
Population Pharmacokinetics of Primaquine in the Korean Population

 , , and
, , and
Abstract
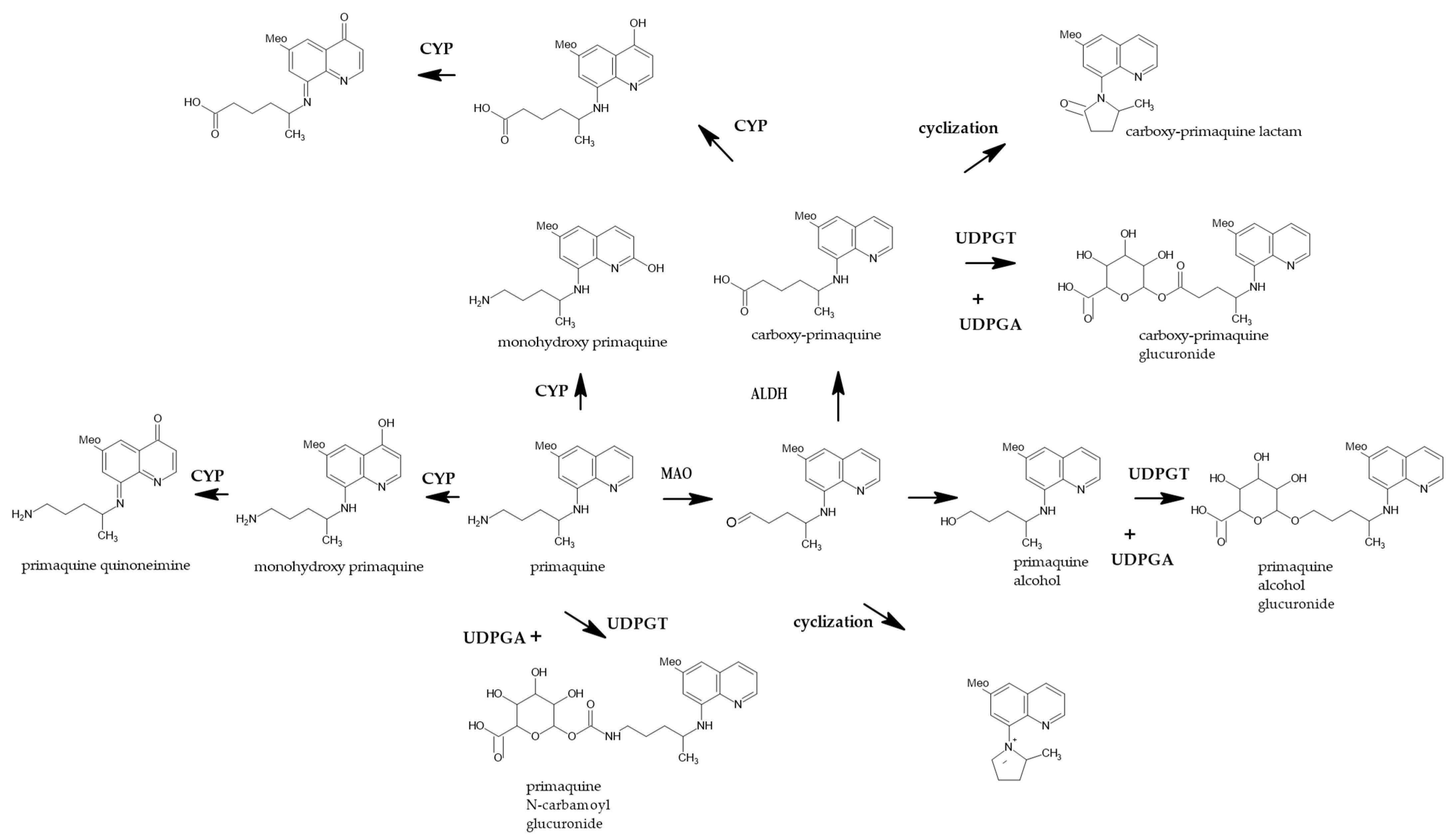
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. PK Data
2.3. Genetic Data
2.4. Bioanalysis
2.5. PK Model Development
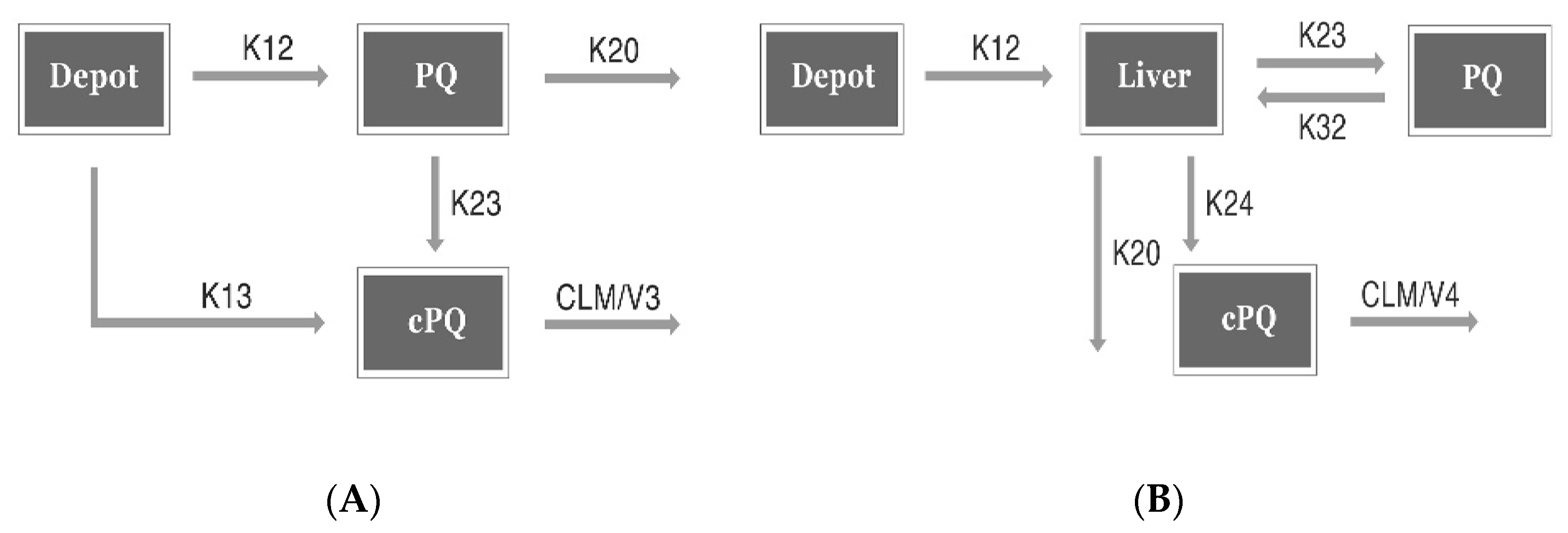
2.5.1. CPK Model
2.5.2. Minimal PBPK Model
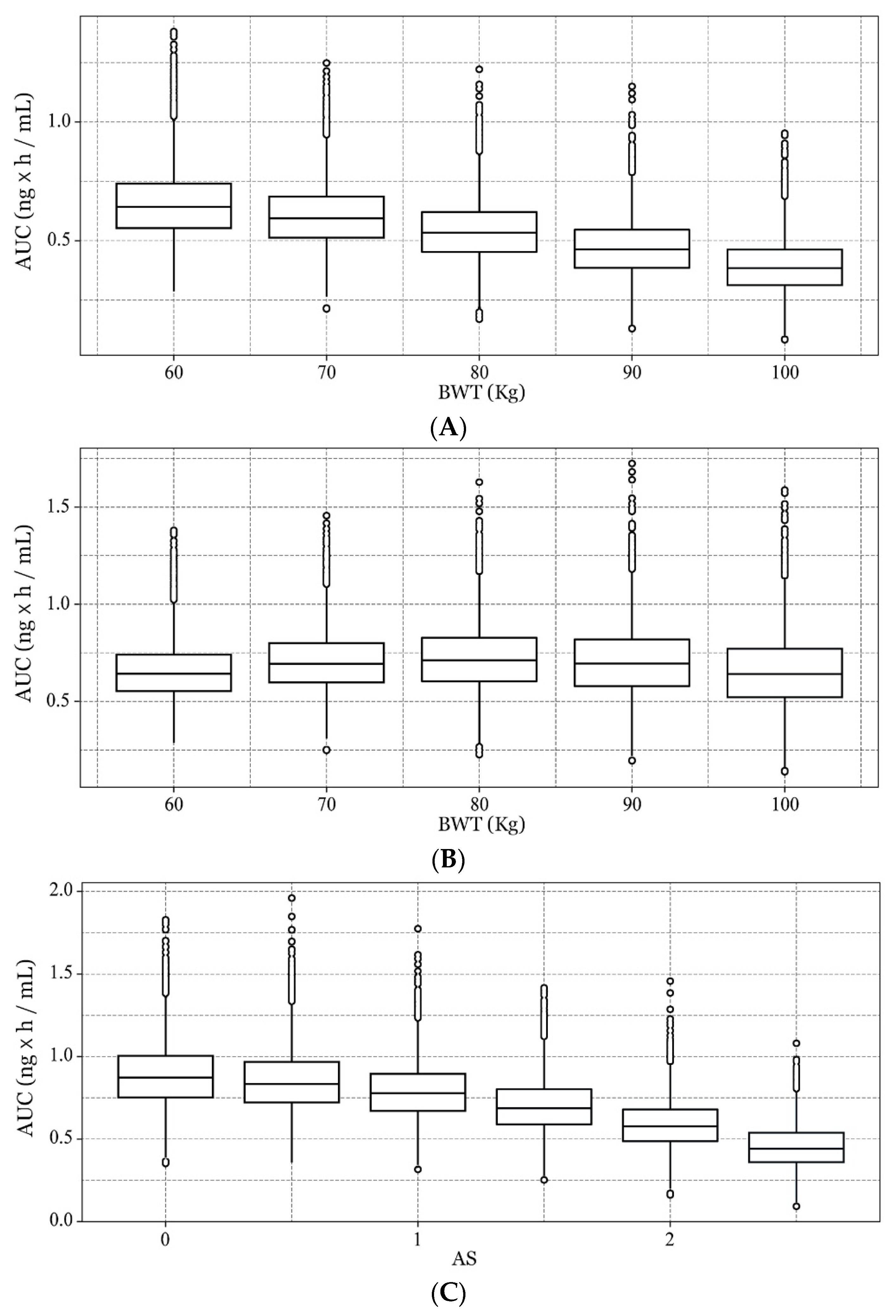
2.5.3. Covariate Analysis
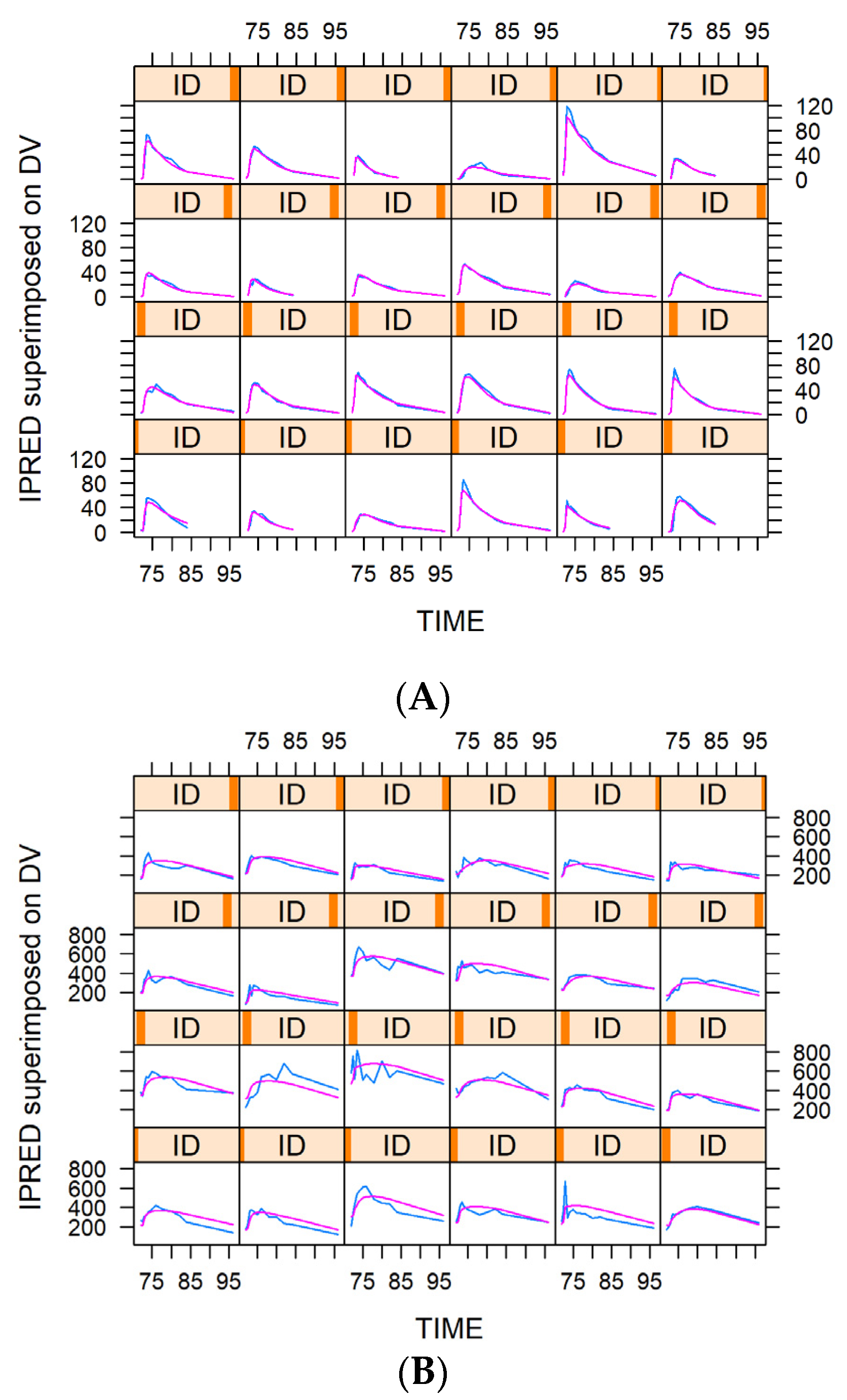
2.6. Model Evaluation and Simulation
2.7. Software
3. Results
3.1. Subject Characteristics
3.2. Model Development
3.2.1. Base Model Development
CPK Model (Conventional PK Model)
Minimal PBPK Model (Minimal Physiology Based PK Model)
3.2.2. Final Model Development
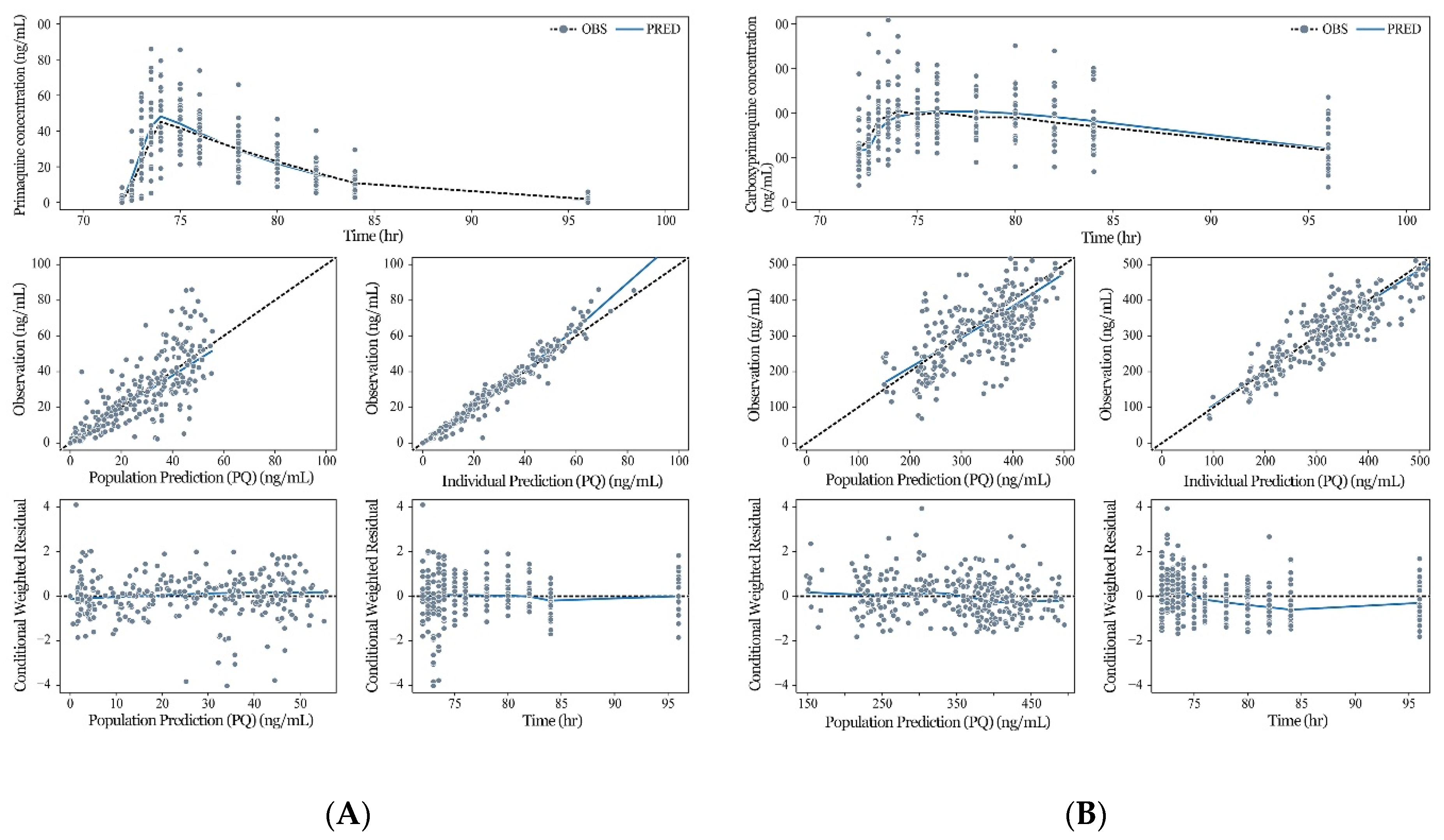
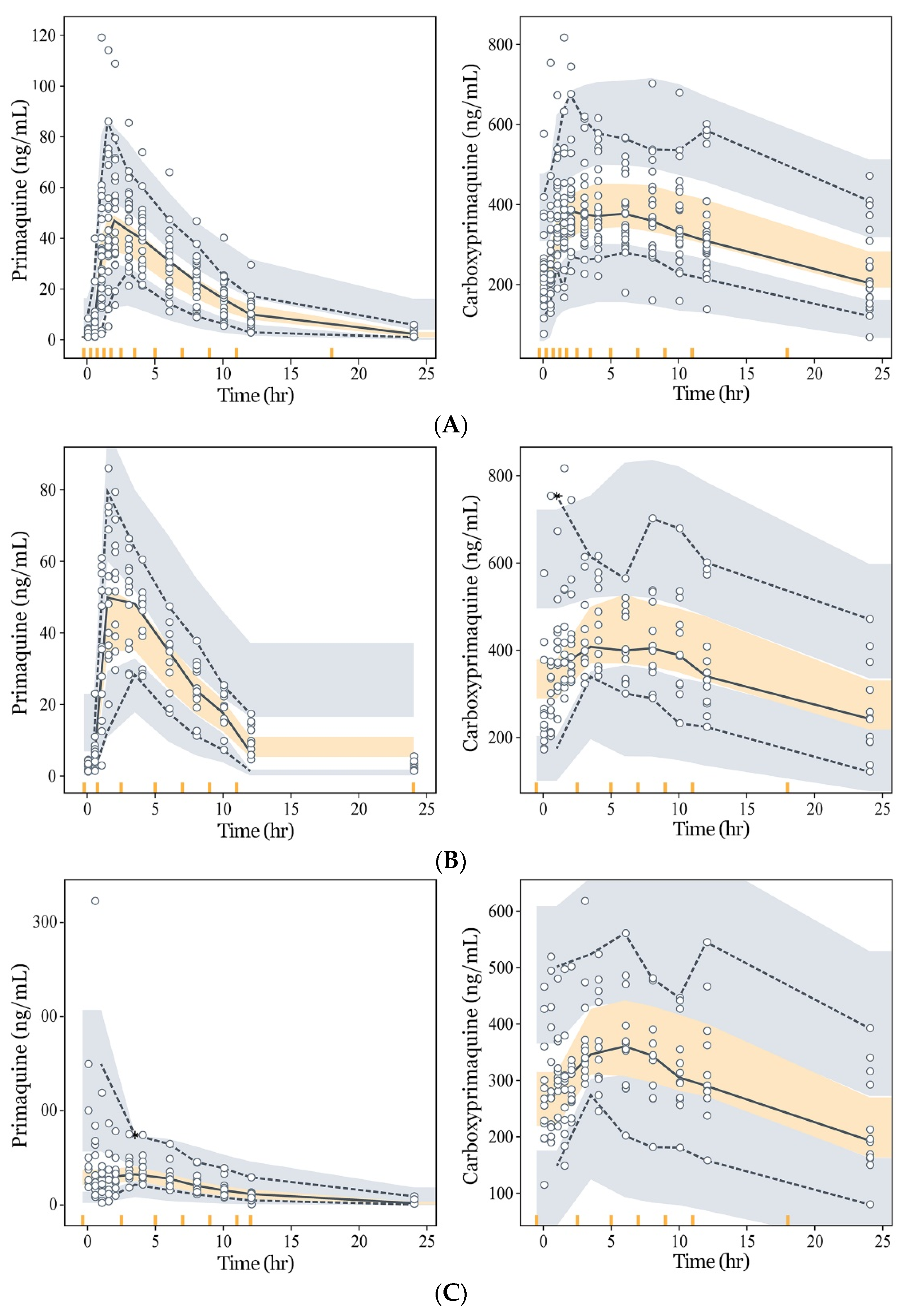
3.3. Model Evaluation and Simulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Bahk, Y.Y.; Lee, H.W.; Na, B.K.; Kim, J.; Jin, K.; Hong, Y.S.; Kim, T.S. Epidemiological characteristics of re-emerging vivax malaria in the republic of korea (1993–2017). Korean J. Parasitol. 2018, 56, 531–543. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Krotoski, W.A.; Krotoski, D.M.; Garnham, P.C.; Bray, R.S.; Killick-Kendrick, R.; Draper, C.C.; Targett, G.A.; Guy, M.W. Relapses in primate malaria: Discovery of two populations of exoerythrocytic stages. Preliminary note. Br. Med. J. 1980, 280, 153–154. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Guddattu, V.; Saravu, K. Therapeutic assessment of primaquine for radical cure of plasmodium vivax malaria at primary and tertiary care centres in southwestern india. Korean J. Parasitol. 2016, 54, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Graves, P.M.; Gelband, H.; Garner, P. Primaquine or other 8-aminoquinoline for reducing plasmodium falciparum transmission. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vale, N.; Moreira, R.; Gomes, P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009, 44, 937–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, J.K.; Hoffman, S.L. Primaquine therapy for malaria. Clin. Infect. Dis. 2004, 39, 1336–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.B.; Luz, F.; Deckers, F.A.; Tauil, P.L. Subdoses of primaquine in overweight patients and malaria vivax relapses: Report of two cases in the federal district, brazil. Rev. Soc. Bras. Med. Trop. 2010, 43, 749–750. [Google Scholar] [CrossRef] [Green Version]
- McFarland, A.P.; Sanchez, J.F.; Mercado, A.; Ventocilla, J.A.; Cavalcanti, S.; Gonzalez, S.; Lescano, A.G. Repeated plasmodium vivax malaria relapses in a peruvian sailor. Malar. J. 2015, 14, 478. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Guidelines for the Treatment of Malaria; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Bennett, J.W.; Pybus, B.S.; Yadava, A.; Tosh, D.; Sousa, J.C.; McCarthy, W.F.; Deye, G.; Melendez, V.; Ockenhouse, C.F. Primaquine failure and cytochrome p-450 2d6 in plasmodium vivax malaria. N. Engl. J. Med. 2013, 369, 1381–1382. [Google Scholar] [CrossRef]
- Leang, R.; Khu, N.H.; Mukaka, M.; Debackere, M.; Tripura, R.; Kheang, S.T.; Chy, S.; Kak, N.; Buchy, P.; Tarantola, A.; et al. An optimised age-based dosing regimen for single low-dose primaquine for blocking malaria transmission in cambodia. BMC Med. 2016, 14, 171. [Google Scholar]
- Fasinu, P.S.; Avula, B.; Tekwani, B.L.; Nanayakkara, N.D.; Wang, Y.-H.; Herath, H.B.; McChesney, J.D.; Reichard, G.A.; Marcsisin, S.R.; Elsohly, M.A.J.M.J. Differential kinetic profiles and metabolism of primaquine enantiomers by human hepatocytes. Malar. J. 2016, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. The Asia-Pacific Perspective: Redefining Obesity and Its Treatment; World Health Organization: Geneva, Switzerland, 2000. [Google Scholar]
- Seo, M.H.; Lee, W.-Y.; Kim, S.S.; Kang, J.-H.; Kang, J.-H.; Kim, K.K.; Kim, B.-Y.; Kim, Y.-H.; Kim, W.-J.; Kim, E.M.; et al. 2018 Korean society for the study of obesity guideline for the management of obesity in Korea. J. Obes. Metab. Syndr. 2019, 28, 40. [Google Scholar] [CrossRef] [PubMed]
- De Gregori, M.; Allegri, M.; De Gregori, S.; Garbin, G.; Tinelli, C.; Regazzi, M.; Govoni, S.; Ranzani, G.N. How and why to screen for cyp2d6 interindividual variability in patients under pharmacological treatments. Curr. Drug Metab. 2010, 11, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Polymorphism of human cytochrome p450 2d6 and its clinical significance: Part ii. Clin. Pharm. 2009, 48, 761–804. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Polymorphism of human cytochrome p450 2d6 and its clinical significance: Part i. Clin. Pharm. 2009, 48, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Simon, S.D.; Pearce, R.E.; Bradford, L.D.; Kennedy, M.J.; Leeder, J.S. The cyp2d6 activity score: Translating genotype information into a qualitative measure of phenotype. Clin. Pharm. 2008, 83, 234–242. [Google Scholar] [CrossRef]
- Goncalves, B.P.; Pett, H.; Tiono, A.B.; Murry, D.; Sirima, S.B.; Niemi, M.; Bousema, T.; Drakeley, C.; Ter Heine, R. Age, weight, and cyp2d6 genotype are major determinants of primaquine pharmacokinetics in African children. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- PubChem. Available online: https://pubchem.Ncbi.Nlm.Nih.Gov/compound/primaquine (accessed on 20 March 2020).
- Pubchem. Available online: https://pubchem.Ncbi.Nlm.Nih.Gov/compound/carboxyprimaquine (accessed on 20 March 2020).
- Richardson, P.D.; Withrington, P.G. Liver blood flow. I. Intrinsic and nervous control of liver blood flow. Gastroenterology 1981, 81, 159–173. [Google Scholar] [CrossRef]
- Yu, H.C.; You, H.; Lee, H.; Jin, Z.W.; Moon, J.I.; Cho, B.H. Estimation of standard liver volume for liver transplantation in the Korean population. Liver Transpl. 2004, 10, 779–783. [Google Scholar] [CrossRef]
- Schwartz, E.; Regev-Yochay, G.; Kurnik, D. Short report: A consideration of primaquine dose adjustment for radical cure of plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 2000, 62, 393–395. [Google Scholar] [CrossRef] [Green Version]
- Vieira, M.; Matos Lopes, T.R.; Mello, A.; de Sena, L.W.P.; Commons, R.J.; Vieira, J.L.F. Doses of primaquine administered to children with plasmodium vivax according to an age-based dose regimen. Pathog. Glob. Health 2020, 114, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; O‘Neil, M.; Xie, L.; Caridha, D.; Zeng, Q.; Zhang, J.; Pybus, B.; Hickman, M.; Melendez, V. Assessment of the prophylactic activity and pharmacokinetic profile of oral tafenoquine compared to primaquine for inhibition of liver stage malaria infections. Malar. J. 2014, 13, 141. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, K.A.; Evans, D.A.; Gilles, H.M.; Greaves, J.; Bunnag, D.; Harinasuta, T. Studies on the pharmacokinetics of primaquine. Bull. World Health Organ. 1981, 59, 407–412. [Google Scholar] [PubMed]
- Zuluaga-Idarraga, L.; Blair, S.; Akinyi Okoth, S.; Udhayakumar, V.; Marcet, P.L.; Escalante, A.A.; Alexander, N.; Rojas, C. Prospective study of plasmodium vivax malaria recurrence after radical treatment with a chloroquine-primaquine standard regimen in turbo, colombia. Antimicrob. Agents Chemother. 2016, 60, 4610–4619. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.T. Special populations: Profiling the effect of obesity on drug disposition and pharmacodynamics. In Drug Discovery and Evaluation: Methods in Clinical Pharmacology, 2nd ed.; Hock, F.J., Gralinski, M.R., Eds.; Springer International Publishing, Cham: New York City, NY, USA, 2020; pp. 723–747. [Google Scholar] [CrossRef]
- Tekwani, B.L.; Avula, B.; Sahu, R.; Chaurasiya, N.D.; Khan, S.I.; Jain, S.; Fasinu, P.S.; Herath, H.B.; Stanford, D.; Nanayakkara, N.D.J.D.M.; et al. Enantioselective pharmacokinetics of primaquine in healthy human volunteers. Drug Metab. Dispos. 2015, 43, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-R.; Kuh, H.-J.; Kim, M.-Y.; Kim, Y.-S.; Chung, W.-C.; Kim, S.-l.; Kang, M.-W.J. Pharmacokinetics of primaquine and carboxyprimaquine in Korean patients with vivax malaria. Arch. Pharmacal Res. 2004, 27, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Saraph, Y.; Revankar, S.; Doshi, K.; Bharucha, E.; Desai, N.; Vaidya, A.; Subrahmanyam, D.; Gupta, K.; Satoskar, R. Pharmacokinetics of primaquine in patients with P. Vivax Malaria. Eur. J. Clin. Pharmacol. 1986, 31, 205–210. [Google Scholar] [CrossRef]
- Constantino, L.; Paixao, P.; Moreira, R.; Portela, M.J.; Do Rosario, V.E.; Iley, J. Metabolism of primaquine by liver homogenate fractions. Evidence for monoamine oxidase and cytochrome p450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxicol Pathol. 1999, 51, 299–303. [Google Scholar] [CrossRef]
- Jin, X.; Pybus, B.S.; Marcsisin, R.; Logan, T.; Luong, T.L.; Sousa, J.; Matlock, N.; Collazo, V.; Asher, C.; Carroll, D.; et al. An lc-ms based study of the metabolic profile of primaquine, an 8-aminoquinoline antiparasitic drug, with an in vitro primary human hepatocyte culture model. Eur. J. Drug Metab. Pharm. 2014, 39, 139–146. [Google Scholar] [CrossRef]
- Mihaly, G.W.; Ward, S.A.; Edwards, G.; Orme, M.L.; Breckenridge, A.M. Pharmacokinetics of primaquine in man: Identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharm. 1984, 17, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Pybus, B.S.; Sousa, J.C.; Jin, X.; Ferguson, J.A.; Christian, R.E.; Barnhart, R.; Vuong, C.; Sciotti, R.J.; Reichard, G.A.; Kozar, M.P.; et al. Cyp450 phenotyping and accurate mass identification of metabolites of the 8-aminoquinoline, anti-malarial drug primaquine. Malar. J. 2012, 11, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcsisin, S.R.; Reichard, G.; Pybus, B.S. Primaquine pharmacology in the context of cyp 2d6 pharmacogenomics: Current state of the art. Pharmacol. Ther. 2016, 161, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, J.K.; Louisa, M.; Noviyanti, R.; Ekawati, L.; Elyazar, I.; Subekti, D.; Chand, K.; Gayatri, A.; Instiaty, I.; Soebianto, S.; et al. Association of impaired cytochrome p450 2d6 activity genotype and phenotype with therapeutic efficacy of primaquine treatment for latent plasmodium vivax malaria. JAMA Netw. Open 2018, 1, e181449. [Google Scholar] [CrossRef] [Green Version]
- Pukrittayakamee, S.; Tarning, J.; Jittamala, P.; Charunwatthana, P.; Lawpoolsri, S.; Lee, S.J.; Hanpithakpong, W.; Hanboonkunupakarn, B.; Day, N.P.J.; Ashley, E.A.; et al. Pharmacokinetic interactions between primaquine and chloroquine. Antimicrob. Agents Chemother. 2014, 58, 3354–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, B.; Duffull, S.B. What is the best size descriptor to use for pharmacokinetic studies in the obese? Br. J. Clin. Pharm. 2004, 58, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajgor, D.; Gogtay, N.; Kadam, V.; Kocharekar, M.; Parulekar, M.; Dalvi, S.; Vaidya, A.; Kshirsagar, N. Antirelapse efficacy of various primaquine regimens for plasmodium vivax. Malar. Res. Treat. 2014, 2014, 347018. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographics | Normal Group (n = 12) | Obese Group (n = 12) | p Value * |
|---|---|---|---|
| Male, n (%) | 12 (100.0) | 12 (100.0) | – |
| Age (years) | 29.1 ± 8.4 | 26.4 ± 7.4 | 0.417 |
| Height (cm) | 175.4 ± 4.6 | 173.1 ± 5.1 | 0.251 |
| Body weight (kg) | 67.6 ± 7.6 | 83.3 ± 6.7 | <0.001 |
| Body mass index (kg/m2) | 21.9 ± 2 | 27.8 ± 1.8 | <0.001 |
| CYP2D6, n (%) | 1.000 | ||
| AS (= 0.5) | 0 (0.0) | 1 (4.2) | |
| AS (= 1.0) | 3 (12.5) | 3 (12.5) | |
| AS (= 1.5) | 6 (25.0) | 6 (25.0) | |
| AS (= 2.0) | 3 (12.5) | 2 (8.3) |
| Parameter | Description | Estimate (Unit) | RSE (%) |
|---|---|---|---|
| Structural Parameters | |||
| V3 | Volume of distribution for PQ | 142.2 (L) | 3.9 |
| V4 | Volume of distribution for CPQ | 30.1 (L) | 8.4 |
| CLMAO | CL of PQ via MAO pathway | 19.1 (L/h) | 7.5 |
| CLCYP | CL of PQ via CYP2D6 pathway | 7.5 (L/h) | 27.6 |
| CLM | CL of CPQ | 1.3 (L/h) | 8.7 |
| KA | Absorption rate constant | 1.7 (h−1) | 12.7 |
| ALAG1 | Absorption lag time | 0.45 (h) | 1.7 |
| COVAS | Covariate effect of AS on CLCYP | 1.254 | 11 |
| COVBW | Covariate effect of BW on CLCYP | 0.041 | 9.1 |
| Inter-Individual Variabilities | |||
| ω2V3 | BSV on V3 | 16.6 (CV%) | 13.4 |
| ω2CLMAO | BSV on CLMAO | 22.7 (CV%) | 23.9 |
| ω2CLCYP | BSV on CLCYP | 55.2 (CV%) | 30 |
| ω2KA | BSV on KA | 82.9 (CV%) | 16.1 |
| ω2CLM | BSV on CLM | 20 (CV%) | 21.4 |
| ω2ALAG | BSV on ALAG | 6.8 (CV%) | 30.6 |
| Residual Error | |||
| σ2pro1 | Proportional error of PQ | 17.9 (CV%) | 13.9 |
| σ2add | Additive error of CPQ | 22.3 (SD) | 31.7 |
| σ2pro2 | Proportional error of CPQ | 15.7 (CV%) | 14.2 |
| Parameter | Normal Weight Group (n = 12) | Obese Group (n = 12) | p Value |
|---|---|---|---|
| AUCPQ (ng·h/mL) | 610.2 ± 149.5 | 538.6 ± 160.2 | <0.01 |
| AUCPQ (ng·h/mL/Kg) | 9.2 ± 2.6 ** | 6.5 ± 2.1 ** | |
| AUCCPQ (ng·h/mL) | 8917 ± 2323.4 | 7903.9 ± 2521.9 | <0.01 |
| AUCCPQ (ng·h/mL/Kg) | 134 ± 40.2 ** | 95.9 ± 32.8 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.-Y.; Chae, D.-W.; Kim, C.-O.; Lee, S.-E.; Kwak, Y.-G.; Yeom, J.-S.; Park, K.-S. Population Pharmacokinetics of Primaquine in the Korean Population. Pharmaceutics 2021, 13, 652. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13050652
Lee W-Y, Chae D-W, Kim C-O, Lee S-E, Kwak Y-G, Yeom J-S, Park K-S. Population Pharmacokinetics of Primaquine in the Korean Population. Pharmaceutics. 2021; 13(5):652. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13050652
Chicago/Turabian StyleLee, Woo-Yul, Dong-Woo Chae, Choon-Ok Kim, Sang-Eun Lee, Yee-Gyung Kwak, Joon-Sup Yeom, and Kyung-Soo Park. 2021. "Population Pharmacokinetics of Primaquine in the Korean Population" Pharmaceutics 13, no. 5: 652. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13050652