was exclusively obtained. This could be shifted towards aromatic ring iodination by changing the reaction medium to acetonitrile. The 100% iodine atom economy and the use of an aqueous reaction medium showcased the promising potential of the SDS-promoted route. From an economic point of view, this route is particularly interesting as it employed cheap and abundant reagents. Nonetheless, the presence of H2SO4 in the reaction medium still limited the substrate scope, as acid-sensitive functional groups (e.g., OH) are not compatible with these experimental conditions.
In 2014, an alternative method was developed using NH4NO3 (0.1–0.25 equiv.), I2 (0.5 equiv.), H2SO4 (0.05–0.125 equiv.) and air in CH3CN (Scheme 11) [56]. Under these conditions, several ketones were iodinated in moderate to excellent yields (58–90%). In addition, OH-substituted aromatic ketones were successfully transformed into their iodinated analogues. However, for di- and tri-methoxy-substituted ketones, selective ring iodination instead of α-iodination occurred; these by-products account for a required and more tedious purification process.
Alternatively to O2 and H2O2, Zhdankin and co-workers demonstrated that the hypervalent m-iodosylbenzoic acid could also act as a suitable oxidation agent [57]. With the reaction conditions shown in Scheme 12, a variety of aromatic and aliphatic ketones and β-dicarbonyls were iodinated. At the end of the reaction, the reduced form of the hypervalent iodine oxidant, m-iodobenzoic acid, could be easily removed by passing the reaction mixture through an anionic exchange resin (Amberlite IRA 900 HCO3−). Afterwards, recovery of the m-iodobenzoic acid was achieved by treating the resin with HCl. With similar drawbacks to earlier strategies, this method still allowed for 100% I-atom economy while also carrying the benefits of recycling the oxidizing reagent.
Over the past two decades, numerous acid-free direct iodination protocols have been developed. These methods tolerated acid-sensitive functional groups, such as amines or alcohols. A remarkable iodination system was developed by Rao and co-workers, which only required the addition of iodine (4 equiv.) to the methyl ketone in dimethylether at 90 °C (Scheme 13) [58]. Several aromatic α-iodoketones were obtained in moderate yields (62–94%). In this reaction, the dual role of I2 was highlighted as it acts as both a Lewis acid and a direct iodine source. The acid-free conditions and short reaction times were this route’s main advantages. However, the use of a highly volatile solvent, the excess of I2 and the inevitable generation of HI negatively impacted this direct iodination route. The corrosive nature of HI involves extra hazards and might lead to product decomposition.
Using neutral conditions, Yin and co-workers reported a metal-mediated iodination of ketones through the addition of CuO (1 equiv.) and I2 (1 equiv.) to the substrate in MeOH (Scheme 14) [59]. The presence of electron-donating (e.g., OMe, Me) and electron-withdrawing substituents (e.g., Cl, Br) on the aryl ketones was well tolerated. The corresponding α-iodoketones were obtained in 83–99% yield. For nitro-substituted aryl ketones, the yield decreased significantly to 53% and formation of the dimethyl ketal product occurred. This protocol stands out due to the use of inexpensive reagents and its excellent iodination yields. Nonetheless, a significant amount of harmful HI was generated.
An iodine- and trimethylorthoformate-mediated iodination was reported in 2008 (Scheme 15), yielding several aromatic ketones with a wide range of substituents (i.e., para-OH, -Me, -F and common protecting groups such as -OBn, -OTBS and -OAc) in good yields (64–78%) [60]. The authors claimed that the work-up drastically altered the outcome of the reaction, since the true reaction mixture contained primarily the α-iodinated dimethoxy ketal product. Upon reaction completion, when the reaction was directly treated with an aqueous Na2S2O3 solution followed by extraction, the α-iodoketal product was mainly obtained (Scheme 15, right). However, if the reaction mixture was first stirred in water, then treated with the Na2S2O3 solution, α-iodoketones were obtained as the main products (Scheme 15, left).
In 2010, Lee and co-workers illustrated the halogenation of ketones using another iodine activator, the hypervalent [hydroxyl(tosyloxy)iodo]benzene (HTIB), in an IL, i.e., [bmim][BF4] (Scheme 16) [61]. Two different iodination agents, iodine (1.2 equiv.) and methyl iodide (3 equiv.), were compared for a set of ketones. Although both generated the corresponding α-iodoketones in similar yields, they required different reaction conditions. The I2-mediated direct halogenation was carried out at 60 °C for 3–4 h. With methyl iodide, the reaction time increased to 20 h and the temperature decreased to room temperature. The protocol yielded five α-iodinated aryl ketones in moderate to excellent yields (61–96%). This route emerged as a relatively mild and green direct halogenation route linked to HTIB, a neutral, highly stable, and relatively safe oxidant. The use of an IL also contributed to simplifying the work-up process by allowing the product to be simply extracted without any further purification needed.
Goswami reported the selective α-iodination route mediated by iodine (0.5 equiv.) and Oxone® (2KHSO5·KHSO4·K2SO4) (0.1 equiv.) [62]. The reaction proceeded by grinding the reagents and the ketone in a mortar with a pestle. A diverse set of 1,3-dicarbonyls and ketones were halogenated in excellent yields. However, only a limited number of aromatic ketones were tested under these conditions to afford the desired products in excellent yields (91%), after grinding for 3 min. The procedure stands out from other direct iodination routes due to its solvent-free approach, involving a more straightforward work-up. So far, the very limited scope of aryl ketones can be considered as its major weakness.
In 2018, Gong and co-workers designed a graphene oxide (GO)-catalysed iodination of arenes and ketones in nitromethane (Scheme 17) [63]. Upon the addition of I2 (1.1 equiv.), several aryl ketones were efficiently iodinated when stirred for 5 min (min) at 120 °C. Substrates bearing electron-donating groups (i.e., OMe and Me) performed well under these conditions (96% for both). In comparison, substrates with electron-withdrawing substituents (i.e., F and CN) gave somewhat lower yields (74% and 68%, respectively). The reaction proceeded via a radical pathway, with an iodine radical being formed under the influence of the unpaired electrons of the GO; single electron transfer (SET) from the iodinated substrate back to the GO regenerated the GO catalyst and allowed its re-entry into the catalytic cycle. The authors were able to scale up the reaction to the gram scale in air, thus adding a major benefit to this approach. Despite the efficiency and high applicability of this protocol, the generation of stoichiometric amounts of HI and the high temperature needed are considered major drawbacks.
Overall, direct halogenation methods, employing Br2 or I2, possess similar intrinsic weaknesses. Bromine is a toxic, corrosive and irritating halogenation agent [46,47]. Although less harmful than bromine, the use of iodine also poses serious safety and health issues [64,65]. Hence, the next sections will focus on halogen sources that are generally less harmful to human health and to the environment.
2.1.3. NXS as Halogenation Agents
The user friendliness, availability, and low cost of N-halosuccinimides (NXS) has enabled its widespread use as a halogenation reagent (Figure 3). NXS can be recycled after the reagent is consumed in the halogenation reaction, demonstrating its sustainable nature.
In 2017, Lim and co-workers reported a direct bromination and chlorination route optimised for aromatic ketones [66]. The procedure involved the addition of NBS (1.05 equiv.) and TMSOTf (0.05 equiv.) to the starting material and stirring the obtained solution in MeCN for three days at room temperature. Five α-bromoketones (i.e., p-H, -F, -Cl, -CN and -NO2) were isolated in moderate to good yields (60–77%). Despite the straightforward work-up procedure, the extremely long reaction times have limited the widespread use of this protocol. Chlorination was also established via the addition of NCS (1 equiv.) and para-toluenesulfonic acid-monohydrate (pTsOH·H2O) (1.5 equiv.) to the ketone. After stirring for 7 h at 80 °C, eight α-chloroketones were obtained (Figure 4). The excess of pTsOH·H2O used renders the protocol incompatible with acid-sensitive groups.
Similar pTsOH-based chlorination and bromination were also described by Lee and co-workers (Scheme 18) [67]. In these reactions, 1.5 equiv. of the acid and 1 equiv. of NXS yielded three α-bromo (90–92%) and three α-chloroketones (74–82%) with three different substituents on the aromatic ring (i.e., p-OMe, -Cl, -NO2). Here, again, the very limited scope puts the general nature of the method into serious doubt.
A more general pTsOH-catalysed bromination protocol was developed by Huang and co-workers using MW irradiation [68]. A solution of the ketone, NBS (1 equiv.) and pTsOH (0.1 equiv.) in dichloromethane (DCM) was irradiated for 30 min at 80 °C. This procedure yielded 11 aromatic α-bromoketones in good to excellent yields (85–95%). The presence of electron-withdrawing, -donating or acid-sensitive substituents (i.e., OH) on the aryl ketones was well tolerated. The position of these substituents on the aromatic ring did not influence the bromination efficiency. The versatility of this route was further highlighted by the bromination of heteroaromatic ketones with high efficacy (93–95%).
KH2PO4 could also promote the direct bromination of aryl ketones as reported by Reddy and co-workers (Scheme 19) [69]. Under mild acidic conditions using 0.1 equiv. of KH2PO4 and 1.1 equiv. of NBS, a wide variety of brominated products were isolated in moderate to excellent yields (46–94%). Nitro- and cyano-substituted aromatic ketones were less efficiently brominated. For substrates bearing OH and NH2 substituents, ring bromination was exclusively observed.
Moving away from Brønsted acid-based conditions, the same group highlighted silica as another efficient promoter for the bromination of aryl ketones [70]. Using the conditions depicted in Scheme 20, a wide variety of aryl ketones was successfully brominated. Substrates bearing highly electron-donating (i.e., p-OMe) and electron-withdrawing (i.e., NO2, CN) groups did not fare well in this protocol, as their corresponding α-bromoketones were obtained in yields of 64%, 42% and 45%, respectively. For some substrates, dibromination also occurred, highlighting the procedure’s modest selectivity. The short reaction times, however, can be considered as the main feature of this route.
It should be mentioned that SiO2-catalysed α-bromination is not a new concept. Paul and co-workers had previously reported the NBS- and HClO4·SiO2-mediated halogenation affording 11 aromatic α-bromoketones in good to excellent yields (77–93%) [71]. Using this process, heteroaromatic (i.e., pyridinyl, thienyl, furyl) ketone substrates were efficiently α-brominated, albeit with longer reaction times (16–18 h) compared to their aromatic counterparts (8–12 h). Similarly to the aforementioned routes, the protocol demonstrated a rather moderate selectivity as the α,α-dibrominated products were observed in some cases. Other protocols involving the use of modified SiO2, such as NaHSO4·SiO2 and HSO3·SiO2, have also demonstrated good catalytic activity in the direct halogenation reaction. Both yielded 2-bromo-1-phenylethanone in 72% and 87% yield, respectively. The authors claimed that HSO3·SiO2 could even be recycled and reused for three consecutive runs without significant loss in catalytic activity. Additionally, Salama reported that SiCl4 efficiently catalysed the direct halogenation of aromatic ketones [72]. In this procedure, SiCl4 (2 equiv.) and NXS (2 equiv.) were added to the aryl ketone in CH3CN. The protocol yielded several α-chloro-, α-bromo- and α-iodoketones in moderate to excellent yields.
Other Lewis-acid-based protocols involved the use of transition metals to efficiently perform the direct halogenation of aryl ketones [73]. Sudalai and co-workers described a Cu(OTf)2-catalysed bromination reaction that afforded α-bromoketones in 65–89% yield (Scheme 21). Next to the mono-brominated product, α,α-dibromoketones were also observed as a minor product, making this route less attractive compared to existing and more selective α-bromination procedures.
Waghmode and Ramaswamy designed a photochemical α-bromination of cyclic and acylic ketones. This route generated seven aromatic bromoketones upon the addition of NBS (1.05 equiv.) to the substrates and subsequent UV-VIS irradiation in diethylether [74]. Thorough assessment of this route’s efficiency is not possible, as bromination yields were not reported. The authors mentioned that α,α-bromination occurred for most of the substrates, which highlights the modest selectivity of the developed procedure.
In 2019, Jangid and co-workers developed a TiO2 nanoparticle (NP)-catalysed bromination and chlorination of aromatic ketones [75]. With the corresponding NXS and t-butyl hydrogen peroxide (TBHP), 12 α-haloketones were obtained in good to excellent yields (Scheme 22). The short reaction times and excellent halogenation yields can be considered as the main advantages of this route.
The use of volatile and hazardous organic solvents counteracted the progress that some NXS-based methods accomplished in terms of sustainability. Thus, Togo and co-workers attempted to provide a greener bromination approach [76]. A set of ketones was halogenated upon addition of NBS (1.2 equiv.) and pTsOH·H2O (0.2 equiv.) in two different ILs, i.e., [bmim][PF6] and N-butyl-N-methyl-pyrrolidinium bis(trifluoromethanesulfonyl)imidate ([bmpy][NTf2]) (Figure 5, left). The use of an IL not only facilitated the work-up, but also allowed the reaction medium to be reused up to seven times without significant loss in bromination efficiency. Both ILs appeared to be effective halogenation reaction media, as eight aromatic α-haloketones were obtained in good to excellent yields (74–96%).
Lee and Park designed an alternative procedure for the α-bromination in an IL, i.e., [bmim]BF4 (Scheme 23) [77]. The UHP (2 equiv.)- and NXS (2 equiv.)-mediated reaction yielded four α-chloro and four α-bromoketones (p-H, -Me, -Cl and -NO2) in moderate to good yields (68–78% and 74–86%, respectively).
Another NXS-mediated halogenation in an IL, i.e., 1-methyl-3-(4-sulfobutyl)imidazolium triflate ([bmim(SO3H)][OTf]), was also developed by Stavber (Figure 5, right) [78]. Remarkably, the selectivity of the procedure was highly dependent on the type of halogenation carried out. In general, α-iodination of aromatic ketones was exclusively obtained, while a mixture of α- and α,α-dihalogenated product was observed for the bromination, with the former species as the major product. Upon increasing the reaction time, this ratio could be shifted to selectively obtain the latter product. The chlorination exclusively yielded the dihalogenated aryl ketones. The authors claim that [bmim(SO3H)][OTf] could be recycled and reused for up to eight consecutive runs without any observable loss in halogenation efficiency. The sustainable character of this route and its versatility make it stand out from other halogenation routes.
Achieving a good reaction performance under solvent-free conditions is an important step forward for any reaction and is good practice for future advancement in sustainable chemistry. Stavber reported the solvent-free halogenation of acetophenone derivatives, mediated by sub-stoichiometric amounts of pTsOH (Scheme 24) [79,80]. The reagents were ground in a mortar with a pestle, yielding α-bromo, α-chloro and α-iodoketones in satisfactory yields. This procedure could be applied to the bromination, chlorination, and iodination of ketones. In the case of the chlorination, dichlorination of the substrates was a serious issue. Note that thus far, this dichlorination issue has been mentioned several times. Furthermore, trimethoxy-substituted ketones generated exclusive ring bromination. Interestingly, performing the bromination reactions in an aqueous solvent shifted the selectivity of this route toward ring bromination.
In 2003, Lee and co-workers published a solvent-free iodination of aromatic ketones mediated by NIS (1.2 equiv.) and pTsOH (1.2 equiv.) [81]. Five α-iodoketones were obtained within 1–1.2 min in good to excellent yields (75–90%) after irradiation of the reaction mixture in a microwave at 700 W. The system stood out from other direct iodination routes mainly due to its short reaction times and excellent performance.
In 2015, Patel and co-workers reported catalyst-free bromination in neat conditions [82]. Under these conditions, the ketone and NBS (1.1 equiv.) were ground in a mortar in order to obtain a homogenous solid mixture, which was subsequently irradiated in the MW at 300 W for 1–4 min. Ten α-bromoketones were obtained in good to excellent yields (82–93%). This protocol tolerated the presence of electron-donating and -withdrawing substituents on the aromatic ring. However, a significant decrease in bromination yield was observed for dimethoxy- and nitro-substituted aryl ketones. The recycling strategy of NBS, developed by Patel, the solvent- and catalyst-free nature as well as the short reaction times, highlighted the sustainable character of this methodology.
2.1.4. Halides (X−) as X2 Sources
Methods using halide anions (X−) are among the most promising methodologies as they offer a cheaper and safer alternative for direct electrophilic halogenation of ketones, especially when combined with environmentally benign oxidants, such as H2O2, molecular oxygen or even an electrochemical cell setup. Typically, these methods involve the use of MX, HX or NR3X in combination with an oxidant to generate in situ the active halogenation reagent, X2. An example using H2O2 in acidic media is shown in Scheme 25.
Metal Halides (MX)
Similarly to their NaNO2-I2 system (Scheme 25), Stavber and co-workers designed a NaNO2-KI-based procedure for the α-iodination of aromatic ketones, which yielded nine α-iodoketones in moderate to excellent yields (Scheme 26) [83]. KI exhibited similar iodination efficiency to molecular iodine when compared to the NaNO2-I2-mediated reaction [55]. Nonetheless, potassium iodide remains a much better choice than I2 as an iodination agent, as it is cheaper and safer to handle. It should be noted that a similar effect of the bromination solvent on the regioselectivity was observed, as described for the I2 reaction. When carried out in aqueous ethanol, selective α-bromination occurred. When the iodination was performed in dry acetonitrile, the aromatic ring was exclusively halogenated.
Another MX-based procedure was developed by Zhang and co-workers using NaCl and K2SO8 to perform the chlorination of arenes in acetonitrile [84]. The selectivity of the method was altered to exclusively lead to the α-chlorination when phenylethanone was used. Since the scope only consisted of one aryl ketone, broader screening is required for a better assessment of this route’s efficacy. A similar sodium chloride-based halogenation was reported in 2004 (Scheme 27) [85], with a broad range of alkanes, arenes and ketones being converted into their chlorinated derivatives. However, the method only showed moderate chlorination efficiency with aryl ketones.
In 2005, Gupta and co-workers reported the synthesis of α-bromoketones mediated by the combination of UHP and NaBr with acetic acid-functionalised silica. After microwave irradiation at 300 W, 12 α-bromoaryl ketones could be isolated in good yields (Scheme 28) [86,87]. The presence of electron-withdrawing, -donating and acid-sensitive groups on the phenyl ring was well tolerated. This route’s main features are its solvent-free conditions and the extremely short reaction times.
Quaternary Ammonium Halides (NH4X)
Over the past two decades, a significant amount of work has been dedicated to the development of NH4X- and Oxone®-mediated halogenation reactions. The high stability, water solubility and non-toxic nature make Oxone® an attractive oxidation agent. In 2012, Narender and Zhou independently described the chlorination of aromatic ketones in MeOH using a NH4Cl and Oxone® system (Scheme 29) [88]. The mechanism follows a similar path to MX-based oxidation approaches, generating in situ the X2 active reagents. Both protocols established a broad substrate scope although Narender’s method showed the best chlorination efficiency. The two procedures reported a similar trend, i.e., electron-poor aryl ketones were less efficiently chlorinated. No polychlorination or ring chlorination was observed for either route, stressing their excellent selectivity.
Using the same system, Narender and co-workers reported the α-bromination of 21 aromatic ketones in low to excellent yields (24–97%) (Scheme 30) [89]. Electron-deficient substrates (i.e., bearing CN and NO2 groups) appeared to be deactivated under these conditions, as they were isolated in only 21 and 35% yield. Furthermore, this route showed a rather moderate selectivity for these substrates, since α-bromo-dimethylketals were obtained as the major products. Additionally, some substrates showed signs of ring halogenation, adding to the system’s selectivity issues.
Narender and co-workers went on to develop another procedure for the α-iodination of ketones, generating a wide variety of α-iodoketones (Scheme 31) [90,91]. Again, a similar trend was observed; aryl ketones bearing electron-withdrawing groups were less efficiently iodinated. This route yielded a significant amount of either the ring iodinated or the α,α-diiodinated product for substrates carrying OH- and OMe-substituents. The iodination thus appeared to be less selective than the chlorination, which ultimately hinders its applicability.
One of the most interesting approaches employing ammonium halide salts is its combination with electrochemical oxidation conditions [92]. In an electrochemical cell equipped with a Pt/Pt electrodes, aryl ketones were efficiently brominated (Scheme 32). Under these conditions, Br− was oxidised to Br2, which then reacted with water to generate the HOBr active species. The broad substrate scope highlighted the versatile nature of this protocol, and despite the acidic conditions, acid-sensitive groups (i.e., OH) were well tolerated.
Makrandi and co-workers developed a solvent-free NH4Br-mediated α-bromination of alkanones [93]. After grinding a mixture of the starting material, NH4Br (2 equiv.) and (NH4)2S2O8 (2.5 equiv.) for 15 min at room temperature, 10 (hetero)aromatic ketones were isolated in good to excellent yields (83–92%). The procedure tolerated the presence of electron-withdrawing (i.e., Cl, NO2), -donating (i.e., OMe, Me) and acid-sensitive (i.e., OH) substituents on the aryl ketones. It should be noted that trace amounts of water were required to ensure complete homogenisation of the solid reaction mixture.
Hydrogen Halides (HX)
At first sight, HX might seem a valuable halogenation alternative to bromine, as it will ensure a high halide atom economy. However, the highly corrosive and toxic nature of this reagent still limits its applicability [94]. Nonetheless, a significant body of work has been reported on this topic and several noteworthy advances have been made.
Akamanchi and co-workers developed an aqueous HBr-based halogenation system for aromatic ketones [95]. In this system, the combination of NaNO2 and KI acted as the bromination catalyst (Scheme 33). The versatility of this route was demonstrated through the bromination of a broad range of aromatic ketones, bearing electron-withdrawing or -donating substituents.
Stavber and co-workers demonstrated the versatility of an NH4NO3- and I2-based system in air [96]. The procedure yielded seven α-chloroketones in moderate to good yields (Scheme 34). This protocol stands out due to its excellent selectivity, as no ring chlorination or dichlorination was observed.
Zhang and co-workers developed an HBr, NaNO2 and O2-mediated ring bromination, yielding a diverse set of halogenated arenes (Scheme 35) [97]. When using aryl ketones, α-bromination occurred instead, generating five α-bromoketones in moderate to excellent yields (68–92%). The NO2-substituted aryl ketones underwent a less efficient bromination, which aligns well with previous observations on the deactivation of electron-poor substrates. The use of molecular oxygen as co-oxidant should also be highlighted.
The use of a more environmentally friendly oxidant system, such as H2O2, has also been reported in combination with HBr, by Stavber and co-workers (Scheme 36) [98]. Interestingly, the bromination was carried out in water, further highlighting the sustainability of this system. 1,3-Diketones, β-ketoesters, and cyclic ketones were brominated in good to excellent yields. However, the scope only consisted of one aromatic ketone, again not making it possible to perform an evaluation of the strength of the protocol. 2-Bromo-1-phenylethanone was isolated in 78% yield. This method exhibited moderate selectivity, since for some of the substrates, unwanted dibromination was a serious issue.
Wakharkar and co-workers performed a comparative study between a H2O2 and a TBHP system with HBr as the bromine source [99]. A set of substrates, bearing electron-withdrawing (i.e., NO2, Cl) and -donating (i.e., OH) groups, were brominated in good to excellent yields (75–95%). Based on the experimental data, both TBHP and H2O2 had similar bromination efficiency. Ring bromination instead of α-bromination exclusively occurred for OH-substituted ketones, highlighting this routes’ selectivity issues.
DMSO was also shown to be an effective oxidant in the HBr-mediated bromination of ketones (Scheme 37) [100]. However, the substrate scope was again limited, making it difficult to evaluate the system’s efficacy and overall applicability.
An alternative HBr-based bromination was reported by Li and co-workers [101], which made use of Cu(NO3)2 (0.5 equiv.) and molecular oxygen to mediate the halogenation. Although the scope is not very broad, the bromination yields were quite good (83–94%). In most cases, dibromination of the substrate was observed, which again complicates purification.
The electrochemical α-bromination of aryl ketones was also reported by Kulandainathan and co-workers [102]. HBr was added to the ketone in acetonitrile in an undivided electrolytic cell containing two Pt electrodes (Scheme 38). A variety of aryl ketones were brominated in good to excellent yields. This method’s fine control over the experimental conditions through its set-up is quite intriguing. Despite the acidic conditions, the presence of acid-sensitive functional groups (i.e., OH) was well tolerated. In addition, this route showed excellent selectivity, as only monobrominated products were observed.
In 2008, a photocatalytic flavin-based method was developed for the chlorination of arenes and aromatic ketones [103]. Halogenases, enzymes catalysing the chlorination of aromatic compounds, provided the inspiration for this catalytic system. Stirring a solution of riboflavin tetraacetate (RFT), methoxy benzyl alcohol (MBA) and HCl in acetonitrile under visible light irradiation, afforded a range of chlorinated arenes. α-chlorination occurred when phenylethanone was used, generating the desired α-bromoketone (Scheme 39). The authors claimed that, under these conditions, the peroxide concentration was kept consistently low, preventing undesired polychlorination of the substrates. Despite the moderate chlorination yields, this route stands out as it is the only catalytic direct halogenation route to date, that mimics an enzymatic process.
ILs are generally considered better alternatives to typical organic solvents as reaction media [104], mainly due to their recyclability and the subsequent ease in product isolation, as biphasic systems can be readily designed. In 2013, Stavber and Laali developed an ethylammonium nitrate ([EtNH3+][NO3−])- and HX-mediated bromination and chlorination, yielding a variety of ring halogenated arenes. Under these conditions, the α-bromoketone was exclusively obtained in 90% yield upon addition of HBr and [EtNH3+][NO3−] to 1-phentylethanone (Scheme 40). However, the recurring problem of ring bromination was observed for the p-OMe substituted phenylethanone.
Another ring halogenation carried out in an IL was reported by Chiappe and co-workers [105]. Herein, the role of 3-methylimidazolium nitrate ([Hmim][NO3]) was shown to be twofold; it acts as a co-solvent and promotor of the halogenation (Figure 6). At this point, it is widely believed that the use of ILs in these reactions will always involve some sort of IL-based activation role. Under these conditions, aryl ketones showed exclusive α-chlorination. 2-Bromo-1-phenylethanone was isolated in 90% yield when the IL (2 equiv.) and an aqueous HCl solution (1 equiv.) were added to the corresponding ketone. Recycling experiments demonstrated that the IL could be reused in four successive runs without a significant loss in activity. The scope was limited to one aryl ketone, which again makes it difficult to assess this route’s applicability.
2.1.5. Polymeric Halogenation Agents
Poly(4-vinyl-N,N-dichlorobenzenesulfonamide) was first used for the halogenation of one aryl ketone, yielding 2-chloro-1-phenylethanone in a moderate 40% yield (Figure 7) [106]. The straightforward reaction conditions and recyclability of the chlorination agent are the main advantages of the protocol. However, prior to halogenation, the ketone deprotonation had to be performed separately using NaH.
In 2019, a simple and green bromination method was developed by Han and co-workers [107], whereby poly(vinylphenyltrimethylammonium tribromide) (PVBMATB) was synthesised by treating the Amberlite 717 resin with HBr and KBrO3 (Scheme 41). The resin proved to be an effective brominating agent as it accommodated the halogenation of eleven aryl ketones in moderate to excellent yields (60–96%). Ortho-substituted ketones were less efficiently brominated, highlighting this route’s limitations with sterically hindered substrates. PVBMATB provided a visual feedback system for the reaction progress, as its colour changed from red to gold when the reaction is completed. The authors claimed that the resin could be recycled and reused up to three consecutive runs with no significant loss in activity. This route is highly valuable from both economic and environmental points of view, associated with the low cost and recyclability of PVBMATB.
Weiss and others showed that a resin, obtained upon treatment of diphenylphosphine-functionalised polystyrene with CH3Br and Br2, exhibited an excellent bromination activity (Figure 8) [108]. The bromination of a variety of alkenes, alkynes and ketones was carried out in DCM, a swelling solvent for the polymeric agent. Only one aromatic ketone was halogenated via this protocol, as 2-bromo-1-phenylethanone was obtained in 98% yield. The α,α-dibromination was always observed, regardless of the nature of the substrate. For comparison, the authors designed a homogenous methyl-triphenylphosphonium bromide-catalysed halogenation in DCM which yielded 2-bromo-1-phenylethanone in 80% yield, which is lower than the heterogeneous conditions used above. This route’s main features are its simple work-up and product isolation, in conjunction with the recyclability of the bromination agent.
Liu and co-workers reported a polymer-supported halogenation agent which mediated the selective α-iodination of four aromatic ketones in excellent yields (Scheme 42) [109]. The polystyrene-bound selenium bromide was regenerated upon treatment with Br2. However, lithium diisopropylamide (LDA) had to be employed prior to halogenation to generate the nucleophilic enolate, which is not ideal, especially when other straightforward one-step options are available.
2.1.6. Ionic Liquids as Halogenation Agent
IL-based halogenation agents are gaining traction in the literature nowadays, mainly due to their contribution to the good practice ethos in terms of recyclability and reusability. In 2015, α-bromination and α-chlorination procedures mediated by, respectively, bis(2-N-methylimidazoliumethyl)-ether dibromochlorate and dichloroiodate, were reported (Figure 9) [110]. Eight α-bromoaryl ketones were isolated in excellent yields (88–95%), upon the addition of bis(2-N-methylimidazoliumethyl)-ether dibromochlorate (0.5 equiv.) to the corresponding ketones under reflux for 8–24 h. Similarly, the reaction with the dichloroiodate analogue generated eight α-chloroketones in 88–93% isolated yield. This procedure tolerated the presence of electron-withdrawing (i.e., Br, Cl, NO2), -donating (i.e., Me, OMe) and acid-sensitive (i.e., OH) groups on the substrates, highlighting its wide applicability. The authors claimed that the ILs can be regenerated and reused; however, no experimental details were provided to support this claim.
Veisi and Sedrpoushan illustrated that 1,4-bis(3-methylimidazolium-1-yl)butane ditribromide was an effective bromination agent for arenes and aryl ketones (Figure 10, left) [111]. Upon addition of the IL (0.5 equiv.) in CH3CN, α-bromination of aryl ketones was achieved, yielding three α-bromoketones (i.e., p-H, -Me, -OMe) in excellent yields (92–96%). The authors demonstrated that the IL could be regenerated and reused for five successive cycles without a significant loss in activity. The straightforward isolation of the brominated products further highlighted this route’s sustainable character (i.e., adding water to the reaction mixture).
Another IL-based brominating agent, immobilised on Fe2O3 support, was successfully utilised for the bromination of a variety of alkenes, alkynes, arenes and aryl ketones (Figure 10, right) [112]. The bromination agent was easily separated from the reaction mixture by using an external magnet. When recovered, the authors claimed that the reagent could be reused up to six consecutive runs without any significant loss in activity. Four α-brominated aromatic ketones were isolated in good to excellent yields (80–90%). The presence of electron-donating substituents decreased the reaction time from eight (for phenylethanone) to six hours (for the p-OMe substituted analogue). Aryl ketones bearing electron-withdrawing groups (i.e., p-Cl and o-NO2) showed a similar trend.
An efficient IL-mediated bromination protocol was also developed by Coa and Hu [113]. [Bmim][Br3] mediated the bromination of six aromatic ketones in good to excellent yields (Scheme 43). The procedure tolerated the presence of both electron-donating and -withdrawing substituents on the phenyl ring. The authors stated that the IL could be recycled several times without any loss in bromination efficacy; however, no experimental data were provided in support of this claim. The short reaction times were an important feature of this IL-based halogenation.
In 2007, Ranu and co-workers designed an acetylmethylimidazolium halide ([Acmim]X) (X = Cl−, Br−) and ceric ammonium nitrate-based α-chlorination and α-bromination of ketones (Figure 11, left) [114,115]. Addition of the IL (1.2 equiv.) and ceric ammonium nitrate (CAN) (2 equiv.) to an aryl ketone, yielded three α-chloro and three α-bromoketones (i.e., p-H, -Me and -OMe substituted) in good yields (80–82% and 80–85%, respectively). The authors mentioned that the selective α-halogenation of this protocol is linked to its radical nature.
Alternatively, a solvent-free protocol mediated by pentyl-pyridinium tribromide was designed and reported by Dorta and Salazar (Figure 11, right) [116]. Only one aryl ketone was tested, as only 2-bromo-1-phenylethanone was isolated in 85% yield. Furthermore, the neat conditions were offset by using ether for extraction purposes during the work-up. The authors stated that the tribromide reagent could easily be regenerated from the reaction mixture using Br2.
Patel and co-workers designed another recyclable bromination agent, i.e., hexamethonium bis(tribromide) (HMBTB) (Figure 12, left) [117]. The procedure involved grinding a solid mixture of HMBTB (0.5 equiv.) and ketones in a mortar for 20–60 min at room temperature. The method demonstrated a broad substrate scope, with aromatic ketones showing exclusive α-bromination. Similarly, the solvent-free α-bromination was also mediated by 1,2-dipyridiniumditri-bromide-ethane (DPTBE) (Figure 12, middle) [118]. DPTBE (0.5 equiv.) was added to the aromatic ketone, and subsequent grinding of the solids in a mortar at room temperature for 1–1.5 h yielded the desired product. This route tolerated the presence of electron-withdrawing (i.e., Cl, NO2) and electron-donating groups (i.e., OMe, OCH2Ph). Five para-substituted α-bromoketones were isolated in good to excellent yields (82–89%). Other tribromides, such as pyridinium hydrotribromide (PHTB), also showed excellent bromination activity (Figure 12, right). PHTB mediated the bromination of several para- and meta-substituted aromatic ketones (i.e., -OMe, Cl, NO2, Br) in good to excellent yields (70–98%) [119].
2.1.7. Miscellaneous Halogenation Agents
Other halogenation strategies include the use of transition metal halides. Sun demonstrated the halogenation activity of copper halides in the direct halogenation of ketones [120]. Addition of Ti-Al binary oxides (0.02 equiv.) and CuX2 (2 equiv.) to phenylethanone, yielded the corresponding α-bromo and α-chloroketones in 42 and 56% isolated yield, respectively. The bromination reaction was carried out in acetonitrile, while the chlorination required formic acid as solvent. For both, the α,α-dihalogenated species were observed as an important side product (isolated in 20% and 10% yield, respectively), highlighting this route’s moderate selectivity.
In contrast to Sun’s route, a broader scope of α-bromoketones were reported by Cai, Peng and An [121,122]. Eleven ketones were brominated upon the addition of 1.5 equiv. of CuBr2 (Scheme 44). At first sight, the mechanism seemed to indicate that at least 2 equiv. of CuBr2 are needed to achieve full conversion; however, the first step involving the formation of the copper enolate could occur with CuBr instead. Additionally, CuBr in the presence of HBr can disproportionate back to CuBr2, thus regenerating the active species. The procedure tolerated the presence of electron-donating and -withdrawing groups on the aryl substrates. NO2-substituted aromatic ketones underwent a less efficient halogenation compared to the unsubstituted aryl ketone. Nevertheless, other electron-poor substrates (i.e., bearing Cl, Br, I substituents) did not exhibit such a decrease in bromination efficiency. An inherent drawback of these CuX2-mediated reactions is the moderate halogen atom economy.
Another transition metal-based protocol for the halogenation of ketones was reported by Zhang and co-workers using FeCl3·6H2O and phenyliodinium diacetate in HOAc [123]. Addition of iron chloride (2 equiv.) and oxidant (1.2 equiv.) to the substrate, yielded 25 α-chlorinated aryl ketones in moderate to good yields. The acidic reaction conditions limited the substrate scope, as it decreased this route’s functional group tolerance (e.g., NHx, OH).
Tian and Sun developed a DMSO and oxalyl bromide ((COBr)2)-based bromination method for a diverse set of alkenes, alkynes and ketones (Scheme 45) [124]. The procedure leads to moderate selectivity, as the α-brominated and α,α-dibrominated products were obtained in a 1:1 ratio. Despite the relatively mild reaction conditions and short reaction times, this route does require further optimisation for more selective α-bromination.
Both Tajbakhsh and Heravi illustrated that 1,4-diazabicyclo[2,2,2]octane (DABCO) can be employed as a halogenation agent (Scheme 46) [125,126]. Heravi and co-workers used tetrameric DABCO-bromine (TBD) supported on silica or alumina to efficiently mediate the α-bromination of four aromatic ketones in good yields. The non-hygroscopic supported solid performed the bromination under mild reaction conditions with an easy work-up (simple filtration). Tajbakhsh and co-workers, on the other hand, employed DABCO as a chlorination agent after treatment with chlorine gas, affording 1,4-dichloro-1,4-diazabicyclo[2,2,2]octane (DCDABCO) (Scheme 46). A set of alkenes, alkynes and ketones were chlorinated via this method, among which stands one, aryl ketone. The authors claim that DCDABCO can be reused for four consecutive reactions without significant loss in activity.
SiCl4 was also shown to be an excellent chlorinating reagent. In 2010, El-Ahl illustrated that a SiCl4 and UHP/iodosylbenzene-based system mediated the chlorination of a broad range of ketones in good to excellent yields (Scheme 47, Figure 13) [127]. Aryl ketones underwent exclusive α-bromination, highlighting the excellent regioselectivity of this route.
An alternative versatile α-halogenation method was reported by Prakash and Mathew in 2011 [128]. An effective halogenation agent was obtained upon the coupling of bromo- or chlorotrimethylsilane with KNO3. The procedure yielded eight α-chloroketones and five α-bromoketones (Scheme 47). Dihalogenation was a burdensome issue in this Si-mediated reaction. The α,α-dihalogenated products were obtained for each substrate, complicating purification.
Zou demonstrated that in the α-chlorination of aromatic ketones, 1,3-dichloro-5,5-dimethyl-hydantoin (DCDMH) acts as an inexpensive chlorination agent (Figure 14) [129]. A silica gel-supported route and a pTsOH-catalysed method were therefore developed. The former converted the aromatic ketones with a significantly higher efficiency than the latter. Together with the shorter reaction times (1 h vs. 8 h, respectively), the SiO2-supported route seemed to be superior. However, this route was not as selective as the pTsOH promoted version. The chlorination of nitro-substituted aromatic ketones yielded a mixture of the α-chlorinated ketone and the ketal. Furthermore, for methoxy-substituted substrates, ring chlorination was exclusively observed.
Alumina-supported α-bromination and α-iodination protocols have been developed [130]. Gupta described a solvent-free hexamethylenetetramine-bromine (HMTAB)-mediated halogenation. Eleven aromatic α-bromoketones were obtained in good yields (70–80%) upon grinding HMTAB (1.6 equiv.), basic alumina and the substrate in a mortar. The homogenous solid mixture was subsequently irradiated in a MW for 5–10 min. Electron-withdrawing and -donating groups were well tolerated, as well as acid-sensitive ones (e.g., OH, NHx). The main advantages of this procedure are its short reaction times and the solvent-free conditions.
CAN demonstrated excellent catalytic activity in the α-chlorination of (a)cylic ketones [131,132]. This route employed the rather unconventional acetyl chloride as the chlorine source, yielding one α-chloroarylketone (i.e., 2-chloro-1-phenylethanone) in 87%. The authors claimed that α-chlorination was exclusively observed.
In 2019, Matsubara and co-workers designed a bis(1,3-dimethyl-2-imidazolidinone)hydrotribromide (DITB)-mediated halogenation [133]. The α-bromination of a set of ketones was established upon the addition of DTIB (1.1 equiv.). Seven α-bromoketones could be isolated among which five aromatic examples could be isolated in moderate to good yields (Scheme 48). Methoxy-substituted aryl ketones were obtained in significantly lower yields, which was ascribed to dibromination of that substrate.
2.2. Nucleophilic Halogenation
The nucleophilic halogenation approach to generate α-halogenated ketones is much less attractive than its electrophilic counterpart, mainly due to the need for an intermediary step to install a leaving group on the carbon atom of the enolate. This in turn generates unnecessary waste. Nonetheless, in this section, we would like to mention a few reports to better clarify this approach.
A clear example of this strategy can be found in the report by Lee and co-workers describing the solvent-free MgX2 and HTIB-based halogenation (Scheme 49) [134]. The generality of this route was demonstrated by the bromination, chlorination and iodination of a set of aryl ketones under the same reaction conditions. Phenylethanone derivatives were brominated, chlorinated, and iodinated in good to excellent yields (82–91%, 80–90% and 65–75%, respectively). The procedure was thus very effective for both the bromination and chlorination; however, a slight decrease in efficiency was observed for the iodination. This route tolerated the presence of both electron-withdrawing (i.e., Br, NO2) and -donating groups (i.e., OMe) on the substrates. The reaction is performed in two steps; the first one using HTIB installs an OT leaving group on the α-carbon atom, and the following step consists of Br− nucleophilic substitution on the same carbon to afford the corresponding α-halogenated ketone.
In an attempt to circumvent the need for stoichiometric amounts of a leaving group, Lal and co-workers designed and reported a dioxidevanadium(V)-catalysed bromination (Scheme 50) [135]. Five ketones were brominated in good to excellent yields. Remarkably, the procedure tolerated acid-sensitive substituents (i.e., OH) on the aromatic ring. However, large amounts of H2O2 as oxidant were needed. The authors gave a plausible mechanism, involving the initial formation of vanadium C-enolate via C-H activation of the ketone, followed by a Br− nucleophilic attack on the same carbon, which liberates the vanadium complex and generates the α-bromoketone, thus closing the catalytic cycle. However, in our opinion, an electrophilic halogenation mechanism could not be excluded, since the protocol operates under harsh oxidative conditions; H2O2 or even the vanadium catalyst can readily oxidise Br− into Br2 (or HOBr after reaction with water), which would then be capable of reacting directly with the ketone.
3. Oxyhalogenation of Hydrocarbons
In the last two decades, alternative protocols have been developed that have targeted the numerous issues raised by the direct halogenation approach. A vast amount of work has been dedicated to the oxyhalogenation of alkenes, alkynes, and secondary alcohols. Nonetheless, these routes either only partially tackled the problems associated with the direct halogenation (e.g., oxidation often required an acidic promotor), or present moderate efficacy and/or selectivity. Herein, we showcase some of the most attractive strategies.
3.1. Alkenes as Substrates
Styrene derivatives have proven to be excellent oxyhalogenation substrates. In 2020, Zhu and co-workers developed a FeX3-catalysed oxyhalogenation mediated by KX in methyl tert-butyl ether (MTBE) (Scheme 51) [136]. Fourteen α-bromo and fourteen α-chloroketones were isolated in moderate to good yields. Electron-poor, -rich and sterically hindered substrates were efficiently functionalised by the developed procedure. However, the protocol failed in the oxyhalogenation of p-OH substituted alkenes, attributed to the use of TsOH. The reaction proceeds via a radical mechanism, where the Br radical is trapped by the styrene derivative. This radical intermediate is then captured by O2 and FeBr2, affording a peroxide intermediate which upon water elimination yielded the corresponding halogenated ketone. Additionally, the oxyhalogenation presents a substrate specific character, as the oxychlorination yields were significantly lower than the oxybromination relatives.
Another metal-catalysed oxybromination of olefins was established using a molybdenum(VI)-based complex (Scheme 52) [137]. 2-Bromo-1-phenylethanone was obtained in 73% yield upon addition of 0.2 mol% of a cis-dioxomolybdenum(VI) complex to a mixture of H2O2, KBr, HClO4 and styrene. The bromohydrin and epoxide were isolated as important side products (in 15 and 11%, respectively), highlighting the moderate catalyst selectivity.
Guo and co-workers reported the oxybromination of 11 styrene derivatives upon the addition of KBr (2 equiv.), K2S2O8 (2.5 equiv.) in H2O (Scheme 53) [138]. After stirring for 12 h at 60 °C, the corresponding aryl bromoketones were isolated in low to excellent yields (24–90%). The method tolerated substrates bearing electron-donating (e.g., Me, t-Bu) or -withdrawing (e.g., F, Cl, Br) substituents on the aromatic ring. A significant decrease in hydration efficiency (i.e., 24%) was observed for the OMe-substituted styrene derivative. Furthermore, this route was not compatible with heteroaromatic substrates. Despite its shortcomings, the developed procedure did benefit from readily available starting materials and a safe halogen source. The mechanism involves the initial K2S2O8-mediated oxidation of Br− to Br2, which leads to the formation of HOBr in the presence of water. Addition of HOBr onto the alkene affords the bromohydrin intermediate. Upon oxidation of the latter, the corresponding bromoketone is obtained. In 2019, the authors slightly altered their oxyhalogenation procedure [139]. A KI- and TBHP-based system transformed styrene into the corresponding α-iodoketone in 97% yield.
Adimurthy and Iskra independently established an aqueous H2O2-based oxyhalogenation method [140,141]. Both authors reported the use of HBr as a bromination agent, which generated the corresponding α-bromoketone and the bromohydrin (Table 2). Iskra and co-workers demonstrated that using NBS instead of HBr considerably shifted the selectivity towards the α-haloketone. Despite enhanced selectivity, the hydrin intermediate was still observed in 19% yield. Furthermore, the reported substrate scope consisted of only styrene. The α-haloketone selectivity appeared to be less problematic in the Adimurthy route (Table 2), and a broader scope was established; eight aromatic α-bromoketones were isolated in low to excellent yields (20–94%). Although electron-donating aromatic substituents (e.g., Me, t-Bu) were well tolerated, electron-withdrawing substituents (e.g., Cl, Br, NO2) were not. Moreover, the presence of steric bulk close to the alkene centre appeared to have a detrimental effect on the system’s efficiency.
Adimurthy and Ranu discussed the use of HOBr in the oxybromination of alkenes (Scheme 54) [142]. Their procedure involved the addition of two equivalents of HOBr to various olefins in 1,4-dioxane. Several α-bromoketones were obtained in moderate to excellent yields (40–87%). Regardless of the electronic properties of the substrate, the bromohydrin analogue was consistently observed in significant amounts.
Moorthy and co-workers designed a NXS oxyhalogenation of olefins in DMSO, which employed 2-iodoxybenzoic acid (IBX) as the oxidant [143]. More recent work involved the in situ generation of an IBX derivative via Oxone® (Scheme 55) [144]. This versatile method generated a set of para- and ortho-substituted α-bromoketones in good to excellent yields (79–94%). Electron-withdrawing and -donating groups on the substrates were well tolerated by the NBS and Oxone® system. Meanwhile, Mal and co-workers demonstrated the solvent-free oxyhalogenation of a variety of alkenes using a NXS (1.1 equiv.) and IBX (2 equiv.) system [145]. The procedure generated 2-bromo- and 2-iodo-1-phenyl-ethanone in, respectively, 86% and 89% yield. The authors claimed that the obtained ketones did not require chromatographic purification, facilitating product isolation significantly. They further stated that the oxidant could be recycled via its waste product 2-iodosobenzoic acid. However, product loss was observed and attributed to the high volatility of the halogenated products.
Phukan and co-workers designed an effective N,N-dibromo-p-toluenesulfonamide (TsNBr2)-mediated transformation of alkenes into their corresponding α-bromoketones [146]. Both electron-poor and -rich styrene derivatives were efficiently converted when adding TsNBr2 (2.2 equiv.) to the starting material in an acetone/H2O mixture at room temperature. The resulting products were isolated in good to excellent yields (80–87%). This route stood out due to its mild reaction conditions and good efficiency, although it should be noted that TsNBr2 is not readily accessed [147,148]. In 2017, Loginova et al. reported a ClO2-mediated oxidative chlorination of styrene [149]. This route displayed poor selectivity issues as next to the desired α-chloroketone, five side-products were obtained, significantly complicating the product isolation process (Scheme 56). Furthermore, chlorine dioxide is an extremely toxic and dangerous reagent; its use thus requires strict safety precautions.
Chen and co-workers designed an electrochemical Mn-catalysed oxychlorination procedure, using a reticulated vitreous carbon (RVC)-based anode and cathode (Scheme 57) [150]. Fourteen electron-poor and -rich α-chloroketones were isolated in low to excellent yields. The developed route was not compatible with sterically hindered substrates, as the ortho-substituted product was obtained in only 37% yield. As the FeBr3-based protocol, the role of a peroxide intermediate was noted.
Itoh and co-workers developed an aerobic photo-oxidative protocol yielding α-haloketones from their styrene derivatives [151]. The reaction conditions and scope are depicted in Table 3.
In 2007, Itoh described the synthesis of α-iodoketones upon irradiation with a fluorescent lamp of a mixture of I2 (1 equiv.), O2 and FSM-16 (mesoporous silica) in EtOAc; with FSM-16 acting as a heterogeneous catalyst (Scheme 58) [152]. Several α-iodoketones were obtained from their styrene derivatives in moderate to good yields (38–77%). The experimental data showed that the system coped well with the presence of electron-withdrawing (e.g., Cl, NO2) and -donating (e.g., Me, t-Bu) substituents on the aromatic ring. However, this route was incompatible with sterically hindered alkenes.
Zhang and co-workers utilised 1,3-dibromo-5,5-dimethylhydantoin (DBH) for the oxybromination of styrene derivatives in H2O [153]. The role of DBH (2 equiv.) was twofold, since it acted as an oxidant and as halogenation agent. This protocol yielded 11 aromatic α-bromoketones bearing a diverse set of aromatic substituents (34–85%). This route was not compatible with sterically hindered alkene centres, as the o-NO2 substituted ketone was isolated in only 34% yield. In 2020, Wang and Zhang reported an elaborate scope (18 examples) of α-iodoketones, obtained in good to excellent yields (73–96%), from corresponding olefins [154]. The reaction was catalysed by visible light under mild experimental conditions (Scheme 59).
3.2. Alkynes as Substrates
Similarly to the alkene systems mentioned above, I2- and IBX-based oxyiodination of aromatics was also applied to alkynes in water (Scheme 60) [155]. The protocol showed good tolerance towards electron-donating and electron-withdrawing substitutions on the aromatic alkynes. The role of IBX is to facilitate the initial addition step of I+ and water to alkyne, most likely to generate the I+ active species from I2. Afterwards, the formed iodo-enol compound would tautomerise to the iodoketone form, affording the target compound.
Ahmed and co-workers described an alternative I2-based oxyhalogenation protocol which generated 11 α-chloroketones in good yields (69–79%) upon the addition of HCl to the corresponding phenylacetylene derivatives in DMSO [156]. The protocol tolerated the presence of electron-withdrawing (i.e., F, Br, Cl, CF3) and -donating (i.e., Me, n-Bu) aromatic substituents. Interestingly, the presence of steric bulk near the alkyne centre (ortho-substitution of the aromatic ring) did not affect its efficacy. Acid-sensitive substrates were not tolerated under these conditions, thus limiting the reaction scope.
Phukan and co-workers reported the oxybromination of aromatic alkynes mediated by a TsNBr2 and Na2SO3 system (Table 4) [147]. A broad scope of substrates were efficiently transformed into the desired products, with exclusive formation of the α-bromoketone observed. Nonetheless, the long reaction times and the use of the non-commercially available TsNBr2 could be a major hindrance for its widespread application. He and co-workers investigated the same reaction using a commercially available halogenating reagent, 1,3-dibromo-5,5-dimethyldantoin (DBDMH) (Table 4) [157]. The authors illustrated this route’s versatility as it yielded α-bromo-, α-chloro- and α-iodoketones without the need to alter the reaction conditions (this was achieved simply by changing the halogenating reagent, 1,3-dihalo-5,5-dimethyldantoin).
Pitchumani and others established a route involving the addition of Br2 to phenylacetylene in cyclodextrin (CD) [158]. An alkyne-CD complex was formed upon the interaction of the substrate in the cavity of the cyclodextrin molecule. The experimental data showed that formation of 2-bromo-1-phenylethanone is enhanced with increased cavity size. In α-CD (6 glucose units), only 13% of the ketone was observed, whereas in γ-CD (8 glucose units), the GC yield of the ketone increased to 46%. This route stands out as it is the first to demonstrate such a complexation effect on the selectivity of oxyhalogenation reaction.
A gold-based oxidative halogenation method was designed by Xiang and Zhang using [Au(NTf2)(IPr)] as a catalyst, 8-methylquinoline N-oxide as the oxidant and methanesulfonic acid (MsOH) [159]. One α-chloroketone and two α-bromoketones were obtained via the halide abstraction of the solvent (i.e., dichloro- or dibromoethane). The results, illustrated in Scheme 61, show the poor catalytic activity of the selected AuI complex in this transformation.
In 2017, Xing and co-workers demonstrated a AuIII system accommodating the oxyhalogenation of terminal alkynes (Scheme 62) [160]. This route yielded aromatic, aliphatic and heteroaromatic α-bromoketones (15 in total) in good to excellent yields (73–91%). The versatility of this route was further highlighted as it also generated five α-chloroketones in good to excellent yields (75–90%) and two α-iodoketones in good yields (81–85%). Although this route seemed to be promising, it brings a number of limitations as the use of silver salts imposed several drawbacks; their limited air- and light-stability, as well as the known interference of Ag in the AuI catalysis [161]. The proposed mechanism involved the formation of the ketone first, followed by the halogenation when NXS is added. The authors proposed that the formation of the ketone is initially driven by the Au-catalysed addition of MeOH to the alkyne; however, in our opinion, this would be improbable. The more plausible route would be the Au-catalysed hydration of the alkyne, most likely using residual water present in the solvents used.
4. Oxidative Halogenation of Secondary Alcohols
Closely related to the typical oxyhalogenation route, this method relies on the prerequisite setup of a secondary alcohol group and offers straightforward access to α-haloketones. Terent’ev and co-workers developed a H2O2/HBr system which converted three secondary benzylic alcohols into their corresponding α-bromoketones in moderate to good yields (52–85%) (Scheme 63) [162]. The system was not compatible with ortho-substituted 1-phenylethanol derivatives, as the reaction yield dropped significantly (52%). The developed method showed outstanding selectivity, as exclusive formation of the α-haloketone was observed. The authors claimed that the role of the ratio HBr/H2O2 (1.2:10) is crucial to obtain these results. H2O2 is most likely responsible for oxidizing the corresponding alcohol to generate the carbonyl moiety and for oxidizing Br− to Br2 (or HOBr), the active bromination agent.
The dual role of a urea-choline chloride-based deep eutectic solvent (DES) in the oxidative bromination of secondary alcohols was demonstrated by Azizi and co-workers (Scheme 64) [163]. 2-Bromo-1-phenylethanone was obtained in 68% yield when adding three equivalents of NBS to the corresponding alcohol in the DES. Remarkably, the DES both acted as a catalyst system and solvent in this transformation. Recent studies illustrated the superiority of DES in comparison to ILs, linked to their non-toxic nature, easy and low-cost synthesis [164]. Although the mechanism was not fully disclosed, NBS was most likely responsible for both the bromination and oxidation, with DES activating the N-Br bond.
Narender and co-workers established an aqueous, green and efficient way to generate α-iodoketones from secondary alcohols via addition of NH4I and Oxone® (Scheme 65) [165]. The procedure generated a yield of 72% of 2-iodo-1-phenylethanone, but the yield dropped significantly for alcohols bearing substituted aromatic rings (i.e., p-Me, p-Br, p-NO2, m-OMe). The developed method proved inefficient for substituted 1-phenylethanol derivatives. In 2017, the authors updated the procedure in order to access α-bromoketones via their corresponding alcohols upon the addition of NH4Br (2 equiv.) and Oxone® (3 equiv.) [166]. This altered procedure yielded 10 α-bromoketones in low to excellent yields (38–94%). Nitro-substituted benzylalcohols appeared to be deactivated in the developed system. Furthermore, undesired ring bromination exclusively occurred for methoxy substituted 1-phenylethanol derivatives. Despite the progress made in terms of sustainability, this route still shows a rather moderate oxyhalogenation efficiency and selectivity.
In 2014, a method for the oxychlorination of 1-phenyl-ethanol was reported by Studer and co-workers (Scheme 66) [167]. Ten α-chloroketones were isolated in good to excellent yields (70–87%) by stirring the alcohol substrates with a mixture of trichloroisocyanuric acid (1 equiv.), TEMPO (0.1 equiv.) and MeOH (2 equiv.) in DCM for 5 h. The presence of electron-donating and -withdrawing groups on the substrates was well tolerated. The protocol stands out as it employs a commonly used bleaching and disinfectant agent (i.e., trichloroisocyanuric acid) in a chemical process.
5. Oxidation of Functionalised Hydrocarbons
A different approach to circumvent the problems associated with the direct halogenation route involved using functionalised hydrocarbons as synthons for α-haloketones. In these cases, the halogen is already affixed to the substrate, thus simplifying the process. However, in some of these cases, the substrates could be found as intermediates in the above-mentioned processes (i.e., direct halogenation and oxyhalogenation). Additionally, overall, the fixation of the halogens on the substrates employed in this section could also be burdensome and in some cases limited.
5.1. Haloalkanes as Substrates
Mohammadi and co-workers described the synthesis of 2-bromo-1-phenylethanone from its corresponding bromoalkane in 85% yield [168]. The unconventional 1,4-bis(triphenyl phosphonium)butane peroxodisulfate was employed as the oxidant (Scheme 67). The reaction time was quite short; however, only one example was shown.
Similarly, Jiang and co-workers established a photocatalytic oxidation procedure for the oxidation of a broad range of benzylic alkanes [169]. The corresponding carbonyl compounds were obtained upon the addition of a dicyanopyrazine-derived chromophore (5 mol%), N-hydroxyimide (0.2 equiv.) and FeC2O4·2H2O (0.1 equiv.), with subsequent irradiation of the obtained mixture using a 3 W blue LED light. The authors showed only two examples of α-bromoketones, i.e., 2-bromo-1-phenyl-ethanone and its para-chloro-substituted derivative, in 83% and 55% yields, respectively. The extremely long reaction times (63–66 h) were also quite limiting.
5.2. Haloalkenes as Substrates
Synthesis of 2-chloro- and 2-bromo-1-phenylethanone in, respectively, 78 and 76% yield using molecular oxygen as oxidant was described by He and co-workers (Scheme 68) [170]. The developed method involved stirring a mixture of n-octyl phenylphosphinate (3 equiv.) and the haloalkene at 130 °C for 15 h in the presence of a O2 balloon. This route generated only one side product, i.e., water, highlighting its sustainability.
Jiang and co-workers developed a photocatalytic oxidation of vinyl halides [171]. Five α-bromoketones and five α-chloroketones were obtained in moderate to excellent yields (Scheme 69). The presence of electron-withdrawing (e.g., F, NO2) and -donating (e.g., Me) substituents on the aromatic ring was well tolerated by the catalytic system. The mechanism involved a Fe(III)-Fe(II)-Fe(III) catalytic cycle for the O2-assisted radical oxidation of the vinylic substrates followed by the halogen rearrangement of the terminal position.
An alternative FeIII catalytic oxidation protocol was designed by Xiao [172]. Two different methods were reported as illustrated in Table 5. Method 1 exclusively generated the desired α-bromoketone, while the second method yielded de-halogenated ketones as important side products for some substrates. The ligand’s properties thus seemed to have a considerable effect on the protocol selectivity. Nonetheless, a detailed comparison between both methods is difficult due to the limited scope of method 1.
5.3. Haloalcohols as Substrates
Haloalcohols or halohydrins were also intermediates in the oxyhalogenation reactions discussed above. Therefore, it is important to note the close relationship between the two sections and that protocols discussed in this section could also help to advance the oxyhalogenation strategies.
Wang and Dong independently developed a micellar catalytic oxidation of alcohols [173,174]. Both authors reported the successful conversion of 2-bromo-1-phenylethanol into its corresponding ketone (Table 6). Wang and co-workers enhanced the substrate’s water solubility upon addition of CTAB, a commercially available cationic surfactant, to the reaction medium. Similarly, Dong and co-workers ensured the sufficient solubility of the substrate by adding GMPGS-2000, a non-ionic surfactant. Both procedures exploited the concept of micellar catalysis, which truly distinguished them from the state-of-the-art. However, one major flaw of these micellar catalytic systems concerns the often tedious work-up, attributed to the difficult separation of the organic and aqueous phase [175].
A copper-catalysed oxidation of secondary alcohols was designed by Liu and co-workers [176]. 2-Bromo-1-phenylethanone was isolated in 95% yield upon addition of Cu(OTf) (0.05 equiv.), TEMPO (0.05 equiv.), (S)-5-(pyrrolidin-2-yl)-1H-tetrazole (0.05 equiv.) and KOtBu (1 equiv.) to the corresponding alcohol in dimethylformamide (DMF) at room temperature. However, no reaction/substrate scope was disclosed. Another limited protocol was developed by Nguyen and co-workers using AlMe3-catalysed Oppenauer oxidation (Scheme 70) [177]. The scope again consisted of one α-haloketone, i.e., 2-bromo-1-phenylethanone, making it difficult to assess the method’s efficiency.
He and co-workers described the bis(methoxypropyl)ether (bMPE)-promoted oxidation of benzylic alcohols [178]. The procedure yielded one α-bromo- and one α-chloroketone in good yields (Scheme 71). Although again the scope was limited, the main advantage of this protocol is its solvent-free conditions.
In 2014, Gotor published a T. versicolor Lacasse and TEMPO system, which converted haloalcohols into their corresponding ketones [179]. T. versicolor Lacasse is an extracellular enzyme excreted by white and brown fungi. A broad scope of α,α-dihalogenated aryl ketones were obtained in good to excellent yields. The chemoenzymatic procedure also yielded one α-halocarbonyl compound, i.e., 2-chloro-1-phenylethanone, at a yield of 62%. Despite the mild reaction conditions, the long reaction times (24 h) and the moderate yield still limited the method’s applicability.
6. Hydration of 1-Haloalkynes
The hydration of haloalkynes is a less commonly described approach to access α-haloketones. Despite the plethora of work that involved the hydration of (terminal) alkynes [180,181,182,183,184,185,186,187,188], this strategy has only scarcely been employed in the synthesis of α-haloketones. The hydration is an excellent example of ideal atom economy. Furthermore, this transformation often only requires the presence of one reagent, i.e., H2O, highlighting its green character. This route thus offers immense value from both economic and environmental perspectives.
6.1. Metal-Catalysed Hydrations
Similarly to the hydration of alkynes [187,188], gold complexes have exhibited an excellent catalytic activity in the hydration of 1-haloalkynes [189]. Cai and co-workers described the hydration of 1-haloalkynes catalysed by a gold catalyst immobilised on mesoporous MCM-41 resin [190]. The versatility of this heterogeneous catalytic route was demonstrated by its broad substrate scope. Sixteen α-bromo, four α-chloro and four α-iodoketones were obtained in excellent yields (Scheme 72). The authors claimed that the catalyst can be recycled and reused for eight consecutive runs without significant loss of catalytic activity. Additionally, they stated that after these eight runs, gold leaching did not occur. The excellent atom economy, mild reaction conditions, the generality and catalyst recyclability elevated the importance of this protocol compared to the state-of-the-art. In 2019, the same group reported an updated procedure relying on the in situ activation of MCM-41-[Au(Cl)(PPh3)] (2 mol%) by AgNTf2 [191]. Despite the decreased catalyst loading, this route’s scope was very limited, as only 2-bromo-1-phenyl-ethanone (92%) was obtained.
Another gold-catalysed protocol using [Au(NTf2)(XPhos)] to perform the hydration of 1-bromo and 1-chloroalkynes was designed by He and Xiang [192]. Ten aromatic α-bromoketones and two aromatic α-chloroketones were isolated in low to excellent yields (up to 97% and 91%, respectively), upon of addition of 3 mol% of the catalyst and water (3 equiv.) to the 1-haloalkyne in dichloroethane. The presence of electron-donating (i.e., Me, t-Bu, OMe) and -withdrawing (i.e., F, Br) groups on the substrates was well tolerated. However, the hydration efficacy of the system decreased significantly for sterically hindered substrates. The hydration of the ortho-substituted 1-haloalkynes yielded the corresponding ketones in trace amounts. Furthermore, the protocol demonstrated a substrate specific nature, as it did not accommodate the hydration of 1-iodoalkynes. More recently, Cazin and co-workers developed an efficient catalytic protocol for the Au(I)-catalysed hydration of 1-iodoalkynes (Scheme 73) [193]. The use of Au(I)-NHC catalysts enabled the straightforward synthesis of several aromatic α-iodomethyl ketones in good to excellent yields. The utility of this simple method was further highlighted by showcasing iodination/hydration and hydration/oxidation sequential protocols leading to the construction of molecular complexity.
A broad scope of α-haloketones was also obtained via the Cu(OAc)2·H2O and trifluoroacetic acid (TFA) promoted hydration system developed by He (Table 7) [194]. This route coped well with substrates bearing electron-withdrawing and -donating groups. The authors claimed that the catalyst can be recycled and reused for five consecutive runs without loss in hydration activity. In 2014, Lui and co-workers reported the remarkable catalytic activity of the AgF/TFA system in the hydration of alkynes (Table 7) [195]. This system tolerated a wide variety of functional groups attached to the phenyl ring (i.e., CF3, Br, Me, NO2, OMe, etc.). The procedure showed a substrate specific character as it did not mediate the hydration of 1-iodoalkynes. The use of the strongly acidic TFA is a major drawback, and negatively impacts the functional group tolerance of both routes.
Lee and Lixin independently described an FeClx·yH2O-catalysed hydration [196,197]. Their systems required the presence of methanesulfonic acid (MsOH) in order to transform 1-haloalkynes in sufficiently high yields (Scheme 74). The substrate scope was rather limited, as Lee’s procedure generated one α-bromoketone and Lixin’s reported only three aromatic α-chloroketones. The acidic nature of these protocols further narrowed the hydration scope, as acid-sensitive functional groups (e.g., OH, NHx) did not tolerate these conditions.
Xiao and co-workers designed an In(OTf)3 (10 mol%)-catalysed hydration optimised for 1-bromoalkynes (Scheme 75) [198]. The hydration reaction was only effective if carried out in an acidic solvent, i.e., HOAc. Together with the high reaction temperature, this route requires harsh hydration conditions, which limits its applicability. Nevertheless, 16 α-bromo, 3 α-chloro and 3 α-iodoketones were obtained in good to excellent yields (78–93%). Despite the acidic solvent, the system mediated the hydration of an OH-substituted 1-bromoalkyne, affording the corresponding ketone in 82% yield.
Recently, Xiang and co-workers designed a highly versatile Ce(SO4)2-catalysed hydration [199]. The addition of sulfuric acid to the 1-haloalkyne with 5 mol% of Ce(SO4)2 in DCM, generated 30 α-haloketones in moderate to excellent yields (Scheme 76). Despite the acidic nature of the protocol, acid-sensitive groups (i.e., OH and NHTs) were well tolerated. The presence of steric bulk close to the triple bond appeared to have a considerable effect on the hydration efficiency.
6.2. Non-Metal-Catalysed Hydrations
In 2016, a metal-free hydration of 1-haloalkynes in 2,2,2-trifluoroethanol was described by Wang and co-workers (Scheme 77) [200]. Thirteen α-bromo, seven α-chloro and eight α-iodoketones were obtained in good to excellent yields. Nonetheless, ortho-substituted 1-haloalkynes were less efficiently hydrated due to the presence of steric bulk close to the alkyne centre.
7. Miscellaneous Routes
7.1. α-Functionalised Ketones as Substrates
In 2018, Baire published the selective monodehalogenation of α,α-dihaloketones [201]. 2-Iodo-1-phenylethanone was isolated in 64% yield upon stirring the α,α-diiodo-product for 10 h in a CH3CN/H2O mixture. The procedure yielded an inseparable mixture of the α-haloketone and α,α-dihaloketone for 2-bromo-1-phenylethanone. Alternatively, Ranu et al. established an IL-mediated selective debromination method in which 1-methyl-3-pentylimidazolium tetra-fluoroborate ([pmIm]BF4) acted as both catalyst and reaction medium [202]. Two aromatic α-bromoketones (R = H, Me) and one α-chloroketone (R = Me) were generated in good yields (Scheme 78). This route exhibited short reaction times, excellent efficacy and selectivity, and the use of a recyclable and readily available catalyst.
In the last decade, Sayyahi and co-workers dedicated considerable effort towards the development of an effective SN2 substitution protocol yielding α-functionalised ketones from their corresponding α-bromoketones [203,204,205,206]. Their most recent work involved an imidazolium poly(ionic liquid)-based substitution (Scheme 79). Four aromatic α-iodoketones were obtained in excellent yields (87–91%) upon the addition of the IL and NaI to the substrate in H2O at 60 °C. The authors claimed that the IL can be recycled and reused in up to five consecutive runs without significant loss in activity. This route has some promising features, i.e., catalyst recycling, aqueous solvent and mild reaction conditions; however, it still suffers from the need to synthesise the α-bromoketone substrate.
An alternative substitution procedure, developed by Zhang and co-workers, generated 14 α-chloroketones in quantitative yields from α-bromoketones (Scheme 80) [207]. This route tolerated the presence of electron-donating and -withdrawing aromatic substituents. Additionally, sterically hindered substrates were tolerated. This procedure afforded quantitative yields, thus avoiding chromatographic purification of the products.
Wang and co-workers designed a synthetic protocol converting α-diazoketones in α-halo-ketones (Scheme 81) [208]. The protocol yielded ten α-chloroaryl ketones in 56–82% when stirring a mixture of FeCl3 (0.5 equiv.), silica gel (100 mg) and the diazo compound in DCM for 10 min. Furthermore, seven α-bromoaryl ketones were obtained in moderate to excellent yields (55–95%) by changing the Lewis acid to FeBr3. This route’s versatility was demonstrated through its tolerance towards electron-donating (e.g., Me, OMe) and -withdrawing (e.g., I, Cl, NO2) aromatic substituents, as well as sterically hindered substrates.
Kappe and Gutmann developed a continuous process for the synthesis of α-chloroketones from their acetyl chloride derivatives (Scheme 82) [209,210]. The protocol involved mixing a N-methyl-N-nitroso-p-toluenesulfonamide (diazald) solution in DMF and a KOH solution in MeOH/H2O with the starting material to afford the α-diazoketone intermediate. Subsequent addition of HCl allowed the isolation of four aromatic α-chloroketones in excellent yields (88–90%). The on-demand generation and direct consumption of diazomethane or its precursor certainly eliminate human exposure to the reagent, thus reducing the risk of explosive decomposition.
Jordan and co-workers aimed to establish a sustainable procedure for the Appel halogenation, which converts alcohols in alkyl halides [211]. Although a broad scope of halides was obtained, the procedure only yielded two aryl α-haloketones (Scheme 83). The authors claimed that O=PPh3 was a recyclable organocatalyst; however, tedious procedures are required to completely remove the oxide from the reaction mixture [212]. Lai and co-workers described an alternative procedure which also accessed 2-choro-1-phenylethanone from its α-hydroxy ketone [213]. Stirring a cooled mixture of the ketol, tosyl chloride (1.1 equiv.), triethylamine (2 equiv.) and 4-dimethylaminopyridine (2 mol%) in DCM yielded the α-chloroketone in 83% yield. This procedure seemed to exhibit a higher efficacy than the previous one; however, its scope was still limited and did not permit a thorough comparison with other routes.
Zhang and Tang designed an SN2 substitution method yielding α-haloketones from α-hydroxy-benzotriazole functionalised ketones (Scheme 84) [214]. Their method is quite practical; however, it relies on the prior placement of an hydroxybenzotriazole group on the starting substrate which is not entirely straightforward, thus limiting the scope significantly.
7.2. Alternative Substrates
Yingpeng, Yulai and co-workers demonstrated that silyl enol ethers are suitable synthons for aromatic α-haloketones (Scheme 86) [216]. One α-bromo- and 12 α-chloroketones were generated upon the addition of (diacetoxyiodo)benzene (1.5 equiv.) and trimethylsilyl halide (2 equiv.) to the silyl enol ether in acetonitrile. 2-Bromo-1-phenylethanone was isolated in 64% yield and a set of α-chloroketones were obtained in moderate to excellent yields (55–90%). The procedure tolerated the presence of electron-donating (e.g., Me, OMe, NMe2) and -withdrawing (e.g., Cl, Br) substituents on its aromatic substrates. The mechanism as described in Scheme 86 highlighted the initial formation of a ketone intermediate bearing an iodo-leaving group, followed by nucleophilic substitution with the corresponding halide to afford the desired compound.
Patil, Adimurthy and Ranu established the synthesis of α-bromoketones from their corresponding vic-1,2-dibromides [217]. As shown in Scheme 87, six haloketones were isolated in moderate to excellent yields. Selectivity issues occurred for the OMe-substituted dibromides, as the brominated arylhydrin was obtained as an important side product. This problem and the relatively long reaction times are this route’s main disadvantages.
α-Chloroketones were also accessed via the chloroacetylation of arenes (Scheme 88) [218]. This FeIII-catalysed procedure exhibits a low efficacy; however, when highly sterically hindered arenes were used, a remarkable increase in yield was observed.
Ram and Manoj designed the CuI promoted synthesis of α-haloketones from trichloromethyl carbinols [219]. Fifteen carbinols were efficiently converted upon addition of two equivalents of CuCl and bipyridine in dichloroethane. Subsequent stirring for 3 h under reflux yielded the corresponding α-chloroketones in 82–96%. This route coped well with the presence of electron-withdrawing (e.g., Cl, Br, NO2) and -donating (e.g., Me, OMe) groups on substrates.
In 2007, Yoshimatsu et al. reported an HCl/EtOH-based system yielding 15 (hetero)aromatic α-chloroketones from their enol ethers in 18–100% (Scheme 89) [220], though it should be noted that harsh reaction conditions are required to access the enol ether compounds.
Enolacetates have also proven to be suitable synthons for α-haloketones [221]. Electron-poor, electron-rich and sterically hindered enolacetates were efficiently transformed upon the addition of N-iodosaccharin (Figure 15). Despite the broad scope, only one aromatic α-iodoketone (i.e., 2-iodo-1-phenylethanone) was isolated in 56% yield.
8. Conclusions and Outlook
Overall, this review analysed several methodologies to access aromatic α-haloketones. In the last two decades, a plethora of halogenation agents have been developed, though X2 remains the most abundantly used. Alternative and more sustainable halogen sources are beginning to have a significant impact in the literature, as demonstrated by the overview given herein. N-halosuccinimides, HX and MX strategies appear to be the most promising, as they showcase a better balance between reactivity and associated hazards. Ionic liquid-based halogenation agents could also be a step forward in the right direction, though it is still too early to properly assess these protocols, since their use is still limited due to their cost and restricted (commercial) availability. It is our hope that the combination of the current knowledge, collated in this review, will undoubtedly lead to improved strategies soon.
The community has strived to tackle the direct halogenation issues. Several oxyhalogenation methods for alkenes, alkynes and secondary alcohols have been described in the last 20 years. However, these only partially addressed the creation of a general and useful method, as some methods require the presence of an acidic promotor or have demonstrated efficacy and/or selectivity limitations. On the other hand, research showed that the oxidation of functionalised hydrocarbons could very well yield the synthetically valuable α-haloketones. This route’s value in organic synthesis is still limited due its moderate activity, selectivity and/or versatility. Additionally, the use of non-readily (commercially) available starting materials further hinders their applicability.
In comparison to most protocols dedicated to the direct halogenation of ketones, the hydration of 1-haloalkynes has only scarcely been explored in the last two decades. The latter route can be considered as the best alternative for the direct halogenation, especially considering its high atom efficiency and the use of only water as reagent.
The current field still possesses a remarkable growth potential, and we therefore hope to see more advances soon that directly address the numerous problems highlighted in this review.
Funding
This work was funded by UGent BOF (Ghent University, Bijzonder OnderzoeksFonds).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aggarwal, V.K.; Fang, G.; Kokotos, C.G.; Richardson, J.; Unthank, M.G. A practical synthesis of a [2.2.1] bicyclic chiral sulfide for asymmetric transformations. Tetrahedron 2006, 62, 11297–11303. [Google Scholar] [CrossRef]
- Kuang, Y.L.; Lu, Y.; Tang, Y.; Liu, X.H.; Lin, L.L.; Feng, X.M. Asymmetric synthesis of spiro-epoxyoxindoles by the catalytic Darzens reaction of isatins with phenacyl bromides. Org. Lett. 2014, 16, 4244–4247. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.; Zaib, S.; Ahmad, M.; Gardiner, J.M.; Ahmad, A.; Hameed, A.; Furtmann, N.; Gutschow, M.; Bajorath, J.; Iqbal, J. Novel structural hybrids of pyrazolobenzothiazines with benzimidazoles as cholinesterase inhibitors. Eur. J. Med. Chem. 2014, 78, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.M.; Basuny, A.M.; Arafat, S.M. Utilization of stearic acid extracted from olive pomace for production of triazoles, thiadiazoles and thiadiazines derivatives of potential biological activities. J. Oleo. Sci. 2015, 64, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Kikui, H.; Moriyama, K.; Togo, H. One-pot preparation of 2,5-disubstituted and 2,4,5-trisubstituted oxazoles from aromatic ketones with molecular iodine, oxone, and trifluoromethanesulfonic acid in nitriles. Tetrahedron 2015, 71, 5267–5274. [Google Scholar] [CrossRef]
- Zheng, H.F.; Wang, Y.; Xu, C.R.; Xu, X.; Lin, L.L.; Liu, X.H.; Feng, X.M. Stereodivergent synthesis of vicinal quaternary-quaternary stereocenters and bioactive hyperolactones. Nat. Commun. 2018, 9, 1968. [Google Scholar]
- Yang, Y.W.; Yang, W.B. Divergent synthesis of N-heterocycles by Pd-catalyzed controllable cyclization of vinylethylene carbonates. Chem. Commun. 2018, 54, 12182–12185. [Google Scholar] [CrossRef]
- Sujatha, K.; Shilpa, R.S.; Gani, R.S. Synthesis, antifungal and anti-bacterial activity of n-(4-chloro-2-trifluoroactyl phenyl)-aminothiazole derivatives. Int. Res. J. Pharm. 2019, 10, 150–155. [Google Scholar]
- Ruggeri, M.; Dombrowski, A.W.; Djuric, S.W.; Baxendale, I.R. Photochemical flow synthesis of 3-hydroxyazetidines. ChemPhotoChem 2019, 3, 1212–1218. [Google Scholar] [CrossRef]
- Li, Y.; Han, J.P.; Luo, H.; An, Q.Y.; Cao, X.P.; Li, B.S. Access to benzylic quaternary carbons from aromatic ketones. Org. Lett. 2019, 21, 6050–6053. [Google Scholar] [CrossRef]
- Bangalore, P.K.; Vagolu, S.K.; Bollikanda, R.K.; Veeragoni, D.K.; Choudante, P.C.; Misra, S.; Sriram, D.; Sridhar, B.; Kantevari, S. Usnic acid enaminone-coupled 1,2,3-triazoles as antibacterial and antitubercular agents. J. Nat. Prod. 2020, 83, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.D.; Batanero, B.; Barba, F. Electrochemical preparation of α,α’-dicarbonylselenides. Tetrahedron 2004, 60, 4609–4612. [Google Scholar] [CrossRef]
- Gouda, M.A.; Sherif, Y.E.; Elsherbini, M.S. Synthesis, anti-inflammatory, and analgesic evaluation of some 2-amino-5-selenothiazoles. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 1633–1643. [Google Scholar] [CrossRef]
- Chen, Z.L.; Hu, F.L.; Huang, S.L.; Zhao, Z.X.; Mao, H.; Qin, W.L. Organocatalytic enantioselective selenosulfonylation of a C-C double bond to form two stereogenic centers in an aqueous medium. J. Org. Chem. 2019, 84, 8100–8111. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Nakamura, H.; Aoki, K.; Suzuki, M.; Yamada, K.; Hirata, Y. Synthetic intermediate potentially useful for synthesis of dendrobine. Tetrahedron 1971, 27, 5157–5162. [Google Scholar] [CrossRef]
- Ikeda, M.; Teranishi, H.; Nozaki, K.; Ishibashi, H. Triethylborane-mediated atom-transfer cyclisation of 2-iodo-N-(prop-2-enyl)acetamides and related compounds. J. Chem. Soc. Perkin Trans. 1998, 1, 1691–1697. [Google Scholar] [CrossRef]
- Campos, K.R.; Chen, C.; Ishibashi, H.; Kato, S.; Klapars, A.; Kohmura, Y.; Pollard, D.J.; Takezawa, A.; Waldman, J.H.; Wallace, D. Process for Making Lactam Tachykinin Receptor Antagonists. Patent WO/2008/021029 A3, 21 February 2008. [Google Scholar]
- Lapointe, G.; Schenk, K.; Renaud, P. Total synthesis of (+/−)-cylindricine C. Org. Lett. 2011, 13, 4774–4777. [Google Scholar] [CrossRef]
- Gryparis, C.; Lykakis, I.N.; Efe, C.; Zaravinos, I.P.; Vidali, T.; Kladou, E.; Stratakis, M. Functionalized 3(2H)-furanones via photooxygenation of (beta-keto)-2-substituted furans: Application to the biomimetic synthesis of merrekentrone C. Org. Biomol. Chem. 2011, 9, 5655–5658. [Google Scholar] [CrossRef]
- Lapointe, G.; Kapat, A.; Weidner, K.; Renaud, P. Radical azidation reactions and their application in the synthesis of alkaloids. Pure Appl. Chem. 2012, 84, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, T.; Lee, B.H.; Yoo, S.E.; Lee, K.; Yi, K.Y. 3-Substituted-(5-arylfuran-2-ylcarbonyl)guanidines as NHE-1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1291–1295. [Google Scholar] [CrossRef]
- Vladimirova, S.; Bijev, A. An access to new N-pyrrolylcarboxylic acids as potential COX-2 inhibitors via Paal-Knorr cyclization. Heterocycl. Commun. 2014, 20, 111–115. [Google Scholar] [CrossRef]
- Jeong, K.W.; Lee, J.H.; Park, S.M.; Choi, J.H.; Jeong, D.Y.; Choi, D.H.; Nam, Y.; Park, J.H.; Lee, K.N.; Kim, S.M.; et al. Synthesis and in-vitro evaluation of 2-amino-4-arylthiazole as inhibitor of 3D polymerase against foot-and-mouth disease (FMD). Eur. J. Med. Chem. 2015, 102, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wu, X.; Geng, X.; Wang, C.; Zhou, Y.; Wu, Y.D.; Wu, A.X. I2/PhI(OAc)2 copromoted amination reaction: Synthesis of α-dicarbonylsulfoximine derivatives by incorporating an intact dimethyl sulfoxide. J. Org. Chem. 2019, 84, 8322–8329. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Xu, Q.; Li, C.; Luo, J.; Li, X.X.; Wang, L.J.; Jiang, B.; Shi, D.Y. Toward a treatment of diabesity: In vitro and in vivo evaluation of uncharged bromophenol derivatives as a new series of PTP1B inhibitors. Eur. J. Med. Chem. 2019, 166, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Demirayak, S.; Sahin, Z.; Ertas, M.; Bulbul, E.F.; Bender, C.; Biltekin, S.N.; Berk, B.; Saglik, B.N.; Levent, S.; Yurttas, L. Novel thiazole-piperazine derivatives as potential cholinesterase inhibitors. J. Heterocycl. Chem. 2019, 56, 3370–3386. [Google Scholar] [CrossRef]
- Ansari, M.; Shokrzadeh, M.; Karima, S.; Rajaei, S.; Hashemi, S.M.; Mirzaei, H.; Fallah, M.; Emami, S. Design, synthesis and biological evaluation of flexible and rigid analogs of 4H-1,2,4-triazoles bearing 3,4,5-trimethoxyphenyl moiety as new antiproliferative agents. Bioorg. Chem. 2019, 93, 103300. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Patel, H.D. Synthesis of α-bromocarbonyl compounds: Recent advances. Tetrahedron 2014, 70, 3949–3961. [Google Scholar] [CrossRef]
- Jagatheesan, R.; Ramesh, P.; Sambathkumar, S. Selective α-bromination of aryl carbonyl compounds: Prospects and challenges. Synth. Commun. 2019, 49, 3265–3289. [Google Scholar] [CrossRef]
- Mphahlele, M.J. Molecular iodine-mediated α-iodination of carbonyl compounds. J. Chem. Res. 2010, 34, 121–126. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Balar, C.R.; Sharma, V.S.; Prajapati, N.P.; Vekariya, M.K.; Sharma, A.S. Preparation of α-iodocarbonyl compounds: An overall development. ChemistrySelect 2018, 3, 9189–9203. [Google Scholar] [CrossRef]
- Erian, A.W.; Sherif, S.M.; Gaber, H.M. The chemistry of α-haloketones and their utility in heterocyclic synthesis. Molecules 2003, 8, 793–865. [Google Scholar] [CrossRef] [Green Version]
- De Kimpe, N.; Verhé, R.; Patai, S.; Rappoport, Z. The Chemistry of α-Haloketones, α-Haloaldehydes and α-Haloimines, 1st ed.; Wiley: Chichester, UK, 1988. [Google Scholar]
- Moiseev, I.K.; Makarova, N.V.; Zemtsova, M.N. α-Halo ketones in C, N-, O-, and S-alkylation reactions. Russ. J. Org. Chem. 2003, 39, 1685–1701. [Google Scholar] [CrossRef]
- Fulopova, V.; Soural, M. Solid-phase synthesis of heterocycles with α-haloketones as the key building blocks. Synthesis 2016, 48, 3684–3695. [Google Scholar]
- Vekariya, R.H.; Patel, K.D.; Prajapati, N.P.; Patel, H.D. Phenacyl bromide: A versatile organic intermediate for the synthesis of heterocyclic compounds. Synth. Commun. 2018, 48, 1505–1533. [Google Scholar] [CrossRef]
- Liu, C.L.; Shi, C.; Mao, F.; Xu, Y.; Liu, J.Y.; Wei, B.; Zhu, J.; Xiang, M.J.; Li, J. Discovery of new imidazole derivatives containing the 2,4-dienone motif with broad-spectrum antifungal and antibacterial activity. Molecules 2014, 19, 15653–15672. [Google Scholar] [CrossRef]
- Becker, R.; van den Broek, S.A.M.W.; Nieuwland, P.J.; Koch, K.; Rutjes, F.P.J.T. Optimisation and scale-up of α-bromination of acetophenone in a continuous flow microreactor. J. Flow. Chem. 2012, 2, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Juneja, S.K.; Choudhary, D.; Paul, S.; Gupta, R. In situ-generated zinc bromide-catalyzed α-bromination of alkanones in water. Synth. Commun. 2006, 36, 2877–2881. [Google Scholar] [CrossRef]
- Reece, R.L.; Dickson, D.B.; Burrowes, P.J. Zinc toxicity (new wire disease) in aviary birds. Aust. Vet. J. 1986, 63, 199. [Google Scholar] [CrossRef]
- Fosmire, G.J. Zinc toxicity. Am. J. Clin. Nutr. 1990, 51, 225–227. [Google Scholar] [CrossRef]
- Rahman, M.T.; Kamata, N.; Matsubara, H.; Ryu, I. Quadraphasic phase-vanishing method: Application to bromination reactions that produce acidic by-products. Synlett 2005, 2664–2666. [Google Scholar] [CrossRef]
- Chen, J.Z.; Liu, D.L.; Butt, N.; Li, C.; Fan, D.Y.; Liu, Y.G.; Zhang, W.B. Palladium-catalyzed asymmetric hydrogenation of α-acyloxy-1-arylethanones. Angew. Chem. Int. Ed. 2013, 52, 11632–11636. [Google Scholar] [CrossRef] [PubMed]
- Gudipudi, G.; Sagurthi, S.R.; Perugu, S.; Achaiah, G.; Krupadanam, G.L.D. Rational design and synthesis of novel 2-(substituted-2H-chromen-3-yl)-5-aryl-1H-imidazole derivatives as an anti-angiogenesis and anti-cancer agent. Rsc Adv. 2014, 4, 56489–56501. [Google Scholar] [CrossRef]
- Elejalde, N.R.; Macias, M.; Castillo, J.C.; Sortino, M.; Svetaz, L.; Zacchino, S.; Portilla, J. Synthesis and in vitro antifungal evaluation of novel N-substituted 4-aryl-2-methylimidazoles. ChemistrySelect 2018, 3, 5220–5227. [Google Scholar] [CrossRef]
- Withers, R.M.J.; Lees, F.P. The assessment of major hazards: The lethal toxicity of bromine. J. Hazard. Mater. 1986, 13, 279–299. [Google Scholar] [CrossRef]
- Kirk, R.E.; Othmer, D.F. Encyclopedia of Chemical Technology, 1st ed.; Wiley: New York, NY, USA, 2003. [Google Scholar]
- Jereb, M.; Iskra, J.; Zupan, M.; Stavber, S. Direct α-iodination of ketones induced by aqueous hydrogen peroxide. Lett. Org. Chem. 2005, 2, 465–468. [Google Scholar] [CrossRef]
- Pavlinac, J.; Zupan, M.; Stavber, S. Solvent-free iodination of organic molecules using the I2/urea-H2O2 reagent system. Org. Biomol. Chem. 2007, 5, 699–707. [Google Scholar] [CrossRef]
- Pavlinac, J.; Laali, K.K.; Zupan, M.; Stavber, S. Iodination of organic compounds with elemental iodine in the presence of hydrogen peroxide in ionic liquid media. Aust. J. Chem. 2008, 61, 946–955. [Google Scholar] [CrossRef]
- Iskra, J.; Stavber, S.; Zupan, M. Aerobic oxidative iodination of organic molecules activated by sodium nitrite. Tetrahedron Lett. 2008, 49, 893–895. [Google Scholar] [CrossRef]
- Reuzel, P.G.J.; Bruijntjes, J.P.; Feron, V.J.; Woutersen, R.A. Subchronic inhalation toxicity of amorphous silicas and quartz dust in rats. Food Chem. Toxicol. 1991, 29, 341–354. [Google Scholar] [CrossRef]
- Lin, W.S.; Huang, Y.W.; Zhou, X.D.; Ma, Y.F. In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol. Appl. Pharm. 2006, 217, 252–259. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Stavber, G.; Iskra, J.; Zupan, M.; Stavber, S. Aerobic oxidative iodination of ketones catalysed by sodium nitrite “on water” or in a micelle-based aqueous system. Green Chem. 2009, 11, 1262–1267. [Google Scholar] [CrossRef]
- Prebil, R.; Stavber, S. Aerobic oxidative α-iodination of carbonyl compounds using molecular iodine activated by a nitrate-based catalytic system. Tetrahedron Lett. 2014, 55, 5643–5647. [Google Scholar] [CrossRef]
- Yusubov, M.S.; Yusubova, R.Y.; Funk, T.V.; Chi, K.W.; Kirschning, A.; Zhdankin, V.V. m-Iodosylbenzoic acid as a convenient recyclable hypervalent iodine oxidant for the synthesis of α-iodo ketones by oxidative iodination of ketones. Synthesis 2010, 3681–3685. [Google Scholar] [CrossRef]
- Rao, M.L.N.; Jadhav, D.N. Metal catalyst-free direct α-iodination of ketones with molecular iodine. Tetrahedron Lett. 2006, 47, 6883–6886. [Google Scholar] [CrossRef]
- Yin, G.D.; Gao, M.; She, N.F.; Hu, S.L.; Wu, A.X.; Pan, Y.J. Highly efficient and clean method for direct α-iodination of aromatic ketones. Synthesis 2007, 3113–3116. [Google Scholar] [CrossRef]
- Yadav, J.S.; Kondaji, G.; Reddy, M.S.R.; Srihari, P. Facile synthesis of α-iodo carbonyl compounds and α-iodo dimethyl ketals using molecular iodine and trimethylorthoformate. Tetrahedron Lett. 2008, 49, 3810–3813. [Google Scholar] [CrossRef]
- Lee, J.C.; Kim, J.; Park, H.J.; Kwag, B.; Lee, S.B. Direct metal-free α-iodination of arylketones induced by iodine or iodomethane with HTIB in ionic liquid. B. Korean Chem. Soc. 2010, 31, 1385–1386. [Google Scholar] [CrossRef] [Green Version]
- Goswami, P.; Ali, S.; Khan, M.M.; Khan, A.T. Selective and effective oxone-catalysed α-iodination of ketones and 1,3-dicarbonyl compounds in the solid state. Arkivoc 2007, XV, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Li, S.G.; Deng, G.J.; Gong, H. Metal-free, oxidant-free, and controllable graphene oxide catalyzed direct iodination of arenes and ketones. ChemCatChem 2018, 10, 376–380. [Google Scholar] [CrossRef]
- Pennington, J.A.T. A review of iodine toxicity reports. J. Am. Diet. Assoc. 1990, 90, 1571–1581. [Google Scholar] [CrossRef]
- Block, S.S. Disinfection, Sterilization, and Preservation, 5th ed.; Lippincott Williams & Wilkins: London, UK, 2001. [Google Scholar]
- Lim, B.; Oh, E.T.; Im, J.; Lee, K.S.; Jung, H.; Kim, M.; Kim, D.; Oh, J.T.; Bae, S.H.; Chung, W.J.; et al. Systematic synthesis of diphenyl-substituted carotenoids as molecular wires. Eur. J. Org. Chem. 2017, 2017, 6390–6400. [Google Scholar] [CrossRef]
- Lee, J.C.; Bae, Y.H.; Chang, S.K. Efficient α-Halogenation of carbonyl compounds by N-bromosuccinimide and N-chlorosuccinimde. B. Korean Chem. Soc. 2003, 24, 407–408. [Google Scholar] [CrossRef]
- Guan, X.Y.; Al-Misba’a, Z.; Huang, K.W. Efficient and selective α-bromination of carbonyl compounds with N-bromosuccinimide under microwave. Arab. J. Chem. 2015, 8, 892–896. [Google Scholar] [CrossRef] [Green Version]
- Mohinuddin, P.M.K.; Reddy, B.M.; Reddy, G.T.; Reddy, N.C.G. KH2PO4 as a novel catalyst for regioselective monobromination of aralkyl ketones using N-bromosuccinimide: A green methodology. Org. Commun. 2015, 8, 60–69. [Google Scholar]
- Reddy, B.M.; Kumar, V.V.R.; Reddy, N.C.G.; Rao, S.M. Silica gel catalyzed α-bromination of ketones using N-bromosuccinimide: An easy and rapid method. Chin. Chem. Lett. 2014, 25, 179–182. [Google Scholar] [CrossRef]
- Gupta, R.; Gupta, M.; Paul, S.; Gupta, R.; Loupy, A. HClO4-SiO2: A highly efficient and recyclable catalyst for the selective α-bromination of carbonyl compounds using N-bromosuccinimide. Lett. Org. Chem. 2008, 5, 153–157. [Google Scholar] [CrossRef]
- Salama, T.A.; Novak, Z. N-Halosuccinimide/SiCl4 as general, mild and efficient systems for the α-monohalogenation of carbonyl compounds and for benzylic halogenation. Tetrahedron Lett. 2011, 52, 4026–4029. [Google Scholar] [CrossRef]
- Jagdale, A.R.; Chouthaiwale, P.V.; Sudalai, A. Cu(OTf)2-catalyzed α-halogenation of ketones with 1,3-dichloro-5,5‘-dimethylhydantoin and N-bromosuccinimide. Indian J. Chem. Sect. B 2009, 48, 1424–1430. [Google Scholar] [CrossRef]
- Arbuj, S.S.; Waghmode, S.B.; Ramaswamy, A.V. Photochemical α-bromination of ketones using N-bromosuccinimide: A simple, mild and efficient method. Tetrahedron Lett. 2007, 48, 1411–1415. [Google Scholar] [CrossRef]
- Jangid, D.K.; Dhadda, S.; Goswami, P.G.; Guleria, A.; Pareek, K.; Jangir, N.; Poonam. Microwave-assisted nanocatalysed synthesis of phenacyl halides as future anti-platelet agents. ChemistrySelect 2019, 4, 9348–9353. [Google Scholar] [CrossRef]
- Izumisawa, Y.; Togo, H. Preparaton of α-bromoketones and thiazoles from ketones with NBS and thioamides in ionic liquids. Green Sustain. Chem. 2011, 1, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Park, H.J. Efficient α-chlorination and α-bromination of carbonyl compounds using N-halosuccinimides/UHP in ionic liquid. Synth. Commun. 2007, 37, 87–90. [Google Scholar] [CrossRef]
- Vrazic, D.; Jereb, M.; Laali, K.K.; Stavber, S. Brønsted acidic ionic liquid accelerated halogenation of organic compounds with N-halosuccinimides (NXS). Molecules 2013, 18, 74–96. [Google Scholar] [CrossRef] [Green Version]
- Pravst, I.; Zupan, M.; Stavber, S. Directed regioselectivity of bromination of ketones with NBS: Solvent-free conditions versus water. Tetrahedron Lett. 2006, 47, 4707–4710. [Google Scholar] [CrossRef]
- Pravst, I.; Zupan, M.; Stavber, S. Halogenation of ketones with N-halosuccinimides under solvent-free reaction conditions. Tetrahedron 2008, 64, 5191–5199. [Google Scholar] [CrossRef]
- Lee, J.C.; Bae, Y.H. Efficient alpha-iodination of carbonyl compounds under solvent-free conditions using microwave irradiation. Synlett 2003, 507–508. [Google Scholar] [CrossRef] [Green Version]
- Vekariya, H.R.; Panchal, S.N.; Patel, D.K.; Patel, H.D. Solvent and catalyst free, regioselective α-bromination of aralkyl ketones using N-bromosuccinimide (NBS) under microwave irradiation: An efficient protocol. Cur. Microw. Chem. 2015, 2, 61–68. [Google Scholar] [CrossRef]
- Stavber, G.; Iskra, J.; Zupan, M.; Stavber, S. Aerobic oxidative iodination of organic compounds with iodide catalyzed by sodium nitrite. Adv. Synth. Catal. 2008, 350, 2921–2929. [Google Scholar] [CrossRef]
- Gu, L.Q.; Lu, T.; Zhang, M.Y.; Tou, L.J.; Zhang, Y.G. Efficient oxidative chlorination of aromatics on saturated sodium chloride solution. Adv. Synth. Catal. 2013, 355, 1077–1082. [Google Scholar] [CrossRef]
- Dewkar, G.K.; Thakur, V.V.; Pardhy, S.A.; Sudalai, A.; Devotta, S. Catalytic Process for Regiospecific Chlorination of Alkanes, Alkenes and Arenes. US Patent US6825383B1, 30 November 2004. [Google Scholar]
- Paul, S.; Nanda, P.; Gupta, R. Synthesis of α-bromoalkanones using urea-hydrogen peroxide complex and sodium bromide over silica gel-acetic acid. Indian J. Chem. Sect. B 2005, 44, 184–187. [Google Scholar] [CrossRef]
- Swamy, P.; Kumar, M.A.; Reddy, M.M.; Narender, N. Mild and efficient α-chlorination of carbonyl compounds using ammonium chloride and oxone (2KHSO5·KHSO4·K2SO4). Chem. Lett. 2012, 41, 432–434. [Google Scholar] [CrossRef]
- Zhou, Z.S.; Li, L.; He, X.H. A simple and convenient method for direct α-chlorination of ketones with ammonium chloride and oxone. Chin. Chem. Lett. 2012, 23, 1213–1216. [Google Scholar] [CrossRef]
- Macharla, A.K.; Nappunni, R.C.; Marri, M.R.; Peraka, S.; Nama, N. Oxidative bromination of ketones using ammonium bromide and oxone. Tetrahedron Lett. 2012, 53, 191–195. [Google Scholar] [CrossRef]
- Reddy, M.M.; Kumar, M.A.; Swamy, P.; Narender, N. Oxidative iodination of carbonyl compounds using ammonium iodide and oxone. Tetrahedron Lett. 2011, 52, 6554–6559. [Google Scholar]
- Jagatheesan, R.; Joseph Santhana Raj, K.; Lawrence, S.; Christopher, C. Electrocatalytic oxidative side chain bromination of alkyl aryl ketones. Int. J. Res. Pharm. Chem. 2016, 6, 843–848. [Google Scholar]
- Jagatheesan, R.; Raj, K.J.S.; Lawrence, S.; Christopher, C. Electroselective α-bromination of acetophenone using in situ bromonium ions from ammonium bromide. Rsc Adv. 2016, 6, 35602–35608. [Google Scholar] [CrossRef]
- Jakhar, K.; Makrandi, J.K. An eco-friendly oxidative bromination of alkanones by an aqueous grinding technique. Green Chem. Lett. Rev. 2008, 1, 219–221. [Google Scholar] [CrossRef]
- Nath, J.; Chaudhuri, M.K. Boric acid catalyzed bromination of a variety of organic substrates: An eco-friendly and practical protocol. Green Chem. Lett. Rev. 2008, 1, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ghorpade, A.K.; Huddar, S.N.; Akamanchi, K.G. Aq HBr-NaNO2-KI/air: A new catalytic system for α-monobromination of ketones. Tetrahedron Lett. 2016, 57, 4918–4921. [Google Scholar] [CrossRef]
- Prebil, R.; Stavber, S. The α-chlorination of aryl methyl ketones under aerobic oxidative conditions. Adv. Synth. Catal. 2014, 356, 1266–1274. [Google Scholar] [CrossRef]
- Zhang, G.F.; Liu, R.H.; Xu, Q.; Ma, L.X.; Liang, X.M. Sodium nitrite-catalyzed oxybromination of aromatic compounds and aryl ketones with a combination of hydrobromic acid and molecular oxygen under mild conditions. Adv. Synth. Catal. 2006, 348, 862–866. [Google Scholar] [CrossRef]
- Podgorsek, A.; Stavber, S.; Zupan, M.; Iskra, J. Bromination of ketones with H2O2-HBr “on water”. Green Chem. 2007, 9, 1212–1218. [Google Scholar] [CrossRef]
- Tillu, V.H.; Shinde, P.D.; Bedekar, A.V.; Wakharkar, R.D. Studies on bromination of active methylene by a mixture of hydrobromic acid and hydrogen peroxide (or TBHP). Synth. Commun. 2003, 33, 1399–1403. [Google Scholar] [CrossRef]
- Song, S.; Li, X.W.; Sun, X.; Yuan, Y.Z.; Jiao, N. Efficient bromination of olefins, alkynes, and ketones with dimethyl sulfoxide and hydrobromic acid. Green Chem. 2015, 17, 3285–3289. [Google Scholar] [CrossRef]
- Wang, J.Q.; Wang, X.L.; Niu, Z.Q.; Wang, J.; Zhang, M.; Li, J.H. Copper nitrate-catalyzed α-bromination of aryl ketones with hydrobromic acid. Synth. Commun. 2016, 46, 165–168. [Google Scholar] [CrossRef]
- Kumar, R.S.; Kulangiappar, K.; Kulandainathan, M.A. Convenient electrochemical method for the synthesis of α-bromo alkyl aryl ketones. Synth. Commun. 2010, 40, 1736–1742. [Google Scholar] [CrossRef]
- Hering, T.; Muhldorf, B.; Wolf, R.; Konig, B. Halogenase-inspired oxidative chlorination using flavin photocatalysis. Angew. Chem. Int. Ed. 2016, 55, 5342–5345. [Google Scholar] [CrossRef] [Green Version]
- Prebil, R.; Laali, K.K.; Stavber, S. Metal and H2O2 free aerobic oxidative aromatic halogenation with [RNH3+][NO3−]/HX and [BMIM(SO3H)][(NO3)x(X)y)] (X = Br, Cl) as multifunctional ionic liquids. Org. Lett. 2013, 15, 2108–2111. [Google Scholar] [CrossRef]
- Chiappe, C.; Leandri, E.; Tebano, M. [Hmim][NO3]—An efficient solvent and promoter in the oxidative aromatic chlorination. Green Chem. 2006, 8, 742–745. [Google Scholar] [CrossRef]
- Li, H.Z.; Xue, W.J.; Wu, A.X. Direct synthesis of α-ketothioamides from aryl methyl ketones and amines via I2-promoted sp3 C-H functionalization. Tetrahedron 2014, 70, 4645–4651. [Google Scholar] [CrossRef]
- Han, B.B.; Zheng, Z.B.; Zheng, D.C.; Zhang, L.; Cui, P.; Shi, J.J.; Li, C.J. Application of poly(vinylphenyltrimethylammonium tribromide) resin as an efficient polymeric brominating agent in the α-bromination and α-bromoacetalization of acetophenones. Synth. Commun. 2019, 49, 2512–2520. [Google Scholar] [CrossRef]
- Cristiano, R.; Walls, A.D.; Weiss, R.G. Sequential bromination reactions from beads with methyltriphenylphosphonium tribromide groups. J. Phys. Org. Chem. 2010, 23, 904–909. [Google Scholar] [CrossRef]
- Mao, X.C.; Liu, X.L.; Deng, Q.; Sheng, S.R. Solid-phase organic synthesis of α-iodo ketones using recyclable resin-bound selenium bromide. J. Chem. Res. 2012, 36, 100–102. [Google Scholar] [CrossRef]
- Eshghi, H.; Bakavoli, M.; Ghasemzadeh, M.; Seyedi, S.M. Ionic liquids bis(2-N-methylimidazoliumethyl)ether dichloroiodate/dibromochlorate as an efficient halogenating reagent for the synthesis of α-haloketones. Res. Chem. Intermed. 2015, 41, 1673–1682. [Google Scholar] [CrossRef]
- Veisi, H.; Sedrpoushan, A.; Mohammadi, P.; Faraji, A.R.; Sajjadifar, S. A new recyclable 1,4-bis(3-methylimidazolium-1-yl) utane ditribromide [bMImB]·(Br3−)2 ionic liquid reagent for selective bromination of anilines or phenols and α-bromination of alkanones under mild conditions. Rsc Adv. 2014, 4, 25898–25903. [Google Scholar] [CrossRef]
- Wu, L.Q.; Yin, Z.K. Magnetic-nanoparticle-supported 2,2′-bis[3-(triethoxysilyl)propyl]imidazolium-substituted diethyl ether bis(tribromide): A convenient recyclable reagent for bromination. Eur. J. Inorg. Chem. 2013, 2013, 6156–6163. [Google Scholar] [CrossRef]
- Cao, G.; Hu, A.X.; Xie, Y.L.; Zhou, Z. Novel synthetic approach toward C-substituted piperazine derivatives via C-N bonds formation of α-bromoarylethanones and ethanolamine. J. Heterocycl. Chem. 2013, 50, E126–E130. [Google Scholar] [CrossRef]
- Ranu, B.C.; Adak, L.; Banerjee, S. Halogenation of carbonyl compounds by an ionic liquid, [AcMIm]X, and ceric ammonium nitrate (CAN). Aust. J. Chem. 2007, 60, 358–362. [Google Scholar] [CrossRef]
- Salazar, J.; Dorta, R. Pentylpyridinium tribromide: A vapor pressure free room temperature ionic liquid analogue of bromine. Synlett 2004, 35, 1318–1320. [Google Scholar] [CrossRef]
- Paul, B.; Bhuyan, B.; Purkayastha, D.D.; Dhar, S.S.; Patel, B.K. Hexamethonium bis(tribromide) (HMBTB) a recyclable and high bromine containing reagent. Tetrahedron Lett. 2015, 56, 5646–5650. [Google Scholar] [CrossRef]
- Kavala, V.; Naik, S.; Patel, B.K. A new recyclable ditribromide reagent for efficient bromination under solvent free condition. J. Org. Chem. 2005, 70, 4267–4271. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Hu, L.X.; Wang, X.W.; Zhu, H.D. A simple, efficient and selective α-monobromination for arylacenones under solvent-free condition. Adv. Mater. Res. 2012, 396–398, 1079–1082. [Google Scholar] [CrossRef]
- Badali, M.; Khalafy, J.; Alidoost, E.; Aghazadeh, M. Ammonium hydrotribromide salts as convenient and selective brominating agents of aryl methyl ketones. J. Chem. Soc. Pak. 2016, 38, 859–863. [Google Scholar]
- Su, P.G.; Fan, C.; Yu, H.; Wang, W.Q.; Jia, X.; Rao, Q.F.; Fu, C.X.; Zhang, D.H.; Huang, B.H.; Pan, C.; et al. Synthesis of Ti-Al binary oxides and their catalytic application for C-H halogenation of phenols, aldehydes and ketones. Mol. Catal. 2019, 475, 110460. [Google Scholar] [CrossRef]
- Su, Q.; Zhao, Z.J.; Xu, F.; Lou, P.C.; Zhang, K.; Xie, D.X.; Shi, L.; Cai, Q.Y.; Peng, Z.H.; An, D.L. One-pot preparation of arylethynyl sulfides and bis(arylethynyl) sulfides. Eur. J. Org. Chem. 2013, 2013, 1551–1557. [Google Scholar] [CrossRef]
- Tang, S.Z.; Zhao, W.S.; Chen, T.; Liu, Y.; Zhang, X.M.; Zhang, F.M. A simple and efficient method for the preparation of α-halogenated ketones using iron(III) chloride and iron(III) bromide as halogen sources with phenyliodonium diacetate as oxidant. Adv. Synth. Catal. 2017, 359, 4177–4183. [Google Scholar] [CrossRef]
- Ding, R.; Li, J.Q.; Jiao, W.Y.; Han, M.R.; Liu, Y.G.; Tian, H.Y.; Sun, B.G. A highly efficient method for the bromination of alkenes, alkynes and ketones using dimethyl sulfoxide and oxalyl bromide. Synthesis 2018, 50, 4325–4335. [Google Scholar]
- Heravi, M.M.; Derikvand, F.; Ghassemzadeh, M. Tetrameric DABCO (TM)-bromine: An efficient and versatile reagent for bromination of various organic compounds. S. Afr. J. Chem. 2006, 59, 125–128. [Google Scholar]
- Tajbakhsh, M.; Habibmileh, S. 1,4-Dichloro-1,4-diazonlabicyclo [2,2,2] octane bis chloride, a useful and reusable electrophilic chlorinating agent. Lett. Org. Chem. 2007, 4, 512–514. [Google Scholar] [CrossRef]
- El-Ahl, A.A.S.; Elbeheery, A.H.; Amer, F.A. New, efficient synthesis of α-chloroketones using SiCl4/Urea-hydrogen peroxide or SiCl4/iodosylbenzene reagent systems. Synth. Commun. 2011, 41, 1508–1513. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Ismail, R.; Garcia, J.; Panja, C.; Rasul, G.; Mathew, T.; Olah, G.A. α-Halogenation of carbonyl compounds: Halotrimethylsilane-nitrate salt couple as an efficient halogenating reagent system. Tetrahedron Lett. 2011, 52, 1217–1221. [Google Scholar] [CrossRef]
- Xu, Z.J.; Zhang, D.Y.; Zou, X.Z. α-Chlorination of acetophenones using 1,3-dichloro-5,5-dimethylhydantoin. Synth. Commun. 2006, 36, 255–258. [Google Scholar] [CrossRef]
- Chen, Z.Z.; Zhou, B.; Cai, H.H.; Zhu, W.; Zou, X.Z. Simple and efficient methods for selective preparation of α-mono or α,α-dichloro ketones and β-ketoesters by using DCDMH. Green Chem. 2009, 11, 275–278. [Google Scholar] [CrossRef]
- Paul, S.; Gupta, V.; Gupta, R. A simple and selective procedure for α-bromination of alkanones using hexamethylenetetramine-bromine complex and basic alumina in solvent-free conditions. Synth. Commun. 2003, 33, 1917–1922. [Google Scholar] [CrossRef]
- Roy, S.C.; Rana, K.K.; Guin, C.; Banerjee, B. Ceric ammonium nitrate catalyzed mild and efficient α-chlorination of ketones by acetyl chloride. Arkivoc 2003, 9, 34–38. [Google Scholar] [CrossRef]
- Nishio, Y.; Yubata, K.; Wakai, Y.; Notsu, K.; Yamamoto, K.; Fujiwara, H.; Matsubara, H. Preparation of a novel bromine complex and its application in organic synthesis. Tetrahedron 2019, 75, 1398–1405. [Google Scholar] [CrossRef]
- Lee, J.C.; Park, J.Y.; Yoon, S.Y.; Bae, Y.H.; Lee, S.J. Efficient microwave induced direct α-halogenation of carbonyl compounds. Tetrahedron Lett. 2004, 45, 191–193. [Google Scholar] [CrossRef]
- Kurbah, S.D.; Asthana, M.; Syiemlieh, I.; Lywait, A.A.; Longchar, M.; Lal, R.A. New dioxido-vanadium(V) complexes containing hydrazone ligands: Syntheses, crystal structure and their catalytic application toward C-H bond functionalization. J. Organomet. Chem. 2018, 876, 10–16. [Google Scholar] [CrossRef]
- Syiemlieh, I.; Asthana, M.; Kurbah, S.D.; Lal, R.A. Synthesis, crystal structure and reactivity of homobimetallic vanadium(V) complexes derived from oxaloyldihydrazone ligands. Polyhedron 2019, 170, 202–216. [Google Scholar] [CrossRef]
- Luo, Z.B.; Meng, Y.E.; Gong, X.C.; Wu, J.; Zhang, Y.L.; Ye, L.W.; Zhu, C.Y. Facile Synthesis of α-haloketones by aerobic oxidation of olefins using KX as nonhazardous halogen source. Chin. J. Chem. 2020, 38, 173–177. [Google Scholar] [CrossRef]
- Kurapati, S.K.; Maloth, S.; Pal, S. Complexes of cis-dioxomolybdenum(VI) with unsymmetrical tripodal NO3-donor ligands: Synthesis, characterization and catalytic applications. Inorg. Chim. Acta 2015, 430, 66–73. [Google Scholar] [CrossRef]
- Jiang, Q.; Sheng, W.B.; Guo, C.C. Synthesis of phenacyl bromides via K2S2O8-mediated tandem hydroxybromination and oxidation of styrenes in water. Green Chem. 2013, 15, 2175–2179. [Google Scholar] [CrossRef]
- Zhou, B.C.; Guo, S.Y.; Fang, Z.; Yang, Z.; Liu, C.K.; He, W.; Zhu, N.; Li, X.; Guo, K. KI-promoted oxidative coupling of styrenes with indoles under metal-free conditions: Facile access to C-3 dicarbonyl indoles. Synthesis 2019, 51, 3511–3519. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.D.; Joshi, G.; Adimurthy, S. HBr-H2O2: A facile protocol for regioselective synthesis of bromohydrins and α-bromoketones and oxidation of benzylic/secondary alcohols to carbonyl compounds under mild aqueous conditions. Ind. Eng. Chem. Res. 2010, 49, 8100–8105. [Google Scholar] [CrossRef]
- Podgorsek, A.; Stavber, S.; Zupan, M.; Iskra, J. Environmentally benign electrophilic and radical bromination ‘on water’: H2O2-HBr system versus N-bromosuccinimide. Tetrahedron 2009, 65, 4429–4439. [Google Scholar] [CrossRef]
- Patil, R.D.; Joshi, G.; Adimurthy, S.; Ranu, B.C. Facile one-pot synthesis of α-bromoketones from olefins using bromide/bromate couple as a nonhazardous brominating agent. Tetrahedron Lett. 2009, 50, 2529–2532. [Google Scholar] [CrossRef]
- Moorthy, J.N.; Senapati, K.; Singhal, N. An expedient protocol for conversion of olefins to α-bromo/iodoketones using IBX and NBS/NIS. Tetrahedron Lett. 2009, 50, 2493–2496. [Google Scholar] [CrossRef]
- Chandra, A.; Parida, K.N.; Moorthy, J.N. One-pot synthesis of α-bromo- and α-azidoketones from olefins by catalytic oxidation with in situ-generated modified IBX as the key reaction. Tetrahedron 2017, 73, 5827–5832. [Google Scholar] [CrossRef]
- Achar, T.K.; Maiti, S.; Mal, P. IBX works efficiently under solvent free conditions in ball milling. Rsc Adv. 2014, 4, 12834–12839. [Google Scholar] [CrossRef]
- Rajbongshi, K.K.; Hazarika, D.; Phukan, P. Metal-free protocol for the synthesis of α-bromo ketones from olefins using TsNBr2. Tetrahedron Lett. 2015, 56, 356–358. [Google Scholar] [CrossRef]
- Rajbongshi, K.K.; Hazarika, D.; Phukan, P. TsNBr2 mediated oxidative functionalization of alkynes. Tetrahedron 2016, 72, 4151–4158. [Google Scholar] [CrossRef]
- Loginova, I.V.; Chukicheva, I.Y.; Kuchin, A.V. Reaction of styrene with chlorine dioxide. Russ. J. Gen. Chem. 2018, 88, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.Y.; Jia, X.F.; Wang, L.; Li, B.Y.; Liu, S.Y.; Ma, L.; Gao, W.; Wei, Y.Q.; Chen, J.B. The Mn-catalyzed paired electrochemical facile oxychlorination of styrenes via the oxygen reduction reaction. Chem. Commun. 2019, 55, 12104–12107. [Google Scholar] [CrossRef] [PubMed]
- Nobuta, T.; Hirashima, S.; Tada, N.; Miura, T.; Itoh, A. Facile aerobic photo-oxidative synthesis of phenacyl iodides and bromides from styrenes using I2 or aqueous HBr. Synlett 2010, 15, 2325–2329. [Google Scholar]
- Tada, N.; Ban, K.; Hirashima, S.; Miura, T.; Itoh, A. Direct synthesis of α-bromoketones from alkylarenes by aerobic visible light photooxidation. Org. Biomol. Chem. 2010, 8, 4701–4704. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Itoh, A. Facile synthesis of phenacyl iodides from styrenes under visible light irradiation with fluorescent lamps. Tetrahedron Lett. 2007, 48, 1131–1133. [Google Scholar] [CrossRef]
- Xu, S.H.; Wu, P.; Zhang, W. 1,3-Dibromo-5,5-dimethylhydantoin (DBH) mediated one-pot syntheses of α-bromo/amino ketones from alkenes in water. Org. Biomol. Chem. 2016, 14, 11389–11395. [Google Scholar] [CrossRef]
- Wang, Z.H.; Wang, L.; Wang, Z.M.; Li, P.H.; Zhang, Y.C. A practical synthesis of α-bromo/iodo/chloroketones from olefins under visible-light irradiation conditions. Chin. Chem. Lett. 2021, 32, 429–432. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Singh, A.P.; Basak, A.K. IBX/I2-mediated oxidation of alkenes and alkynes in water: A facile synthesis of α-iodoketones. Tetrahedron Lett. 2008, 49, 5880–5882. [Google Scholar] [CrossRef]
- Rather, S.A.; Kumar, A.; Ahmed, Q.N. Iodine-DMSO-promoted divergent reactivities of arylacetylenes. Chem. Commun. 2019, 55, 4511–4514. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xin, X.; Fu, Z.M.; Xie, L.Y.; Liu, K.J.; Wang, Z.; Li, W.Y.; Yuan, Z.H.; He, W.M. Water-controlled selective preparation of α-mono or α,α’-dihalo ketones via catalytic cascade reaction of unactivated alkynes with 1,3-dihalo-5,5-dimethylhydantoin. Green Chem. 2017, 19, 1983–1989. [Google Scholar] [CrossRef]
- Manickam, M.C.D.; Annalakshmi, S.; Pitchumani, K.; Srinivasana, C. Effect of cyclodextrin complexation in bromine addition to unsymmetrical olefins: Evidence for participation of cyclodextrin hydroxyl groups. Org. Biomol. Chem. 2005, 3, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- He, W.M.; Xie, L.Y.; Xu, Y.Y.; Xiang, J.N.; Zhang, L.M. Electrophilicity of α-oxo gold carbene intermediates: Halogen abstractions from halogenated solvents leading to the formation of chloro/bromomethyl ketones. Org. Biomol. Chem. 2012, 10, 3168–3171. [Google Scholar] [CrossRef]
- Xing, Y.L.; Zhang, M.; Ciccarelli, S.; Lee, J.; Catano, B. AuIII-Catalyzed formation of α-halomethyl ketones from terminal alkynes. Eur. J. Org. Chem. 2017, 2017, 781–785. [Google Scholar] [CrossRef]
- Nolan, S.P. The development and catalytic uses of N-heterocyclic carbene gold complexes. Acc. Chem. Res. 2011, 44, 91–100. [Google Scholar] [CrossRef]
- Nikishin, G.I.; Kapustina, N.I.; Sokova, L.L.; Bityukov, O.V.; Terent’ev, A.O. A H2O2/HBr system—several directions but one choice: Oxidation-bromination of secondary alcohols into mono- or dibromo ketones. Rsc Adv. 2018, 8, 28632–28636. [Google Scholar] [CrossRef] [Green Version]
- Azizi, N.; Khajeh, M.; Alipour, M. Rapid and selective oxidation of alcohols in deep eutectic solvent. Ind. Eng. Chem. Res. 2014, 53, 15561–15565. [Google Scholar] [CrossRef]
- Unlu, A.E.; Arikaya, A.; Takac, S. Use of deep eutectic solvents as catalyst: A mini-review. Green Process. Synth. 2019, 8, 355–372. [Google Scholar] [CrossRef]
- Reddy, M.M.; Swamy, P.; Naresh, M.; Srujana, K.; Durgaiah, C.; Rao, T.V.; Narender, N. One-pot synthesis of α-iodoketones from alcohols using ammonium iodide and oxone in water. Rsc Adv. 2015, 5, 12186–12190. [Google Scholar] [CrossRef]
- Rammurthy, B.; Swamy, P.; Naresh, M.; Srujana, K.; Durgaiah, C.; Sai, G.K.; Narender, N. A new and versatile one-pot strategy to synthesize α-bromoketones from secondary alcohols using ammonium bromide and oxone. New J. Chem. 2017, 41, 3710–3714. [Google Scholar] [CrossRef]
- Jing, Y.Y.; Daniliuc, C.G.; Studer, A. Direct conversion of alcohols to α-chloro aldehydes and α-chloro ketones. Org. Lett. 2014, 16, 4932–4935. [Google Scholar] [CrossRef] [PubMed]
- Badri, R.; Adlu, M.; Mohammadi, M.K. Synthesis and application of 1,4-bis(triphenyl phosphonium)butane peroxodisulfate for conversion of alkyl benzenes to their corresponding acylbenzenes. B. Kor. Chem. Soc. 2015, 8, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.F.; Lin, L.; Ye, X.Y.; Tan, C.H.; Jiang, Z.Y. Aerobic oxidation of benzylic sp3 C-H bonds through cooperative visible-light photoredox catalysis of N-hydroxyimide and dicyanopyrazine. Asian J. Org. Chem. 2017, 6, 422–425. [Google Scholar] [CrossRef]
- Liu, K.J.; Duan, Z.H.; Zeng, X.L.; Sun, M.; Tang, Z.L.; Jiang, S.; Cao, Z.; He, W.M. Clean oxidation of (hetero)benzylic C-sp3-H bonds with molecular oxygen. Acs Sus. Chem. Eng. 2019, 7, 10293–10298. [Google Scholar] [CrossRef]
- Li, S.L.; Zhu, B.; Lee, R.; Qiao, B.K.; Jiang, Z.Y. Visible light-induced selective aerobic oxidative transposition of vinyl halides using a tetrahalogenoferrate(III) complex catalyst. Org. Chem. Front. 2018, 5, 380–385. [Google Scholar] [CrossRef]
- Gonzalez-de-Castro, A.; Xiao, J.L. Green and efficient: Iron-catalyzed selective oxidation of olefins to carbonyls with O2. J. Am. Chem. Soc. 2015, 137, 8206–8218. [Google Scholar] [CrossRef]
- Xie, A.; Zhou, X.X.; Feng, L.D.; Hu, X.Y.; Dong, W. The oxidation of alcohols with O-iodoxybenzoic acid (IBX) in aqueous nanomicelles at room temperature. Tetrahedron 2014, 70, 3514–3519. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, B.L. 2-Iodoxybenzenesulfonic acid-catalysed oxidation of primary and secondary alcohols with oxone in cetyl trimethylammonium bromide micelles at room temperature. J. Chem. Res. 2014, 38, 427–431. [Google Scholar] [CrossRef]
- Dwars, T.; Paetzold, E.; Oehme, G. Reactions in micellar systems. Angew. Chem. Int. Ed. 2005, 44, 7174–7199. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Xie, A.M.; Li, J.J.; Xu, X.; Dong, W.; Wang, B.L. Heterocycle-substituted tetrazole ligands for copper-catalysed aerobic oxidation of alcohols. Tetrahedron 2014, 70, 9791–9796. [Google Scholar] [CrossRef]
- Graves, C.R.; Zeng, B.S.; Nguyen, S.T. Efficient and selective Al-catalyzed alcohol oxidation via Oppenauer chemistry. J. Am. Chem. Soc. 2006, 128, 12596–12597. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J.; Jiang, S.; Lu, L.H.; Tang, L.L.; Tang, S.S.; Tang, H.S.; Tang, Z.L.; He, W.M.; Xu, X.H. Bis(methoxypropyl) ether-promoted oxidation of aromatic alcohols into aromatic carboxylic acids and aromatic ketones with O2 under metal- and base-free conditions. Green Chem. 2018, 20, 3038–3043. [Google Scholar] [CrossRef]
- Kedziora, K.; Diaz-Rodriguez, A.; Lavandera, I.; Gotor-Fernandez, V.; Gotor, V. Laccase/TEMPO-mediated system for the thermodynamically disfavored oxidation of 2,2-dihalo-1-phenylethanol derivatives. Green Chem. 2014, 16, 2448–2453. [Google Scholar] [CrossRef] [Green Version]
- Utimoto, K. Palladium catalyzed synthesis of heterocycles. Pure Appl. Chem. 1983, 55, 1845–1852. [Google Scholar] [CrossRef] [Green Version]
- Imi, K.; Imai, K.; Utimoto, K. Regioselective hydration of alkynones by palladium catalysis. Tetrahedron Lett. 1987, 28, 3127–3130. [Google Scholar] [CrossRef]
- Baidossi, W.; Lahav, M.; Blum, J. Hydration of alkynes by a PtCl4-CO catalyst. J. Org. Chem. 1997, 62, 669–672. [Google Scholar] [CrossRef]
- Tokunaga, M.; Wakatsuki, Y. The first anti-Markovnikov hydration of terminal alkynes: Formation of aldehydes catalyzed by a ruthenium(II)/phosphane mixture. Angew. Chem. Int. Ed. 1998, 37, 2867–2869. [Google Scholar] [CrossRef]
- Beller, M.; Seayad, J.; Tillack, A.; Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004, 43, 3368–3398. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-metal-catalyzed addition of heteroatom-hydrogen bonds to alkynes. Chem Rev. 2004, 104, 3079–3159. [Google Scholar] [CrossRef]
- Hintermann, L.; Labonne, A. Catalytic hydration of alkynes and its application in synthesis. Synthesis 2007, 1121–1150. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, Y.; Utimoto, K. Effective transformation of unactivated alkynes into ketones or acetals by means of Au(III) catalyst. J. Org. Chem. 1991, 56, 3729–3731. [Google Scholar] [CrossRef]
- Teles, J.H.; Brode, S.; Chabanas, M. Cationic gold(I) complexes: Highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem. Int. Ed. 1998, 37, 1415–1418. [Google Scholar] [CrossRef]
- Starkov, P.; Rota, F.; D’Oyley, J.M.; Sheppard, T.D. Catalytic electrophilic halogenation of silyl-protected and terminal alkynes: Trapping gold(I) acetylides vs. a Brønsted acid-promoted reaction. Adv. Synth. Catal. 2012, 354, 3217–3224. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.F.; Liu, D.Y.; Yan, C.Y.; Cai, M.Z. An efficient heterogeneous gold(I)-catalyzed hydration of haloalkynes leading to α-halomethyl ketones. Synth. Commun. 2018, 48, 2983–2991. [Google Scholar] [CrossRef]
- Liu, D.Y.; Nie, Q.; Zhang, R.L.; Cai, M.Z. Heterogeneous gold-catalyzed oxidative cross-coupling of propargylic acetates with arylboronic acids leading to (E)-α-arylenones. Tetrahedron Lett. 2019, 60, 29–34. [Google Scholar] [CrossRef]
- Xie, L.Y.; Wu, Y.D.; Yi, W.G.; Zhu, L.; Xiang, J.N.; He, W.M. Gold-catalyzed hydration of haloalkynes to α-halomethyl ketones. J. Org. Chem. 2013, 78, 9190–9195. [Google Scholar] [CrossRef]
- Gomez-Herrera, A.; Hashim, I.I.; Porre, M.; Nahra, F.; Cazin, C.S.J. Au(I)-catalyzed hydration of 1-iodoalkynes leading to α-iodoketones. Eur. J. Org. Chem. 2020, 2020, 6790–6794. [Google Scholar] [CrossRef]
- Zou, H.X.; He, W.B.; Dong, Q.Z.; Wang, R.J.; Yi, N.N.; Jiang, J.; Pen, D.M.; He, W.M. First catalyzed hydration of haloalkynes by a recyclable catalytic system. Eur. J. Org. Chem. 2016, 2016, 116–121. [Google Scholar] [CrossRef]
- Chen, Z.W.; Ye, D.N.; Ye, M.; Zhou, Z.G.; Li, S.H.; Liu, L.X. AgF/TFA-promoted highly efficient synthesis of α-haloketones from haloalkynes. Tetrahedron Lett. 2014, 55, 1373–1375. [Google Scholar] [CrossRef]
- Park, J.; Yeon, J.; Lee, P.H.; Lee, K. Iron-catalyzed indirect hydration of alkynes in presence of methanesulfonic acid. Tetrahedron Lett. 2013, 54, 4414–4417. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.G.; Yan, D.; Wu, C.; Lan, L.X. Synthesis of α-chloromethyl ketones by iron-catalyzed hydration reaction of alkynyl chlorides. Chem. J. Chin. Univ. 2014, 35, 2563–2566. [Google Scholar]
- Zeng, M.; Huang, R.X.; Li, W.Y.; Liu, X.W.; He, F.L.; Zhang, Y.Y.; Xiao, F. In(OTf)3/acid co-catalyzed hydration of 1-haloalkynes to α-halomethyl ketones. Tetrahedron 2016, 72, 3818–3822. [Google Scholar] [CrossRef]
- Zou, H.X.; Jiang, J.; Yi, N.N.; Fu, W.Q.; Deng, W.; Xiang, J.N. Highly Efficient Synthesis of alpha-Halomethylketones via Ce(SO4)2/Acid Co-Catalyzed Hydration of Alkynes. Chin. J. Chem. 2016, 34, 1251–1254. [Google Scholar] [CrossRef]
- Ye, M.; Wen, Y.L.; Li, H.F.; Fu, Y.J.; Wang, Q.H. Metal-free hydration of aromatic haloalkynes to α-halomethyl ketones. Tetrahedron Lett. 2016, 57, 4983–4986. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Baire, B. Lewis basicity of water for a selective monodehalogenation of α,α-dihalo ketones to α-halo ketones and mechanistic study. Adv. Synth. Catal. 2018, 360, 298–304. [Google Scholar] [CrossRef]
- Ranu, B.C.; Chattopadhyay, K.; Jana, R. Ionic liquid promoted selective debromination of α-bromoketones under microwave irradiation. Tetrahedron 2007, 63, 155–159. [Google Scholar] [CrossRef]
- Kakesh, N.; Sayyahi, S.; Badri, R.; Tahanpesar, E. Imidazolium-based ionic network as a robust heterogeneous catalyst in synthesis of phenacyl derivatives. Russ. J. Gen. Chem. 2019, 89, 1218–1220. [Google Scholar] [CrossRef]
- Amini, A.; Sayyahi, S.; Saghanezhad, S.J.; Taheri, N. Integration of aqueous biphasic with magnetically recyclable systems: Polyethylene glycol-grafted Fe3O4 nanoparticles catalyzed phenacyl synthesis in water. Catal. Commun. 2016, 78, 11–16. [Google Scholar] [CrossRef]
- Sayyahi, S.; Saghanezhad, J. An efficient method for synthesis of phenacyl derivatives under homogeneous phase transfer catalyst condition in aqueous media. Chin. Chem. Lett. 2011, 22, 300–302. [Google Scholar] [CrossRef]
- Kiasat, A.R.; Sayyahi, S. A simple and rapid protocol for the synthesis of phenacyl derivatives using macroporous polymer-supported reagents. Mol. Divers. 2010, 14, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Liu, L.; Li, C.B. High-yielding aqueous synthesis of chloroacetophenones and aroyl chlorohydrins. Rsc Adv. 2016, 6, 25339–25345. [Google Scholar] [CrossRef]
- Shi, X.X.; Zhang, L.Q.; Yang, P.F.; Sun, H.; Zhang, Y.L.; Xie, C.S.; Zhen, O.Y.; Wang, M. Facile and efficient preparation of α-halomethyl ketones from α-diazo ketones catalyzed by iron(III) halides and silica gel. Tetrahedron Lett. 2018, 59, 1200–1203. [Google Scholar] [CrossRef]
- Dallinger, D.; Pinho, V.D.; Gutmann, B.; Kappe, C.O. Laboratory-scale membrane reactor for the generation of anhydrous diazomethane. J. Org. Chem. 2016, 81, 5814–5823. [Google Scholar] [CrossRef]
- Mastronardi, F.; Gutmann, B.; Kappe, C.O. Continuous flow generation and reactions of anhydrous diazomethane using a teflon AF-2400 tube-in-tube reactor. Org. Lett. 2013, 15, 5590–5593. [Google Scholar] [CrossRef]
- Jordan, A.; Denton, R.M.; Sneddon, H.F. Development of a more sustainable Appel reaction. Acs Sus. Chem. Eng. 2020, 8, 2300–2309. [Google Scholar] [CrossRef]
- Batesky, D.C.; Goldfogel, M.J.; Weix, D.J. Removal of triphenylphosphine oxide by precipitation with zinc chloride in polar solvents. J. Org. Chem. 2017, 82, 9931–9936. [Google Scholar] [CrossRef] [Green Version]
- Lai, G.F.; Tan, P.Z.; Ghoshal, P. A one-pot method for the efficient conversion of aryl- and acyl-substituted methyl alcohols into chlorides. Synth. Commun. 2003, 33, 1727–1732. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Tang, Y. Iron-catalyzed regioselective oxo- and hydroxy-phthalimidation of styrenes: Access to α-hydroxyphthalimide ketones. Adv. Synth. Catal. 2016, 358, 752–764. [Google Scholar] [CrossRef]
- Monticelli, S.; Rui, M.; Castoldi, L.; Missere, G.; Pace, V. A practical guide for using lithium halocarbenoids in homologation reactions. Mon. Chem. 2018, 149, 1285–1291. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.W.; Chong, S.Y.; Du, G.G.; Huang, D.F.; Wang, K.H.; Su, Y.P.; Hu, Y.L. Diacetoxyiodobenzene promoted chlorination of silyl enol ether of aryl ketones. Chin. J. Org. Chem. 2016, 36, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.D.; Adimurthy, S.; Ranu, B.C. Easy access to α-bromoketones and epoxides from vic-dibromides under aqueous conditions. Synth. Commun. 2010, 40, 3233–3239. [Google Scholar] [CrossRef]
- Paranjape, T.B.; Gokhale, G.D.; Samant, S.D. Chloroacetylation of arenes using chloroacetyl chloride in the presence of FeCl3 modified montmorillonite K10. Indian J. Chem. Sect. B 2008, 47, 310–314. [Google Scholar]
- Ram, R.N.; Manoj, T.P. Copper(I)-promoted synthesis of chloromethyl ketones from trichloromethyl carbinols. J. Org. Chem. 2008, 73, 5633–5635. [Google Scholar] [CrossRef]
- Yoshimatsu, M.; Sakai, M.; Moriura, E. Novel building blocks: 1-aryl-2-chloro-1-ethoxyethenes-Preparations and transformations. Eur. J. Org. Chem. 2007, 2007, 498–507. [Google Scholar] [CrossRef]
- Dolenc, D. Iodination of enol acetates and 1,3-diones using N-iodosaccharin. Synth. Commun. 2003, 33, 2917–2924. [Google Scholar] [CrossRef]
Figure 1.
α-Haloketones as versatile building blocks in organic synthesis.

Scheme 1.
Moderate selectivity depicted by some direct halogenation protocols.

Scheme 2.
Acid-promoted bromination mechanism.

Figure 2.
Scope of MW radiated bromination reported by Li and Xiang.

Scheme 3.
Continuous flow procedure for the α-bromination of acetophenone.

Scheme 4.
ZnBr2-mediated α-bromination of aromatic carbonyls.

Scheme 5.
Phase-vanishing α-bromination.

Scheme 6.
Acid-free α-bromination of aromatic carbonyls by Chen et al.

Scheme 7.
Acid-free α-bromination of aromatic carbonyls by Portilla and co-workers.

Scheme 8.
Scope of the I2, H2O2- and H2SO4/POM-mediated iodination.

Scheme 9.
I2/H2O2- and I2/UHP-mediated iodination in Ils.

Scheme 10.
Stavber’s NaNO2-catalysed α-iodination reaction and mechanism.

Scheme 11.
NH4NO3 and I2-based direct halogenation protocol.

Scheme 12.
m-Iodosylbenzoic acid- and I2-mediated iodination.

Scheme 13.
Iodination system developed by Rao and co-workers.

Scheme 14.
Copper-mediated iodination of ketones by Yin and co-workers.

Scheme 15.
Iodine- and trimethylorthoformate-mediated iodination.

Scheme 16.
The halogenation of ketones by Lee and co-workers.

Scheme 17.
GO-catalysed iodination of ketones.

Figure 3.
Structure of N-halosuccinimide.

Figure 4.
Chlorination scope by Lim and co-workers.

Scheme 18.
pTsOH-based chlorination and bromination by Lee and co-workers.

Scheme 19.
Direct bromination of aryl ketones as reported by Reddy and co-workers.

Scheme 20.
SiO2-catalysed bromination of aryl ketones.

Scheme 21.
Cu(OTf)2- and NH4OAc-catalysed bromination.

Scheme 22.
Jangdid’s NXS-mediated direct bromination and chlorination.

Figure 5.
Ionic liquids as halogenation solvents.

Scheme 23.
UHP/NXS α-bromination in [bmim]BF4.

Scheme 24.
Solvent-free halogenation of aromatic ketones.

Scheme 25.
Mechanism of the direct electrophilic bromination of ketones using a Br−/H2O2 system.

Scheme 26.
NaNO2-catalysed iodination of aromatic ketones.

Scheme 27.
NaIO4-NaCl α-chlorination of aryl ketones.

Scheme 28.
Gupta’s solvent-free NaBr-mediated halogenation.

Scheme 29.
NH4Cl- and Oxone®-mediated halogenation.

Scheme 30.
The Oxone-based bromination of aromatic ketones by Narender and co-workers.

Scheme 31.
Oxone-based iodination of aromatic ketones by Narender and co-workers.

Scheme 32.
Electrochemical α-bromination of ketones.

Scheme 33.
Aqueous HBr-mediated bromination by Akamanchi and co-workers.

Scheme 34.
Stavber’s NH4NO3- and I2-catalysed chlorination.

Scheme 35.
NaNO2- and O2-mediated bromination by Zhang and co-workers.

Scheme 36.
H2O2/HBr system by Stavber and co-workers.

Scheme 37.
DMSO- and HBr-mediated bromination of acetophenone.

Scheme 38.
Electrochemical HBr-mediated direct bromination.

Scheme 39.
Photocatalysed direct halogenation of acetophenone.

Scheme 40.
[EtNH3+][NO3−]/HX-mediated bromination of 1-phentylethanone.

Figure 6.
[Hmim][NO3], as co-solvent and promotor of the direct halogenation.

Figure 7.
Poly(4-vinyl-N,N-dichlorobenzenesulfonamide) as an effective chlorination agent.

Scheme 41.
Synthesis of the PVBMATB resin.

Figure 8.
Methyl-triphenylphosphonium bromide supported on polystyrene.

Scheme 42.
Liu’s polymer-supported iodination.

Figure 9.
Effective IL-based halogenation agents.

Figure 10.
Bis(imidazolium)-based (left) and Fe2O3-supported (right) ditribromide ILs as effective halogenation agents.
Figure 10.
Bis(imidazolium)-based (left) and Fe2O3-supported (right) ditribromide ILs as effective halogenation agents.

Scheme 43.
Coa and Hu’s α-bromination procedure.

Figure 11.
[Acmim]X (left) and pentylpyridinium tribromide (right).

Figure 12.
Other tribromide-based halogenation agents.

Scheme 44.
Straightforward CuBr2-based halogenation.

Scheme 45.
DMSO- and (COBr)2-mediated bromination.

Scheme 46.
DABCO-mediated halogenations.

Figure 13.
Scope of the SiCl4- and UHP-mediated bromination.

Scheme 47.
Halogenation protocol by Prakash and Mathew.

Figure 14.
Alternative halogenation agents.

Scheme 48.
DTIB-mediated bromination of aromatic ketones.

Scheme 49.
MgX2- and HTIB-based halogenation of ketones.

Scheme 50.
V(V)-catalysed bromination.

Scheme 51.
FeX3- and KX-mediated oxyhalogenation of styrenes.

Scheme 52.
Molybdenum(VI)-catalysed oxybromination of olefins.

Scheme 53.
Oxybromination of styrene derivatives by Guo and co-workers.

Scheme 54.
The use of HOBr in the oxybromination of alkenes.

Scheme 55.
NBS/Oxone system for the oxyhalogenation of olefins by Moorthy and co-workers.

Scheme 56.
Selectivity issues of the ClO2-mediated oxychlorination.

Scheme 57.
Chen’s electrochemical oxychlorination.

Scheme 58.
Itoh’s procedure using irradiation.

Scheme 59.
Zhang’s versatile oxyiodination.

Scheme 60.
I2- and IBX-based oxyiodination of alkynes.

Scheme 61.
[Au(NTf2)(IPr)] catalysed oxyhalogenation.

Scheme 62.
AuIII-catalysed oxyhalogenation of terminal alkynes.

Scheme 63.
H2O2/HBr system for the oxidation of secondary benzylic alcohols.

Scheme 64.
Urea-choline chloride-based deep eutectic solvent (DES) in the oxidative bromination of secondary alcohols.
Scheme 64.
Urea-choline chloride-based deep eutectic solvent (DES) in the oxidative bromination of secondary alcohols.

Scheme 65.
Synthesis of iodomethyl ketones from secondary alcohols.

Scheme 66.
Oxychlorination of 1-phenyl-ethanol by Studer and co-workers.

Scheme 67.
Oxidation of (2-bromoethyl)benzene into the corresponding bromoketone.

Scheme 68.
n-Octyl phenylphosphinate-mediated oxidation of benzyl haloalkanes.

Scheme 69.
Photocatalytic oxidation of vinyl halides.

Scheme 70.
AlMe3-catalysed Oppenauer oxidation.

Scheme 71.
Solvent-free oxidation of functionalised benzylalcohols.

Scheme 72.
The AuI hydration reaction by Cai and co-workers.

Scheme 73.
The AuI hydration iodoalkynes by Cazin and co-workers.

Scheme 74.
Iron chloride-catalysed hydration of 1-bromo and 1-chloroalkynes.

Scheme 75.
In(OTf)3-catalysed hydration of 1-bromoalkynes by Xiao and co-workers.

Scheme 76.
Ce(SO4)2-catalysed hydration of 1-bromoalkynes by Xiao and co-workers.

Scheme 77.
HBF4-catalysed hydration of 1-bromoalkynes by Wang and co-workers.

Scheme 78.
Selective debromination of α,α-dihaloketones.

Scheme 79.
Access to α-iodoketones using a poly(ionicliquid)/NaI system.

Scheme 80.
PhSO2Cl-mediated substitution of α-bromoketones.

Scheme 81.
Halogenation of α-diazoketones by Wang and co-workers.

Scheme 82.
Continuous process for the synthesis of α-chloroketones.

Scheme 83.
Appel halogenation of α-hydroxy ketone.

Scheme 84.
α-Haloketones accessed via α-hydroxybenzotriazole functionalised ketones.

Scheme 85.
ICH2Cl- and MeLi-mediated synthesis of 2-chloro-1-phenylethanone.

Scheme 86.
Silyl enol ethers as suitable synthons for aromatic α-haloketones.

Scheme 87.
Synthesis of α-bromoketones via vic-1,2-dibromides.

Scheme 88.
Chloroacetylation of substituted arenes.

Scheme 89.
Yoshimatsu’s oxidation protocol for enolethers.

Figure 15.
Structure of N-iodosaccharin.


{kind=link}
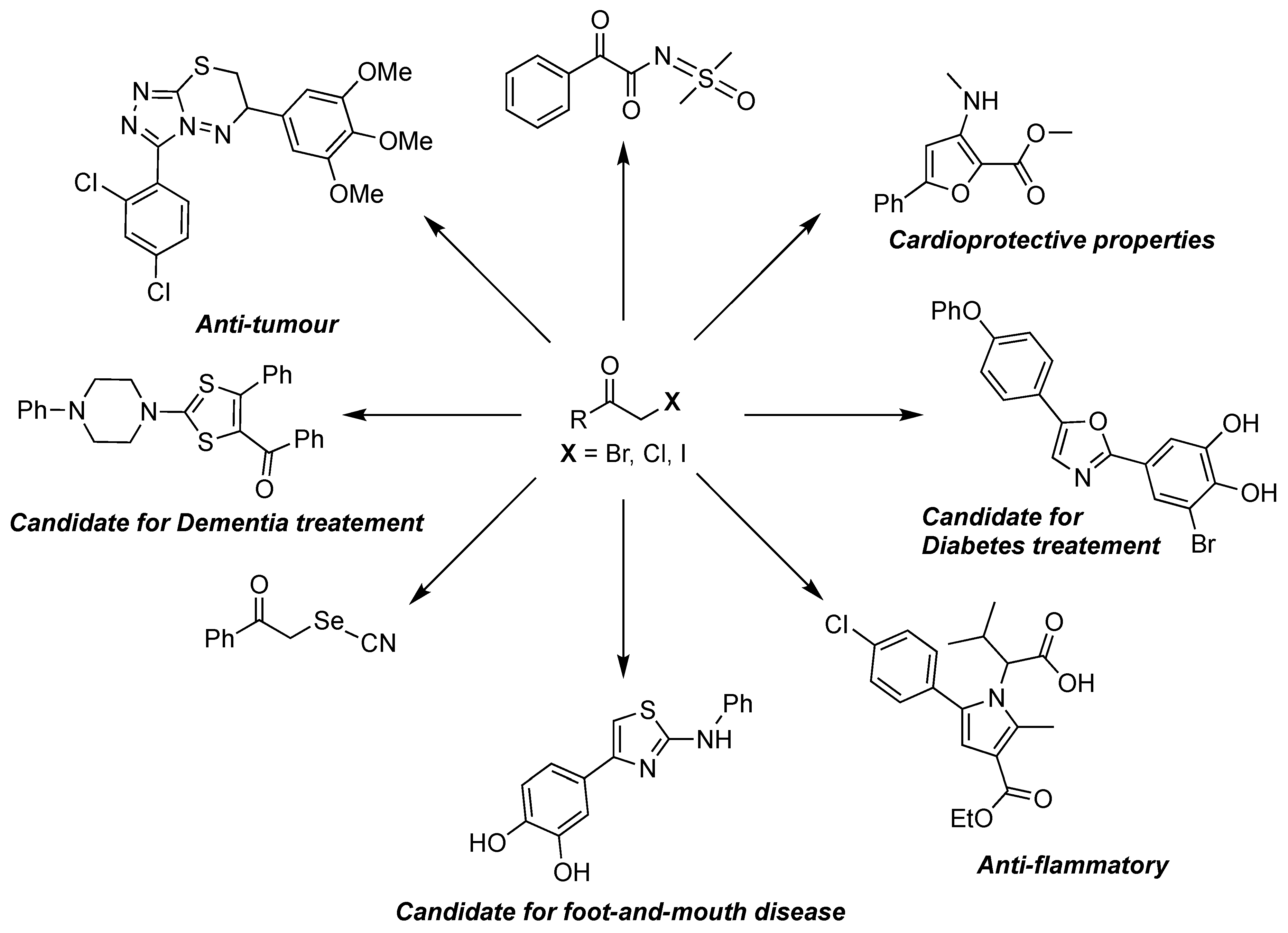{kind=link}
{kind=link}
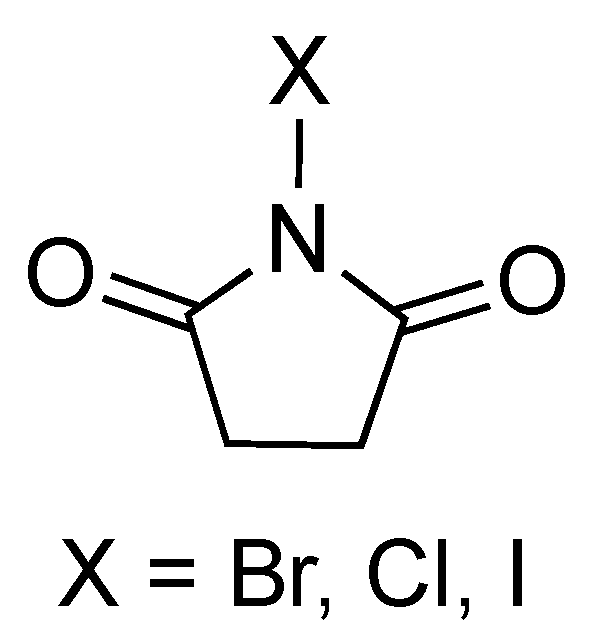{kind=link}
{kind=link}
{kind=link}
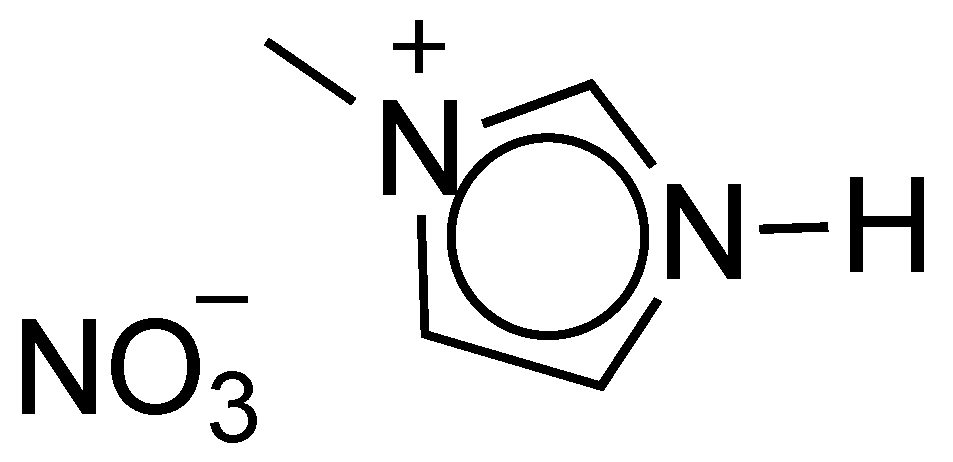{kind=link}
{kind=link}
{kind=link}
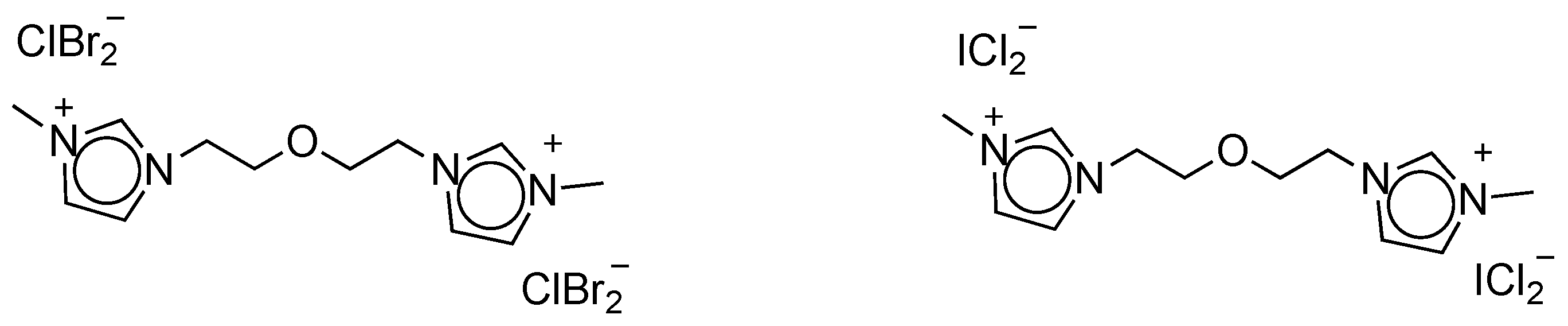{kind=link}
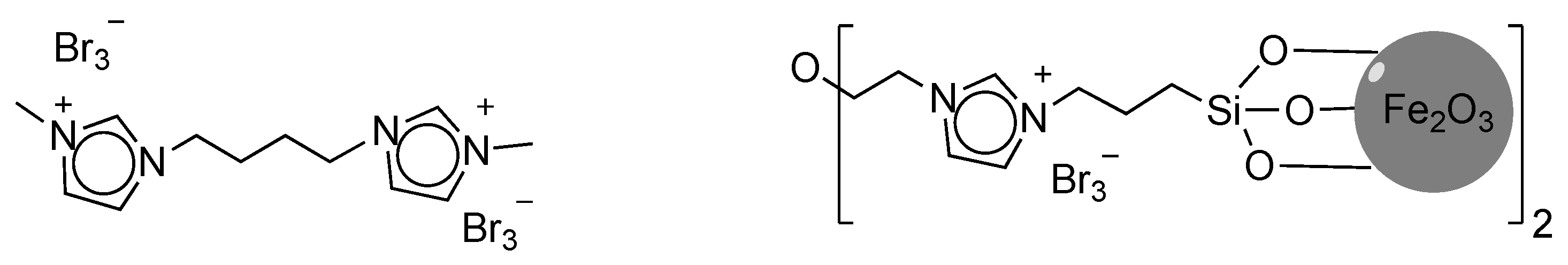{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
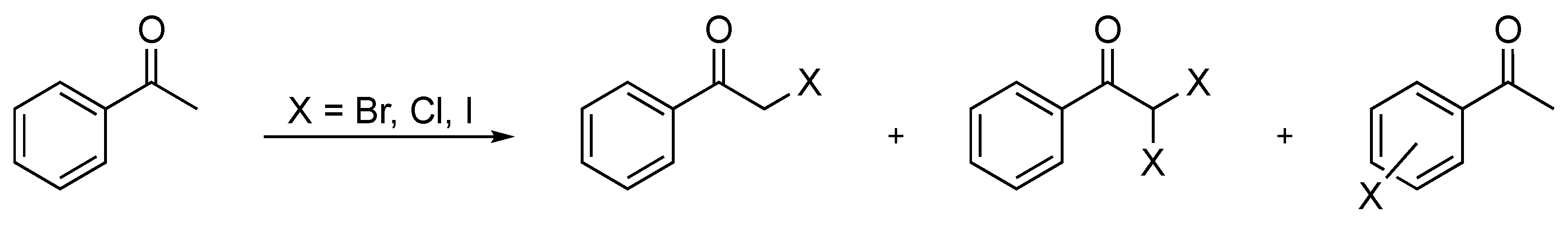{kind=link}
{kind=link}
{kind=link}
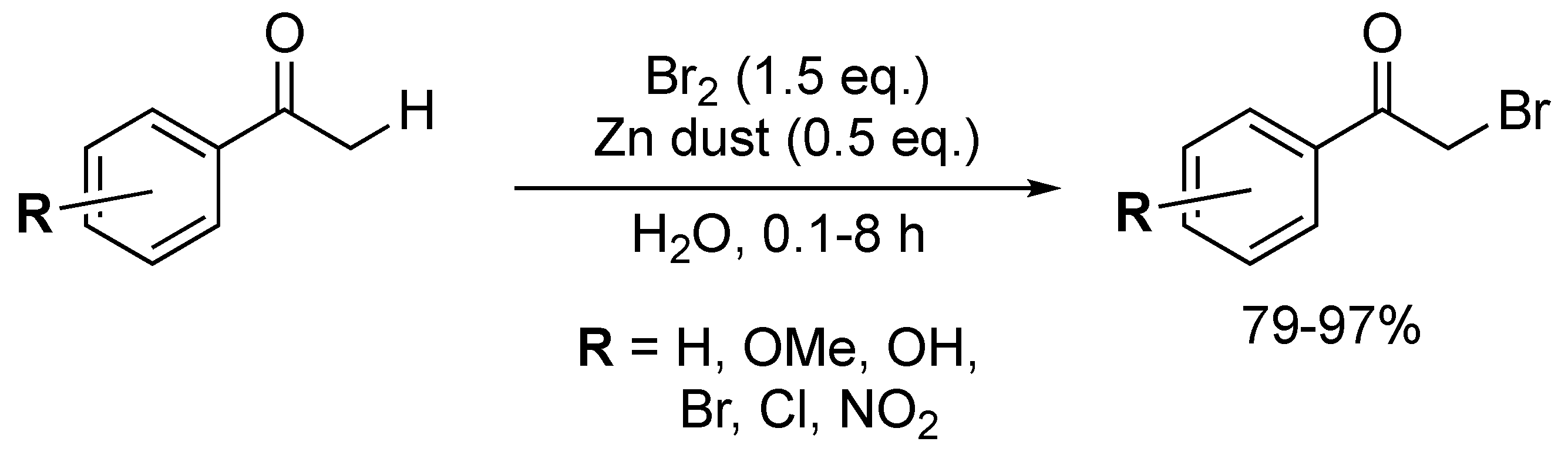{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
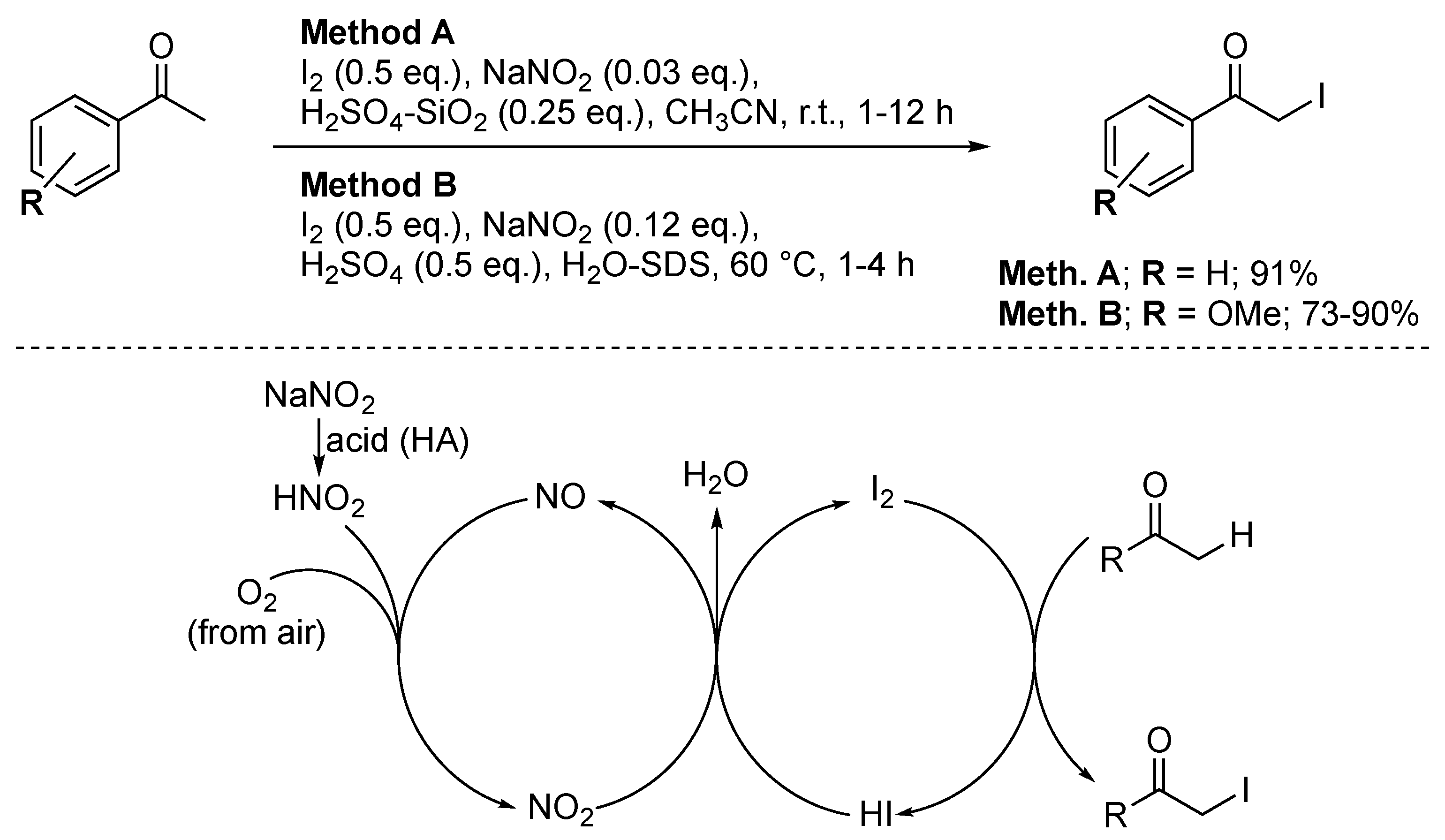{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
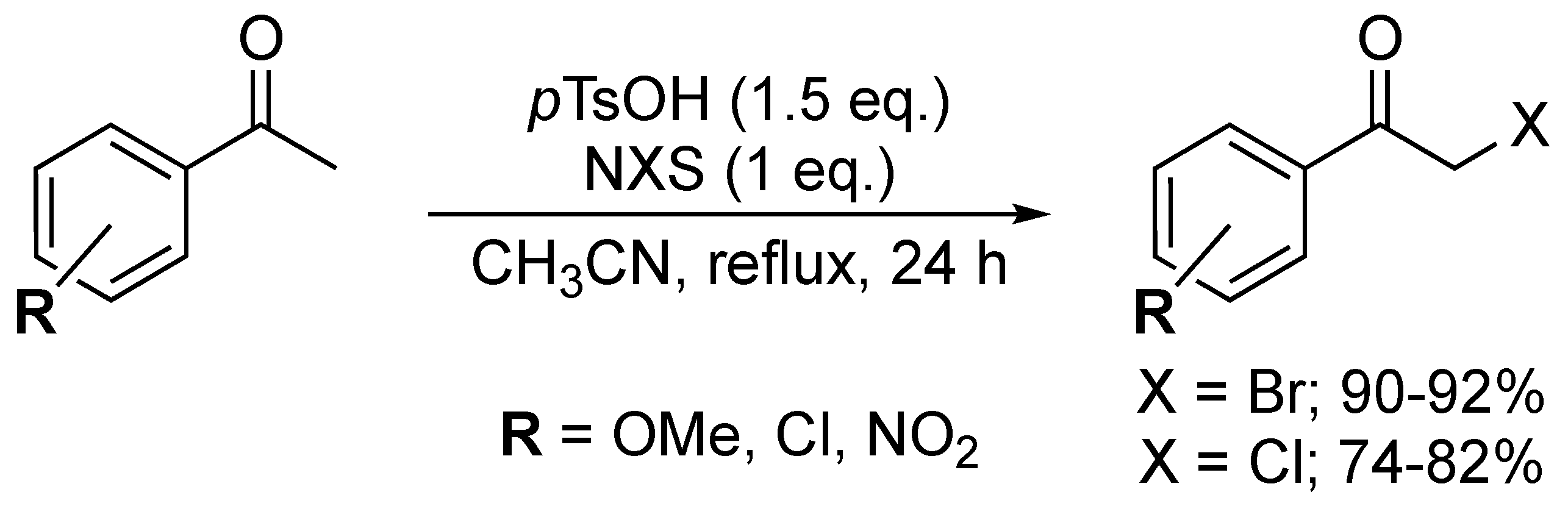{kind=link}
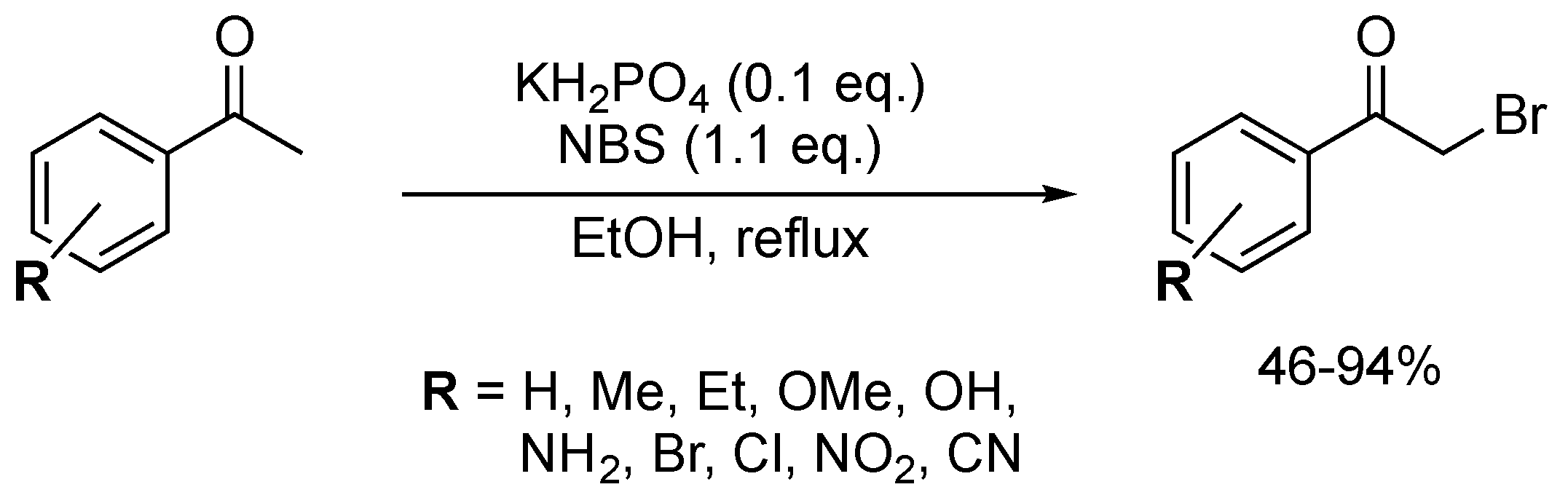{kind=link}
{kind=link}
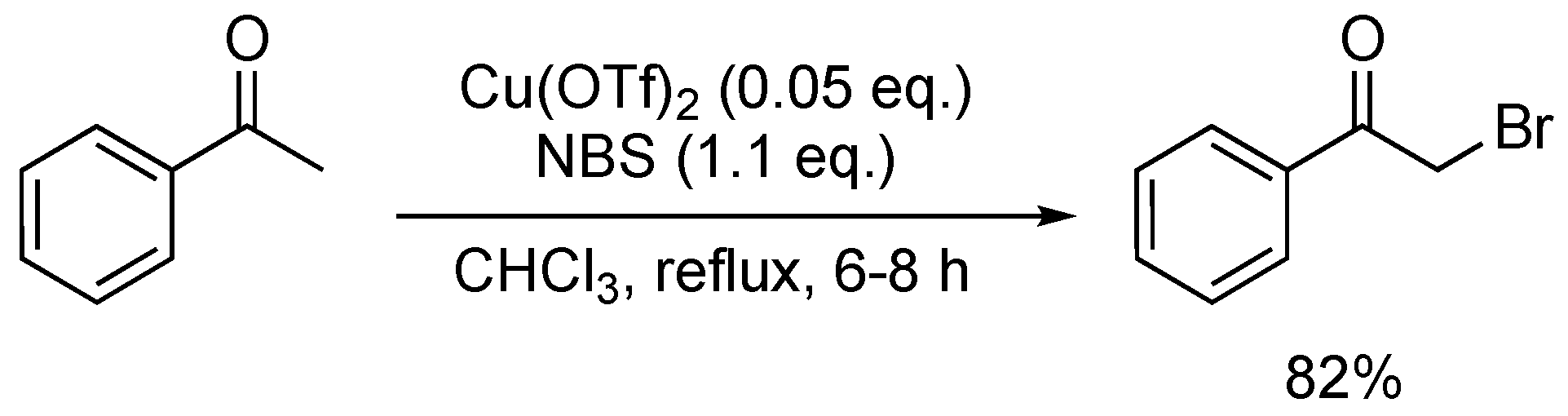{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
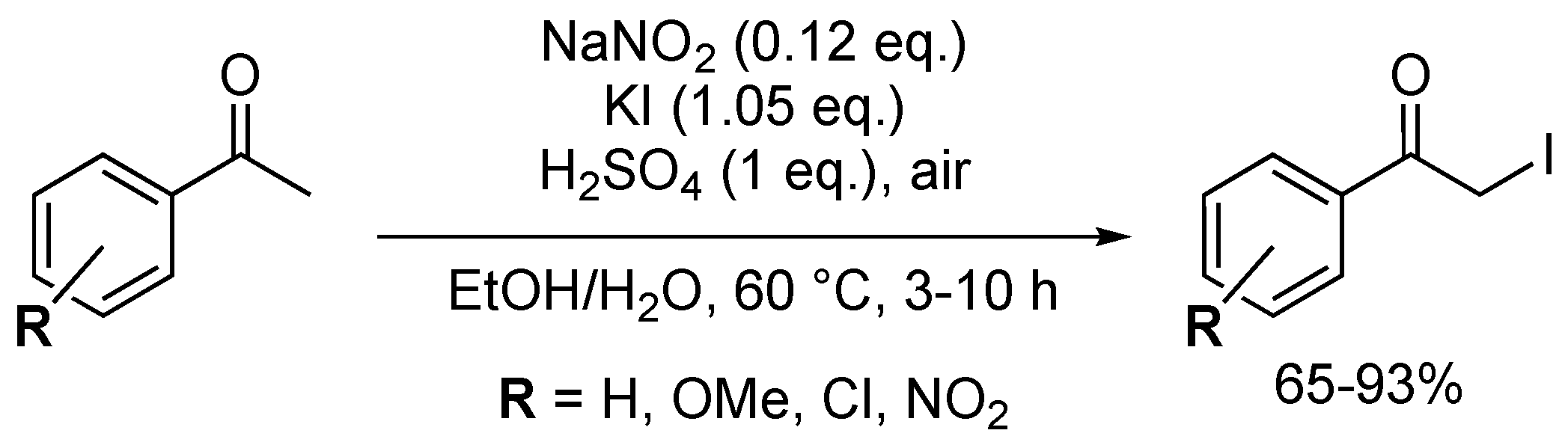{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
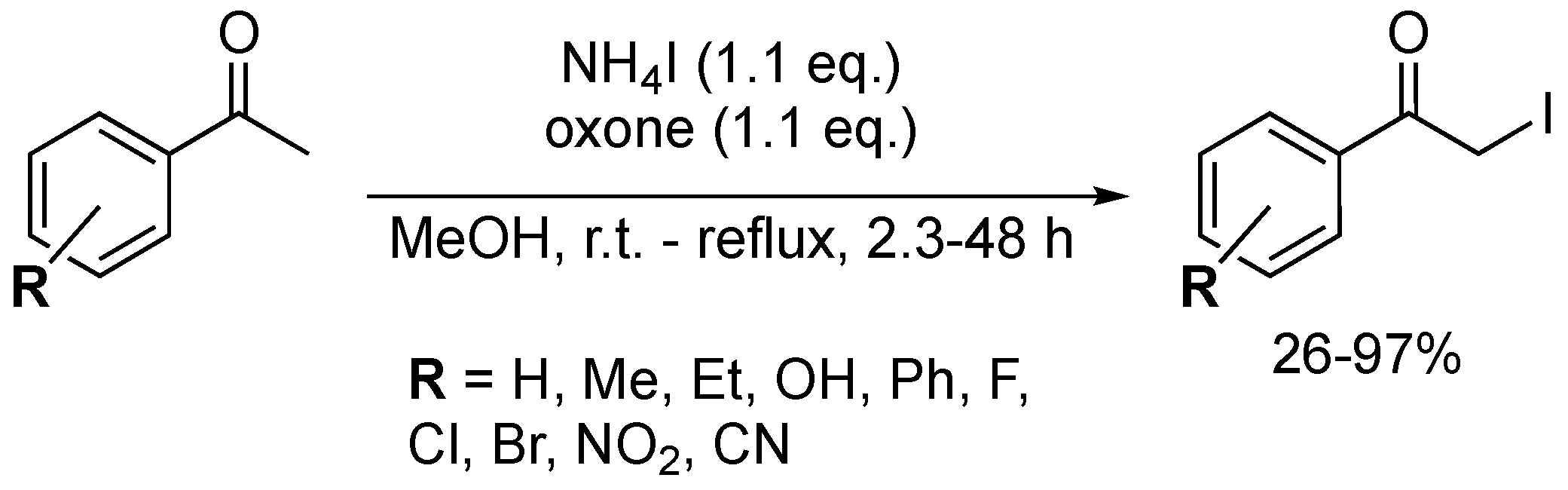{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
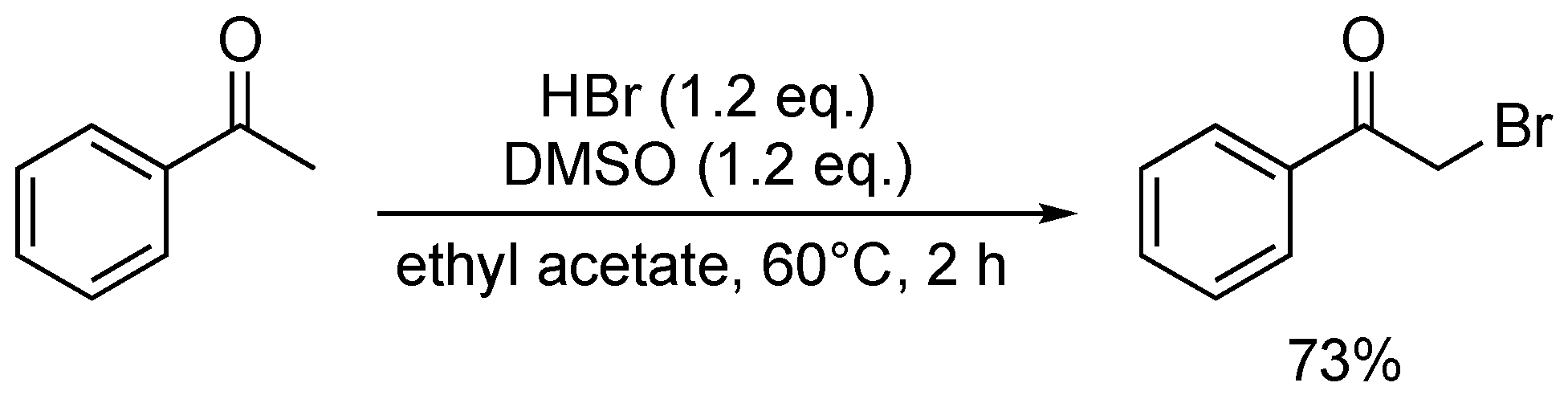{kind=link}
{kind=link}
{kind=link}
{kind=link}
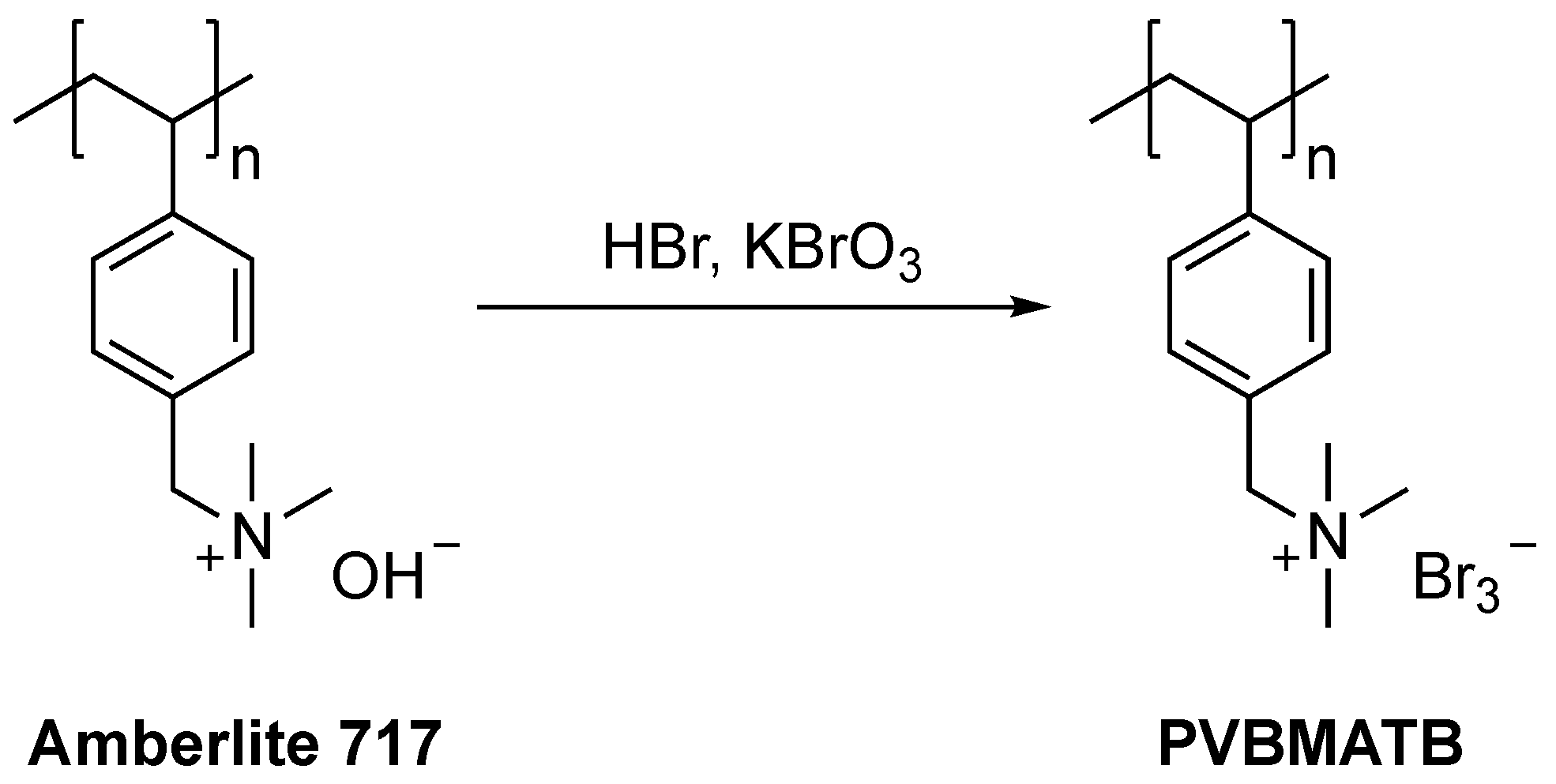{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
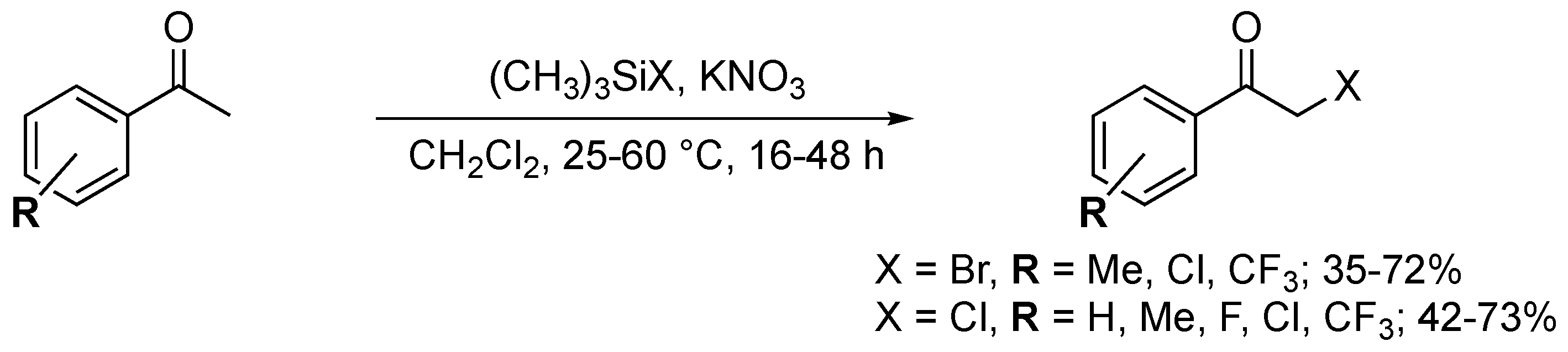{kind=link}
{kind=link}
{kind=link}
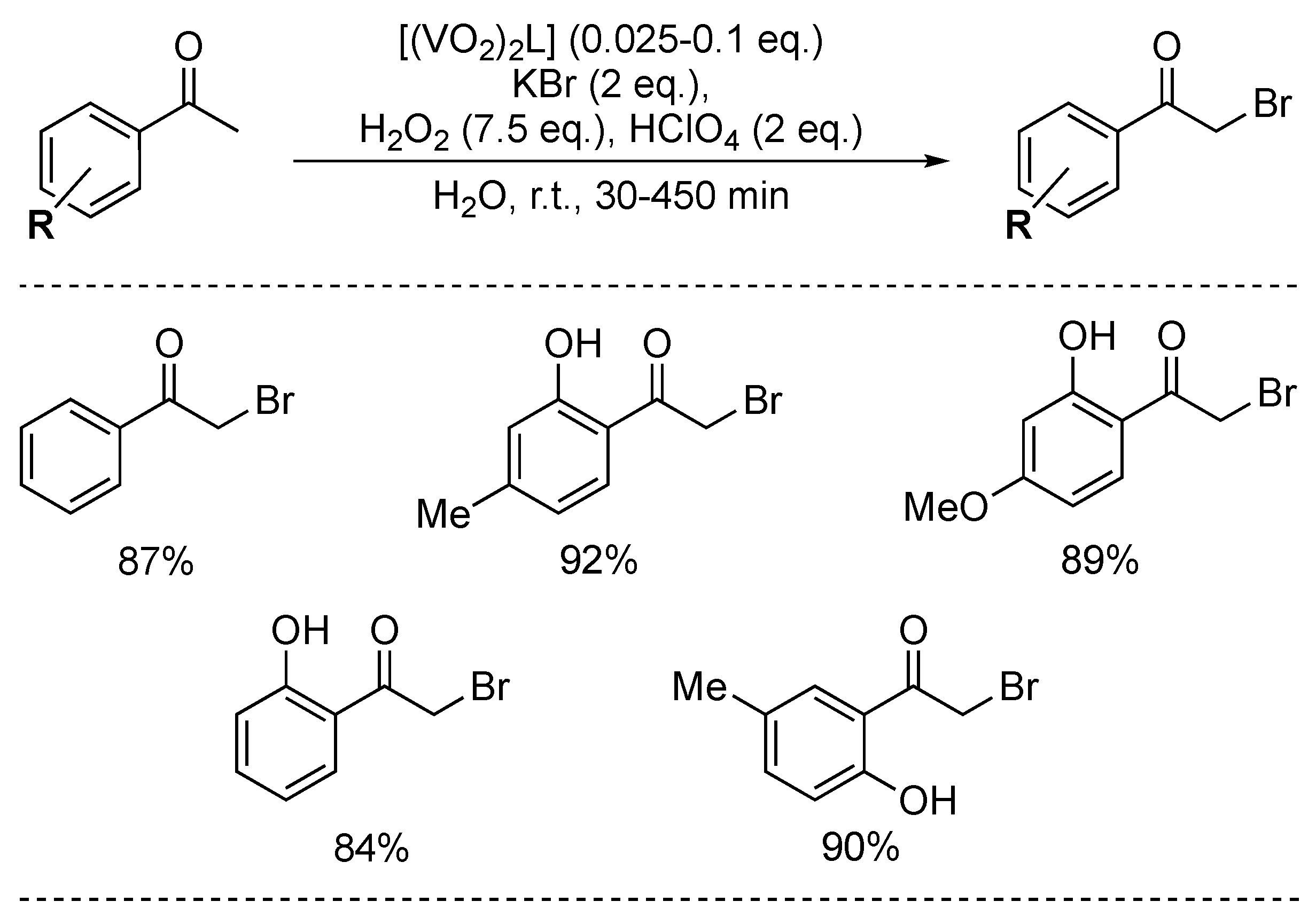{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
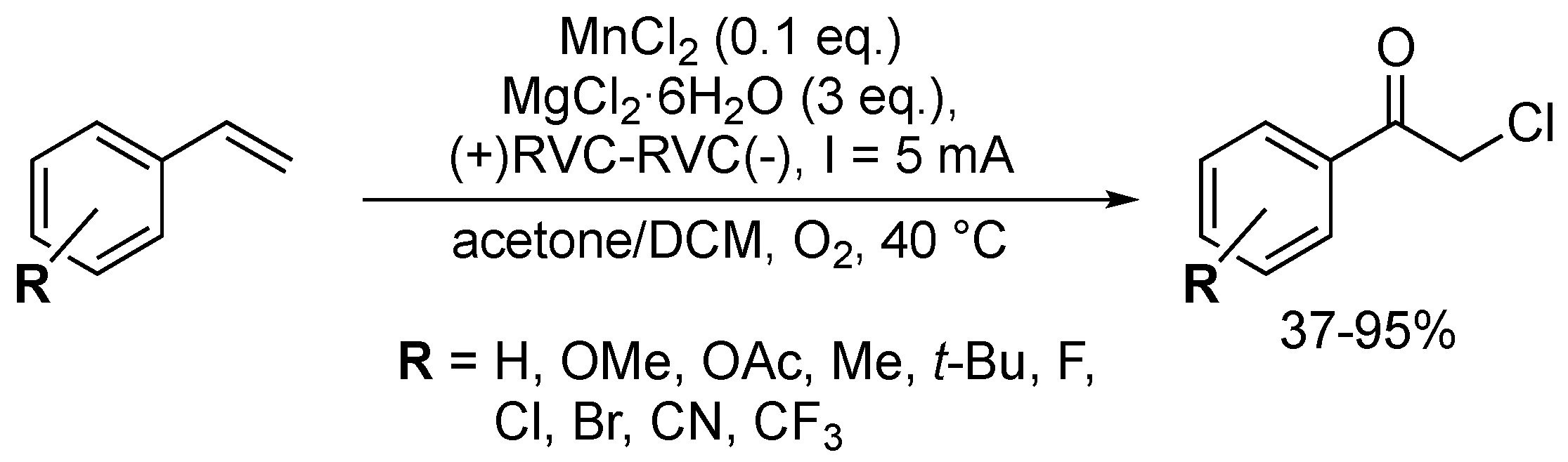{kind=link}
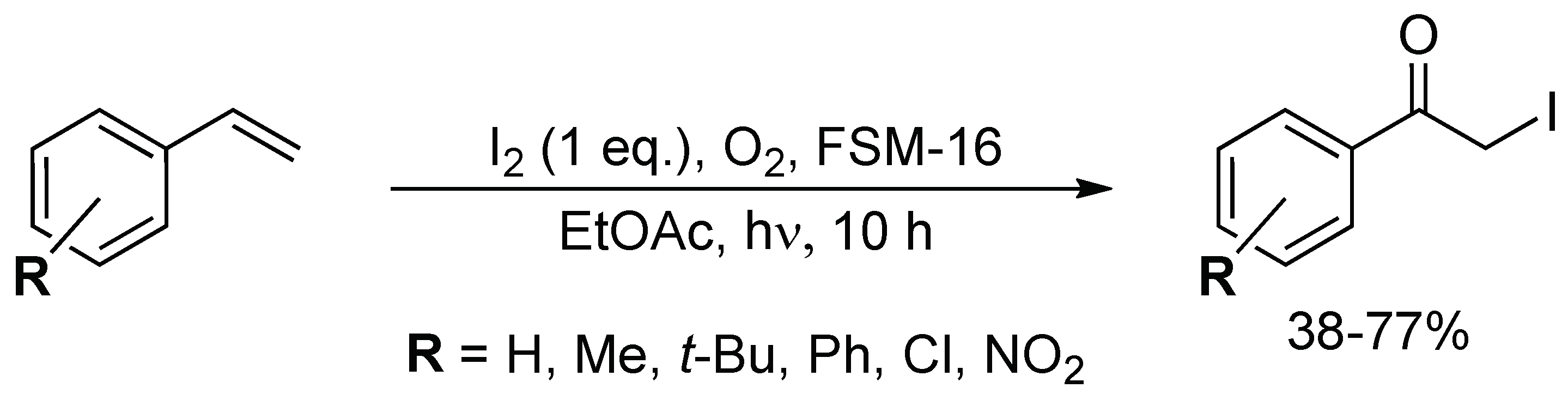{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
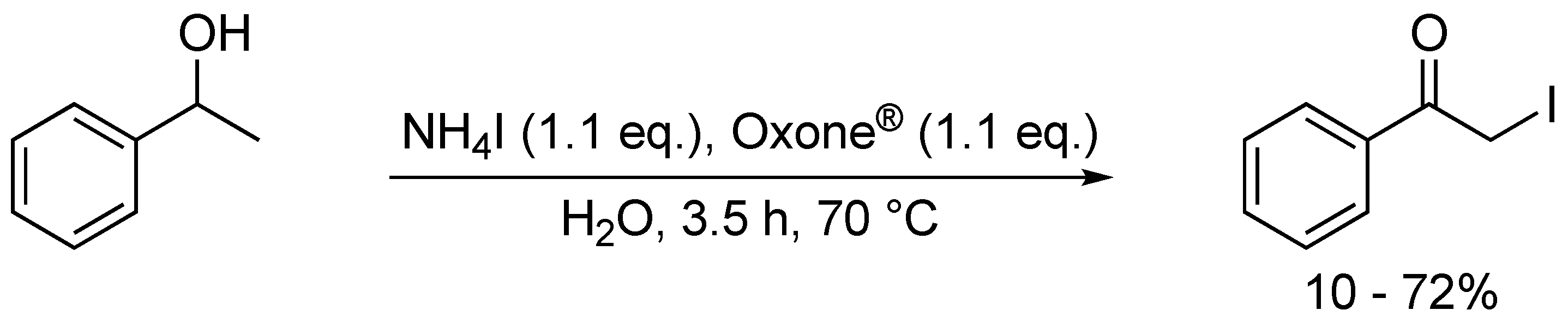{kind=link}
{kind=link}
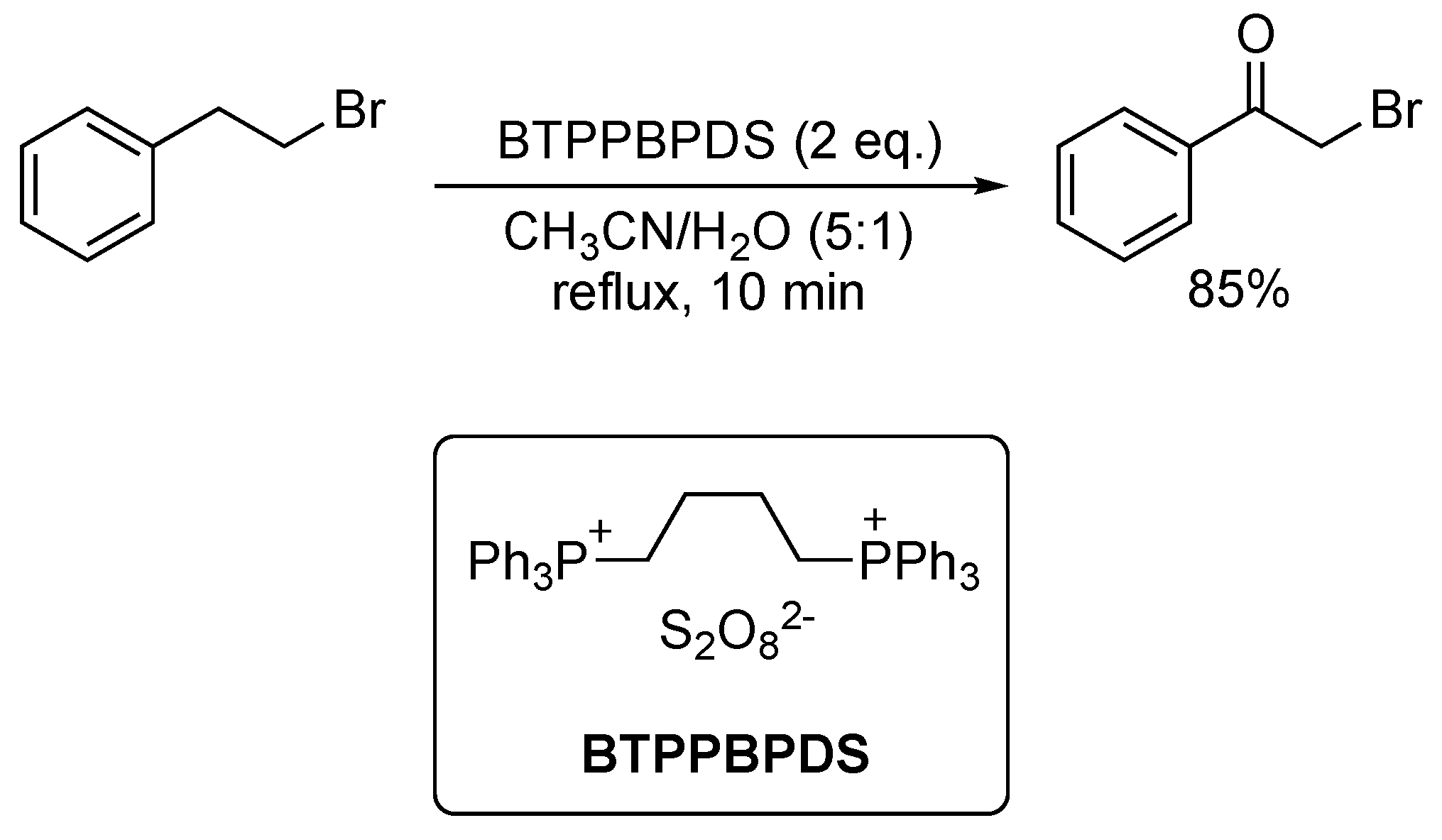{kind=link}
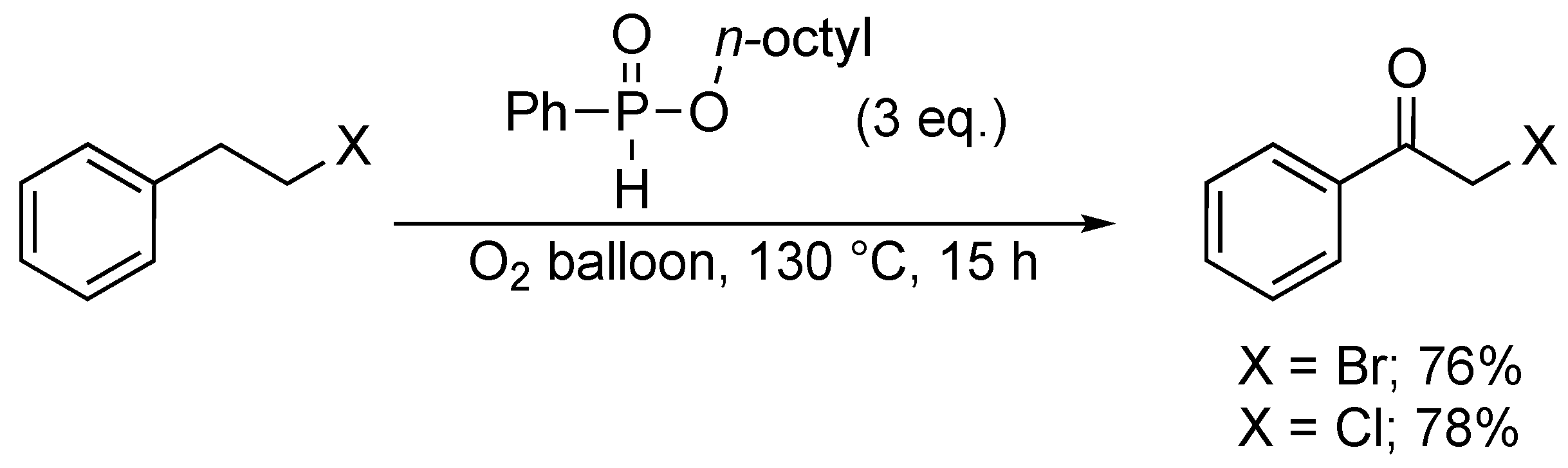{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
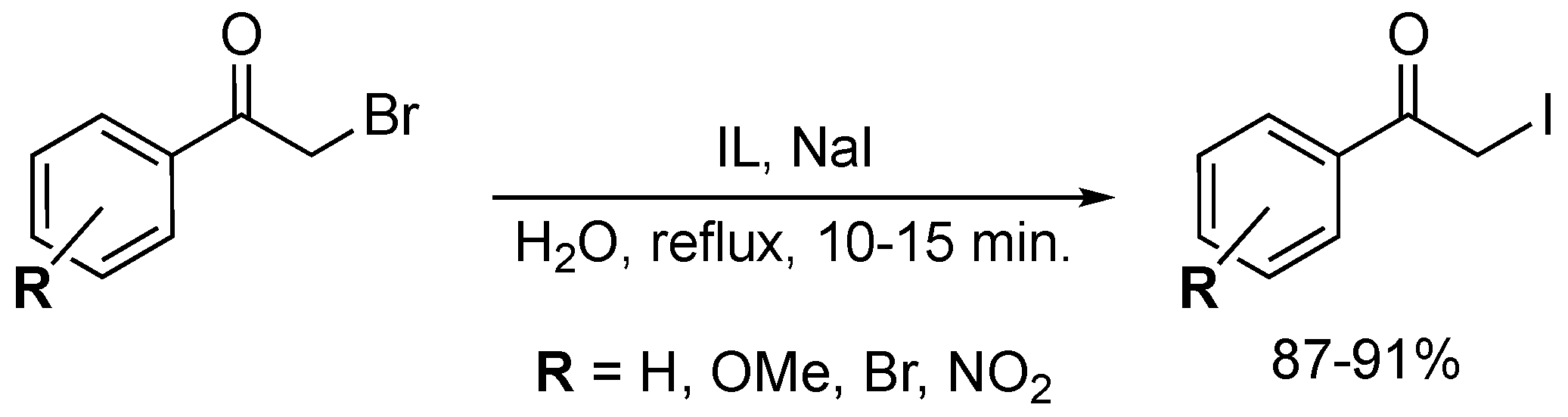{kind=link}
{kind=link}
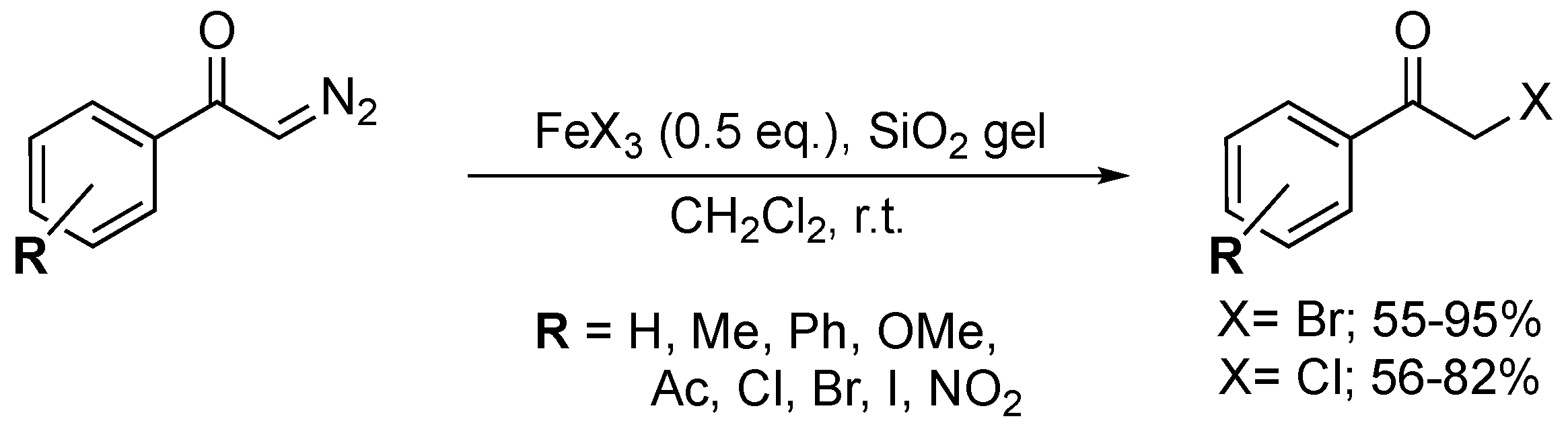{kind=link}
{kind=link}
{kind=link}
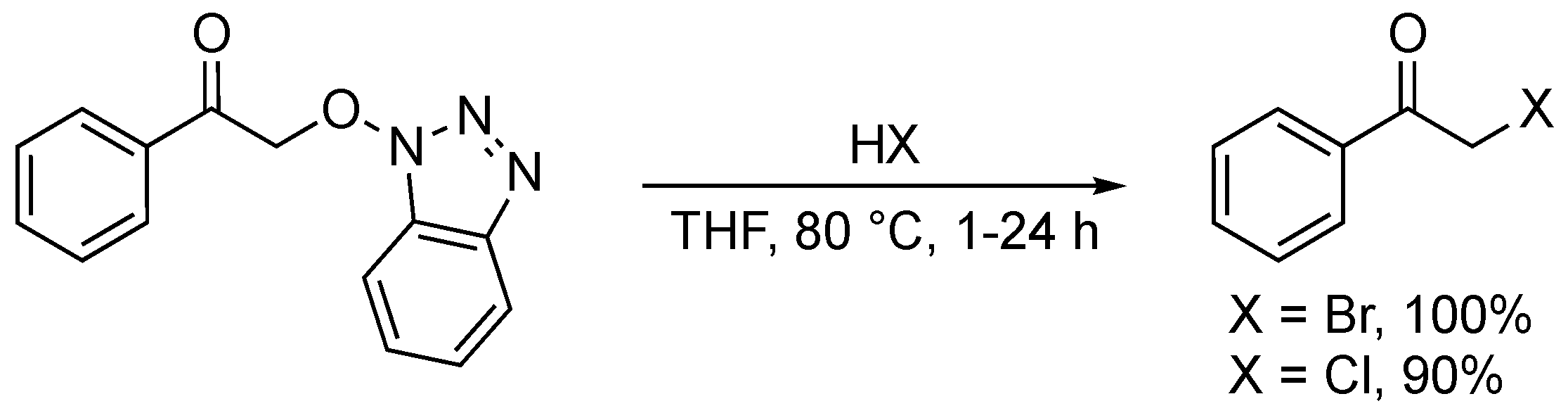{kind=link}
{kind=link}
{kind=link}
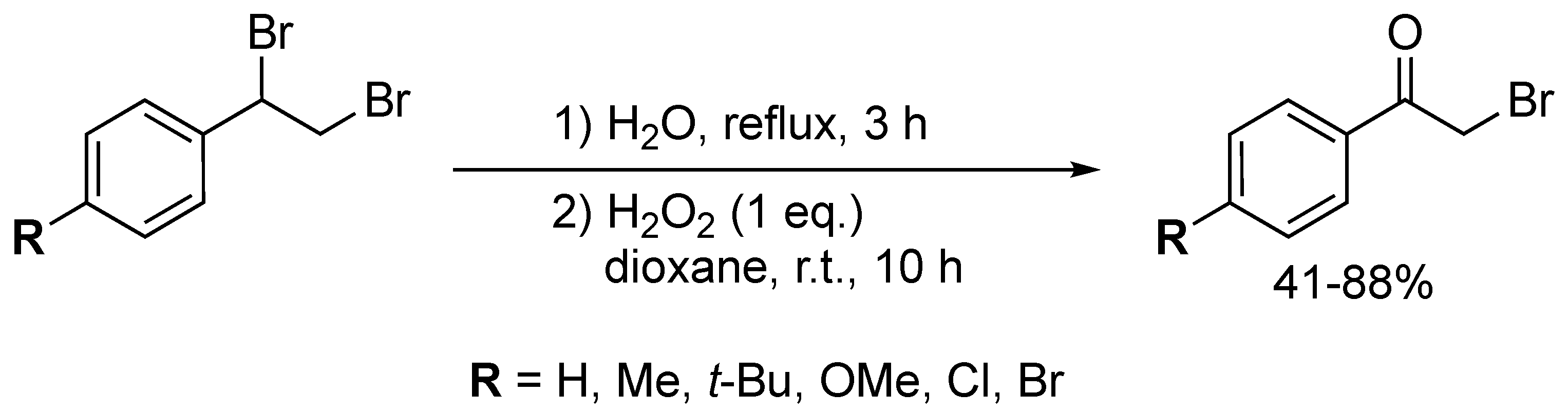{kind=link}
{kind=link}
{kind=link}
Table 1.
The bromination scope reported by Krupadanam.
| R | R′ | R″ | Yield (%) | |
|---|---|---|---|---|
| H | H | H | 85 |  |
| H | H | Cl | 70 | |
| Cl | H | Cl | 60 | |
| H | NO2 | H | 70 | |
| H | H | Br | 75 | |
| H | H | F | 85 | |
| H | H | OMe | 70 | |
| H | H | OCH2Ph | 65 |
Table 2.
Aqueous H2O2-based oxyhalogenation methods.
 | |||
|---|---|---|---|
| Reaction Conditions | α-Bromoketone (%) | Bromohydrin (%) | Ref. |
| HBr (2 equiv.), H2O2 (1 equiv.), H2O, r.t. | 12 | 53 | [141] |
| NBS (2 equiv.), H2O, r.t. | 19 | 81 | [141] |
| 30% aq. HBr (1.7 equiv.), H2O2 (3 equiv.), H2O, r.t. | 94 | 4 | [140] |
Table 3.
Photo-oxidative synthesis of phenylacylhalides.
| X | Reaction Conditions | Scope (Yield) | Aromatic Substituents |
|---|---|---|---|
| I | I2 (0.6 equiv.), hv, H2O/EtOAc | 7 α-iodoketones (43–88%) | H, t-Bu, Me, OMe, Cl, Ph |
| Br | 48% aq. HBr (1.1 equiv.), hv, H2O/EtOAc | 7 α-bromoketones (trace–86%) | H, t-Bu, Me, OMe, Cl, Ph |
Table 4.
Phukan and He’s oxyhalogenation procedures.
| Reaction Conditions | Scope (Yield) | Aromatic Substituents | Refs. |
|---|---|---|---|
| TsNBr2 (2 equiv.), Na2SO3 (8 equiv.), EtOAc/acetone/H2O, r.t, 24 h | 17 α-bromoketones (75–80%) | H, Me, t-Bu, n-Pr, F, Br, Cl | [147] |
| DBDMH (1.2 equiv.), ethylene thiourea (0.15 equiv.), acetone/H2O, 45 °C | 17 α-bromoketones (77–91%) 1 α-chloroketone (78%) 1 α-iodoketones (69%) | H, Me, OMe, OBn, SMe, F, Cl, Br, I, CF3 | [157] |
Table 5.
FeIII-catalysed oxidation of haloalkenes.
| Method | Reaction Conditions | Scope (Yield) | Aromatic Substituents |
|---|---|---|---|
| 1 | Fe(OTf)3 (1.15 mol%), L4 (1.15 mol%), DCE, 75 °C, 8 h | 2 α-Bromoketones (91–92%) | H, Cl |
| 2 | Fe(OTf)3 (1.15 mol%), L1 (1.15 mol%), O2 (1 atm), DCE/DBE 75 °C, 8 h | 4 α-Bromoketones (65–99%) 2 α-Chloroketone (50–70%) 1 α-Iodoketones (70%) | H, Me, OMe, F, Cl, Br |
 | |||
Table 6.
Micellar catalytic oxidation of 2-bromo-1-phenylethanol.
| Reaction Conditions | Scope (Yield) | Refs. |
|---|---|---|
| 2-iodosylbenzoic acid (0.01 equiv.), Oxone® (0.525 equiv.), CTAB (3 wt%), H2O, r.t., 2 h | 2-bromo-1-phenylethanone (95%) | [173] |
| IBX (1.2 equiv.), GMPGS-2000 (2 wt%), H2O, r.t., 24 h | 2-bromo-1-phenylethanone (64%) | [174] |
Table 7.
CuII- and AgI-catalysed hydrations.
| Route | Catalyst (mol%) | Reaction Conditions | Scope (Yield) |
|---|---|---|---|
| He (2016) [194] | Cu(OAc)2·H2O (10) | H2O (1 equiv.) in TFA at 70 °C for 6 h | 15 α-bromoketones (77–96%) 3 α-chloroketones (91–92%) 4 α-iodoketones (90–96%) |
| Liu (2014) [195] | AgF (5) | H2O (1 equiv.) in TFA at 40 °C for 6–16 h | 15 α-bromoketones (61–95%) 3 α-chloroketones (81–90%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Porré, M.; Pisanò, G.; Nahra, F.; Cazin, C.S.J. Synthetic Access to Aromatic α-Haloketones. Molecules 2022, 27, 3583. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113583
AMA Style
Porré M, Pisanò G, Nahra F, Cazin CSJ. Synthetic Access to Aromatic α-Haloketones. Molecules. 2022; 27(11):3583. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113583
Chicago/Turabian StylePorré, Marre, Gianmarco Pisanò, Fady Nahra, and Catherine S. J. Cazin. 2022. "Synthetic Access to Aromatic α-Haloketones" Molecules 27, no. 11: 3583. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27113583