
Changes in the Nasal Microbiota of Pigs Following Single or Co-Infection with Porcine Reproductive and Respiratory Syndrome and Swine Influenza A Viruses

 , , and
, , and 
Abstract
:1. Introduction
2. Results
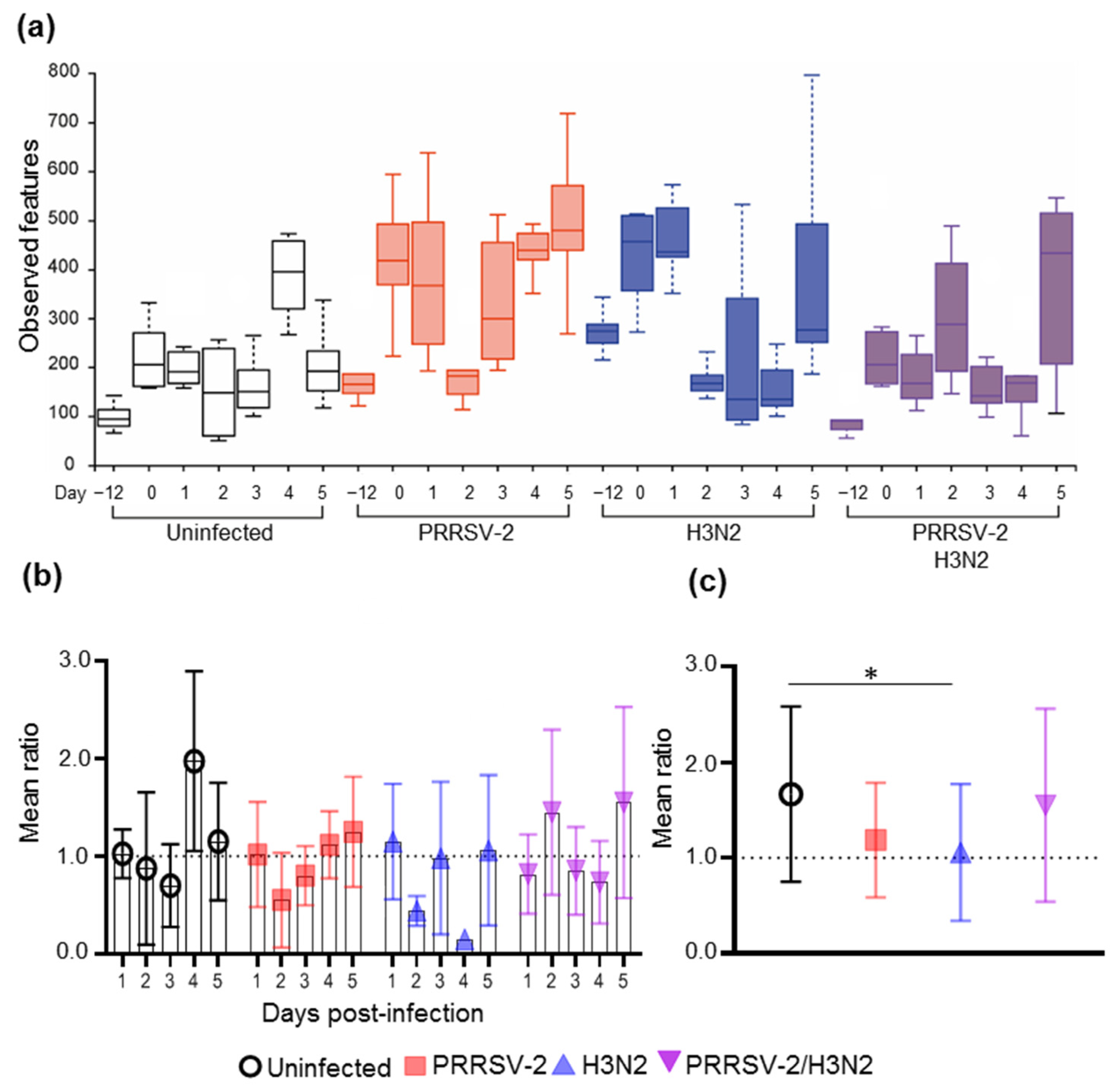
2.1. Effects of Single and Viral Co-Infection on Nasal Microbiota Richness
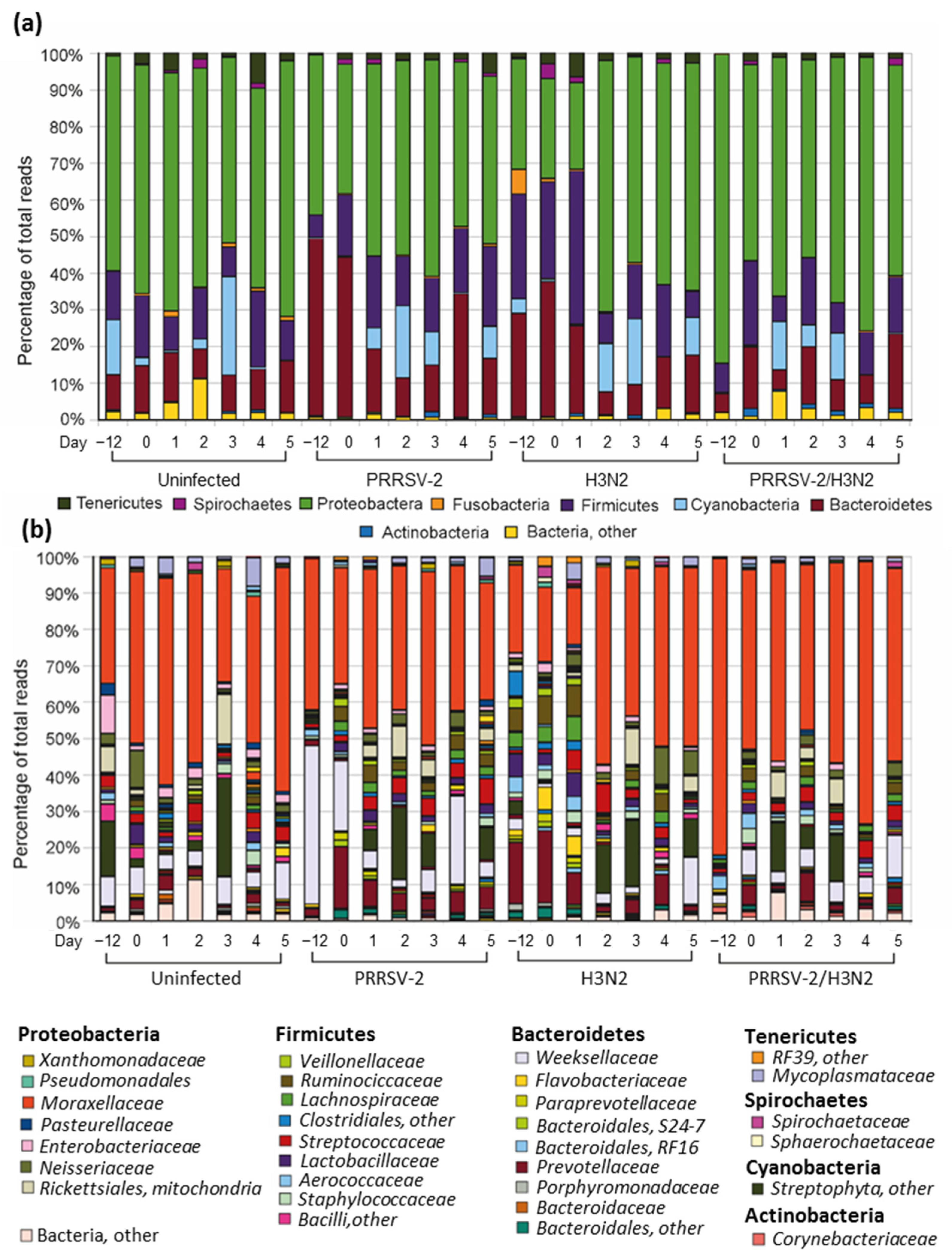
2.2. Description of Nasal Microbiota Communities in Healthy Pigs
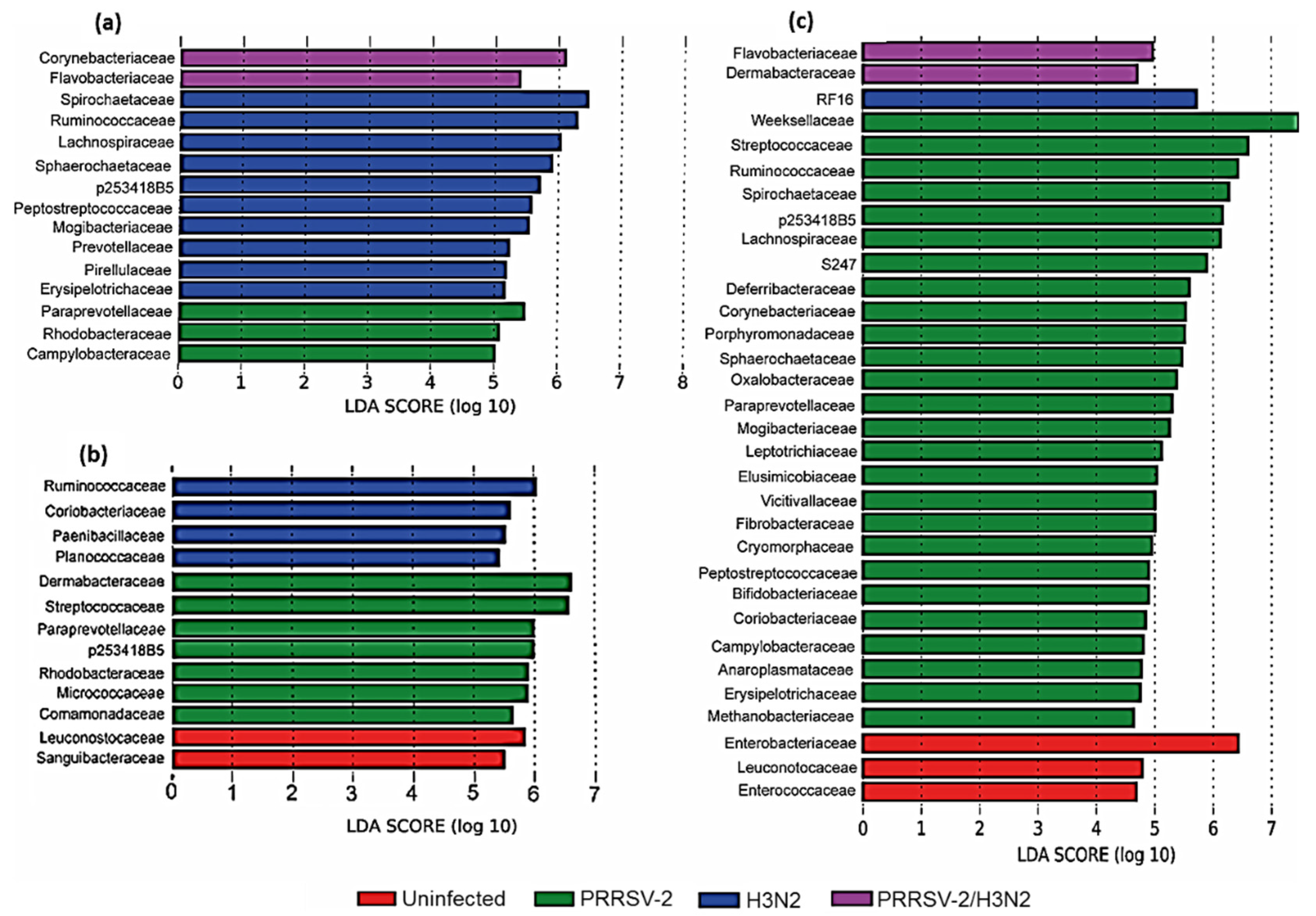
2.3. The Nasal Microbiota Community Changes after Viral Infection
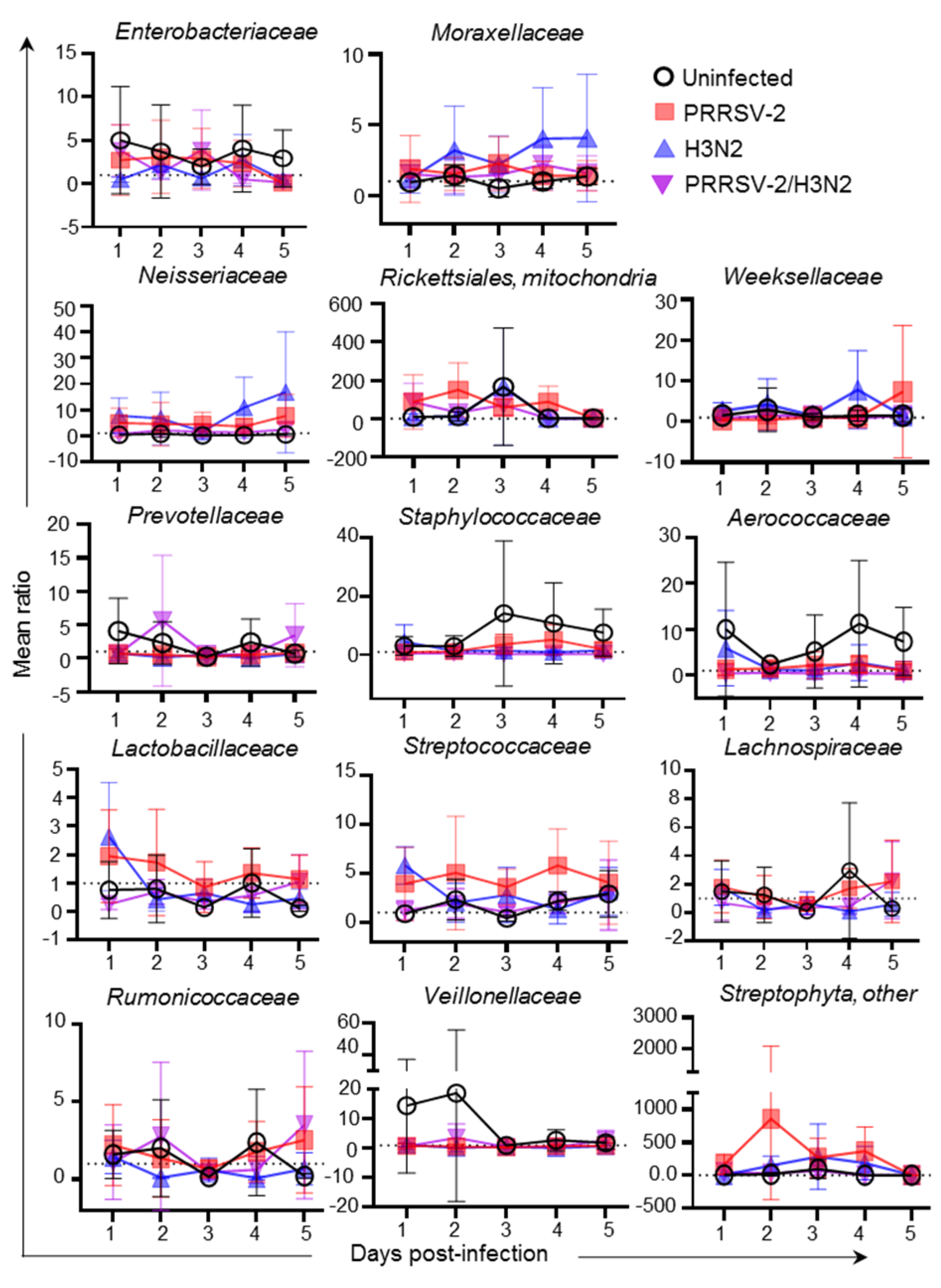
2.4. Correlation Analysis of H3N2 Titre and Relative Abundance of Bacterial Species in Nasal Swabs
3. Discussion
4. Materials and Methods
4.1. Challenge and Nasal Swab Samples
4.2. DNA Extraction by TRIzol and Column Purification
4.3. 16S rRNA Gene Amplicon Sequencing
4.4. Microbiota Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niederwerder, M.C. Role of the Microbiome in Swine Respiratory Disease. Vet. Microbiol. 2017, 209, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal Bacteria Calibrate the Activation Threshold of Innate Antiviral Immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota Regulates Immune Defense against Respiratory Tract Influenza a Virus Infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Vidyarthi, A.; Nadeem, S.; Negi, S.; Nair, G.; Agrewala, J.N. Alteration in the Gut Microbiota Provokes Susceptibility to Tuberculosis. Front. Immunol. 2016, 7, 529. [Google Scholar] [CrossRef]
- Schachtschneider, K.M.; Yeoman, C.J.; Isaacson, R.E.; White, B.A.; Schook, L.B.; Pieters, M. Modulation of Systemic Immune Responses through Commensal Gastrointestinal Microbiota. PLoS ONE 2013, 8, e53969. [Google Scholar] [CrossRef]
- Tsuruta, T.; Inoue, R.; Tsushima, T.; Watanabe, T.; Tsukahara, T.; Ushida, K. Oral Administration of EC-12 Increases the Baseline Gene Expression of Antiviral Cytokine Genes, IFN-γ and TNF-α, in Splenocytes and Mesenteric Lymph Node Cells of Weaning Piglets. Biosci. Microbiota Food Health 2013, 32, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saade, G.; Deblanc, C.; Bougon, J.; Marois-Créhan, C.; Fablet, C.; Auray, G.; Belloc, C.; Leblanc-Maridor, M.; Gagnon, C.A.; Zhu, J.; et al. Coinfections and Their Molecular Consequences in the Porcine Respiratory Tract. Vet. Res. 2020, 51, 80. [Google Scholar] [CrossRef]
- Wang, Q.; Cai, R.; Huang, A.; Wang, X.; Qu, W.; Shi, L.; Li, C.; Yan, H. Comparison of Oropharyngeal Microbiota in Healthy Piglets and Piglets With Respiratory Disease. Front. Microbiol. 2018, 9, 3218. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, X.; Di, D.; Pan, R.; Gao, Y.; Xiao, C.; Li, B.; Wei, J.; Liu, K.; Qiu, Y.; et al. Comparative Analysis of the Pulmonary Microbiome in Healthy and Diseased Pigs. Mol. Genet. Genom. 2021, 296, 21–31. [Google Scholar] [CrossRef]
- Slifierz, M.J.; Friendship, R.M.; Weese, J.S. Longitudinal Study of the Early-Life Fecal and Nasal Microbiotas of the Domestic Pig. BMC Microbiol. 2015, 15, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Gongora, C.; Larsen, N.; Schønning, K.; Fredholm, M.; Guardabassi, L. Differential Analysis of the Nasal Microbiome of Pig Carriers or Non-Carriers of Staphylococcus aureus. PLoS ONE 2016, 11, e0160331. [Google Scholar] [CrossRef] [Green Version]
- Correa-Fiz, F.; Neila-Ibáñez, C.; López-Soria, S.; Napp, S.; Martinez, B.; Sobrevia, L.; Tibble, S.; Aragon, V.; Migura-Garcia, L. Feed Additives for the Control of Post-Weaning Streptococcus suis Disease and the Effect on the Faecal and Nasal Microbiota. Sci. Rep. 2020, 10, 20354. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Fuertes, M.; Correa-Fiz, F.; Fraile, L.; Sibila, M.; Aragon, V. Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis. Pathogens 2021, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Fablet, C.; Marois-Créhan, C.; Simon, G.; Grasland, B.; Jestin, A.; Kobisch, M.; Madec, F.; Rose, N. Infectious Agents Associated with Respiratory Diseases in 125 Farrow-to-Finish Pig Herds: A Cross-Sectional Study. Vet. Microbiol. 2012, 157, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Goyal, S.M.; Joo, H.S. Retrospective Analysis of Etiologic Agents Associated with Respiratory Diseases in Pigs. Can. Vet. J. 2003, 44, 735–737. [Google Scholar]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to Virus Taxonomy and the Statutes Ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef]
- Lunney, J.K.; Fang, Y.; Ladinig, A.; Chen, N.; Li, Y.; Rowland, B.; Renukaradhya, G.J. Porcine Reproductive and Respiratory Syndrome Virus (PRRSV): Pathogenesis and Interaction with the Immune System. Annu. Rev. Anim. Biosci. 2016, 4, 129–154. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, D.J.; Kliebenstein, J.B.; Neumann, E.J.; Zimmerman, J.J.; Rotto, H.F.; Yoder, T.K.; Wang, C.; Yeske, P.E.; Mowrer, C.L.; Haley, C.A. Assessment of the Economic Impact of Porcine Reproductive and Respiratory Syndrome Virus on United States Pork Producers. J. Swine Health Prod. 2013, 21, 72–84. [Google Scholar]
- Chae, C. Commercial Prrs Modified-live Virus Vaccines. Vaccines 2021, 9, 185. [Google Scholar] [CrossRef]
- Zeller, M.A.; Anderson, T.K.; Walia, R.W.; Vincent, A.L.; Gauger, P.C. ISU FLUture: A Veterinary Diagnostic Laboratory Web-Based Platform to Monitor the Temporal Genetic Patterns of Influenza A Virus in Swine. BMC Bioinform. 2018, 19, 397. [Google Scholar] [CrossRef]
- Holzer, B.; Martini, V.; Edmans, M.; Tchilian, E. T and B Cell Immune Responses to Influenza Viruses in Pigs. Front. Immunol. 2019, 10, 98. [Google Scholar] [CrossRef]
- Chrun, T.; Maze, E.A.; Vatzia, E.; Martini, V.; Paudyal, B.; Edmans, M.; McNee, A.; Manjegowda, T.; Salguero, F.J.; Wanasen, N.; et al. Simultaneous Infection with Porcine Reproductive and Respiratory Syndrome and Influenza Viruses Abrogates Clinical Protection Induced by Live Attenuated Porcine Reproductive and Respiratory Syndrome Vaccination. bioRxiv 2021. [Google Scholar] [CrossRef]
- Faure, E.; Kwong, K.; Nguyen, D. Pseudomonas Aeruginosa in Chronic Lung Infections: How to Adapt Within the Host? Front. Immunol. 2018, 9, 2416. [Google Scholar] [CrossRef] [Green Version]
- Correa-Fiz, F.; Gonçalves Dos Santos, J.M.; Illas, F.; Aragon, V. Antimicrobial Removal on Piglets Promotes Health and Higher Bacterial Diversity in the Nasal Microbiota. Sci. Rep. 2019, 9, 6545. [Google Scholar] [CrossRef]
- Correa-Fiz, F.; Fraile, L.; Aragon, V. Piglet Nasal Microbiota at Weaning May Influence the Development of Glässer’s Disease during the Rearing Period. BMC Genom. 2016, 17, 404. [Google Scholar] [CrossRef] [Green Version]
- Mou, K.T.; Allen, H.K.; Alt, D.P.; Trachsel, J.; Hau, S.J.; Coetzee, J.F.; Holman, D.B.; Kellner, S.; Loving, C.L.; Brockmeier, S.L. Shifts in the Nasal Microbiota of Swine in Response to Different Dosing Regimens of Oxytetracycline Administration. Vet. Microbiol. 2019, 237, 108386. [Google Scholar] [CrossRef] [PubMed]
- Strube, M.L.; Hansen, J.E.; Rasmussen, S.; Pedersen, K. A detailed investigation of the porcine skin and nose microbiome using universal and Staphylococcus specific primers. Sci. Rep. 2018, 8, 12751. [Google Scholar] [CrossRef] [PubMed]
- Pirolo, M.; Espinosa-Gongora, C.; Bogaert, D.; Guardabassi, L. The Porcine Respiratory Microbiome: Recent Insights and Future Challenges. Anim. Microbiome 2021, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Hemmink, J.D.; Morgan, S.B.; Aramouni, M.; Everett, H.; Salguero, F.J.; Canini, L.; Porter, E.; Chase-Topping, M.; Beck, K.; Loughlin, R.M.; et al. Distinct Immune Responses and Virus Shedding in Pigs Following Aerosol, Intra-Nasal and Contact Infection with Pandemic Swine Influenza A Virus, A(H1N1)09. Vet. Res. 2016, 47, 103. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the Immune System. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Crisci, E.; Fraile, L.; Montoya, M. Cellular Innate Immunity against PRRSV and Swine Influenza Viruses. Vet. Sci. 2019, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, N.J. Neisseria Models of Infection and Persistence in the Upper Respiratory Tract. Pathog. Dis. 2017, 75, ftx031. [Google Scholar] [CrossRef]
- Saade, G.; Ménard, D.; Hervet, C.; Renson, P.; Hue, E.; Zhu, J.; Dubreil, L.; Paillot, R.; Pronost, S.; Bourry, O.; et al. Porcine Reproductive and Respiratory Syndrome Virus Interferes with Swine Influenza A Virus Infection of Epithelial Cells. Vaccines 2020, 8, 508. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, J.; Cheng, L.; Xu, K.; Yang, Y.; Su, X. Deficiency of HIF-1α Enhances Influenza A Virus Replication by Promoting Autophagy in Alveolar Type II Epithelial Cells. Emerg. Microbes Infect. 2020, 9, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Bertho, N.; Meurens, F. The Pig as a Medical Model for Acquired Respiratory Diseases and Dysfunctions: An Immunological Perspective. Mol. Immunol. 2021, 135, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Borey, M.; Blanc, F.; Lemonnier, G.; Leplat, J.J.; Jardet, D.; Rossignol, M.N.; Ravon, L.; Billon, Y.; Bernard, M.; Estellé, J.; et al. Links between Fecal Microbiota and the Response to Vaccination against Influenza A Virus in Pigs. NPJ Vaccines 2021, 6, 92. [Google Scholar] [CrossRef]
- Ellis, R.J.; Bruce, K.D.; Jenkins, C.; Stothard, J.R.; Ajarova, L.; Mugisha, L.; Viney, M.E. Comparison of the Distal Gut Microbiota from People and Animals in Africa. PLoS ONE 2013, 8, e54783. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Qian, P.Y. Conservative Fragments in Bacterial 16S rRNA Genes and Primer Design for 16S Ribosomal DNA Amplicons in Metagenomic Studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S RRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Family | Spearman r Correlation |
|---|---|
| Enterobacteriaceae | −0.07 |
| Moraxellaceae | −0.23 |
| Neisseriaceae | 0.61 * |
| Rickettsiales, mitochondria | −0.17 |
| Prevotellaceae | 0.20 |
| Weeksellaceae | 0.38 |
| Staphylococcaceae | 0.41 |
| Aerococcaceae | 0.56 |
| Lactobacillaceae | 0.40 |
| Streptococcaceae | 0.27 |
| Lachnospiraceae | −0.42 |
| Ruminococcaceae | 0.27 |
| Veillonellaceae | −0.39 |
| Streptophyta, other | −0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrun, T.; Leng, J.; La Ragione, R.M.; Graham, S.P.; Tchilian, E. Changes in the Nasal Microbiota of Pigs Following Single or Co-Infection with Porcine Reproductive and Respiratory Syndrome and Swine Influenza A Viruses. Pathogens 2021, 10, 1225. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101225
Chrun T, Leng J, La Ragione RM, Graham SP, Tchilian E. Changes in the Nasal Microbiota of Pigs Following Single or Co-Infection with Porcine Reproductive and Respiratory Syndrome and Swine Influenza A Viruses. Pathogens. 2021; 10(10):1225. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101225
Chicago/Turabian StyleChrun, Tiphany, Joy Leng, Roberto M. La Ragione, Simon P. Graham, and Elma Tchilian. 2021. "Changes in the Nasal Microbiota of Pigs Following Single or Co-Infection with Porcine Reproductive and Respiratory Syndrome and Swine Influenza A Viruses" Pathogens 10, no. 10: 1225. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10101225