
Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview


1
Department of Pharmaceutical Sciences, Texas Tech University Health Sciences Center, Amarillo, TX 79106, USA
2
Department of Foundational Medical Studies, Oakland University William Beaumont School of Medicine, Rochester, MI 48309, USA
*
Author to whom correspondence should be addressed.
Pharmaceutics 2021, 13(11), 1779; https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111779
Submission received: 23 September 2021
/
Revised: 15 October 2021
/
Accepted: 20 October 2021
/
Published: 26 October 2021
(This article belongs to the Special Issue Biological Barriers in Health and Disease)
Abstract
:The blood-brain barrier (BBB) is a fundamental component of the central nervous system (CNS). Its functional and structural integrity is vital to maintain the homeostasis of the brain microenvironment by controlling the passage of substances and regulating the trafficking of immune cells between the blood and the brain. The BBB is primarily composed of highly specialized microvascular endothelial cells. These cells’ special features and physiological properties are acquired and maintained through the concerted effort of hemodynamic and cellular cues from the surrounding environment. This complex multicellular system, comprising endothelial cells, astrocytes, pericytes, and neurons, is known as the neurovascular unit (NVU). The BBB strictly controls the transport of nutrients and metabolites into brain parenchyma through a tightly regulated transport system while limiting the access of potentially harmful substances via efflux transcytosis and metabolic mechanisms. Not surprisingly, a disruption of the BBB has been associated with the onset and/or progression of major neurological disorders. Although the association between disease and BBB disruption is clear, its nature is not always evident, specifically with regard to whether an impaired BBB function results from the pathological condition or whether the BBB damage is the primary pathogenic factor prodromal to the onset of the disease. In either case, repairing the barrier could be a viable option for treating and/or reducing the effects of CNS disorders. In this review, we describe the fundamental structure and function of the BBB in both healthy and altered/diseased conditions. Additionally, we provide an overview of the potential therapeutic targets that could be leveraged to restore the integrity of the BBB concomitant to the treatment of these brain disorders.
1. Introduction
Biological barriers perform a significant role in maintaining the integrity and function of many vertebrate organs. Intercellular protein complexes of the plasma membrane form paracellular diffusion barriers that separate internal and external fluid compartments, which is a crucial process for the development and function of all organs [1]. Different biological barriers are present in different body organs such as the skin, intestine, kidney, reproductive system, lung, liver, mouth mucosa, and central nervous system (CNS). The CNS is protected from the external environment by three biological barriers at three interfaces. These are the blood-brain barrier (BBB), the blood–CSF barrier (BCB), and the arachnoid barrier [2]. The BBB is formed by the endothelial cells lining cerebral microvessels and separates the blood from brain interstitial fluid. The choroid plexus epithelium is positioned between the blood and ventricular cerebrospinal fluid (CSF) and forms the blood-CSF barrier. The arachnoid barriers are formed by epithelium positioned between the blood and subarachnoid CSF. These three barrier layers participate in limiting and regulating molecular exchange at the interfaces between the blood and the neural tissue or its fluid spaces. Among these CNS barriers, the BBB exerts the tightest control over the surrounding brain microenvironment. More specifically, the BBB provides barrier functions at three different levels.
The first is a physical barrier that blocks the paracellular pathway of polar substances (including ions) between adjacent endothelial cells. Tight junctional proteins form this barrier through homotypic binding with their homologous counterpart on the adjacent endothelial cells. Different efflux transporters provide the second barrier, with a wide range of affinity for lipophilic substances. These include a P-glycoprotein (P-gp), breast cancer resistant protein (BCRP), and multidrug resistance-associated proteins (MRPs) [2]. The third is a multi-enzymatic barrier that provides the BBB with a certain degree of drug metabolism capabilities [3]. The brain capillary contains a wide range of neurotransmitter-metabolizing enzymes, including cholinesterases, GABA transaminase, aminopeptidases, and endopeptidases, as well as several drug- and toxin-metabolizing enzymes [4]. Thus, the enzymatic blood-brain barrier protects the brain from circulating neurotransmitters as well as from many toxins.
Considering the pivotal role the BBB plays in maintaining brain homeostasis and protecting the CNS, it is understandable that dysfunctions of the BBB can be prodromal to the onset of neurological and spine disorders and/or the worsening of brain disorders. Therefore, it is not surprising that an impairment of the BBB has been associated with severe and detrimental outcomes in the context of many neurological disorders (see Figure 1) [2,5,6]. Since impairment of the BBB is closely linked to many CNS disorders, therapeutic targets that aim to restore the BBB’s viability could be a feasible method to reduce the burden of diseases and improve the outcome for a patient. Hence, this review highlights the pathological conditions associated with dysfunction of the BBB and the therapeutic targets currently exploited to promote BBB restoration.
2. The Function of the BBB
The overall components of the BBB participate in maintaining a stable microenvironment which is important for sustaining complex neural functions and protecting the CNS. The BBB contains different ion channels and transporters, which help maintain the ionic balance for synaptic signaling activity. Potassium (K+), magnesium (Mg2+) and calcium (Ca2+) ions are regulated at the BBB and BCSFB [2,5,6]. For instance, the concentration of potassium (K+) in mammalian plasma is around 4.5 mM. In contrast, in CSF and ISF, it is approximately 2.5–2.9 mM, and it is not affected by external factors such as exercise, meal intake, pathological state, or experimental condition [7,8].
Moreover, the BBB plays a pivotal role in sustaining brain nutrition through a plethora of mechanisms. The BBB has a low passive permeability for water-soluble nutrients and metabolites essential for the nervous tissue. On the other hand, specific transporters present in the BBB allow for the transportation of other essential substances, for instance, glucose and amino acids that cannot pass through the BBB. Expression of these selective and region-specific (luminal and abluminal surfaces of the ECs) transporters ensures BBB endothelium polarization [3,9]. Further, the BBB plays a significant role in regulating the neurotransmitter in CNS by keeping the central and peripheral neurotransmitter pool separated. Uncontrolled release of neurotransmitters such as glutamate into the brain CSF during hypoxia in ischemic stroke conditions may cause severe and permanent neurotoxic damage [3,10].
Additionally, the movement of macromolecules between blood and brain parenchyma is controlled and maintained by the BBB. CSF contains a lower concentration of proteins than in plasma as the BBB prevents the entry of macromolecules into the CNS. Different plasma proteins, such as albumin, prothrombin, and plasminogen, may cause cellular apoptosis, damaging nerve tissues [11,12,13]. Factor Xa converts prothrombin to thrombin, and the tissue plasminogen activator converts plasminogen to plasmin. Both factor Xa and the tissue plasminogen activator are present in the brain. Leakage of these large macromolecular proteins into the brain through a disrupted BBB may result in severe pathological consequences, including seizures, glial cell activation and division, scarring, and cell death [13]. The BBB also serves as a shield to protect the CNS from various toxins circulating in the blood. These toxic molecules can be endogenous proteins, metabolites, or xenobiotics obtained through diet or environmental pollutants. If they gain entry into the brain, they may compromise neuronal activity and/or promote cell death [2].
3. Structure of the Blood-Brain Barrier: An Overview
The development of the BBB begins during the fetal stage, and it is well constructed by the point of birth, especially for macromolecules and proteins [14,15,16,17,18,19,20,21]. At the cellular level, the BBB consists of microvascular endothelial cells (EC) lining the luminal walls of brain microvessels alongside closely associated pericytes embedded within the basal membrane and surrounded by astrocytic end-feet processes (see Figure 2) that support EC’s phenotypic differentiation and the maintenance of BBB features [2,3]. The microcapillary endothelium is characterized by the expression of tight junctions (TJs), a lack of fenestrations, negligible pinocytotic trafficking, and distinct asymmetrical distribution patterns of the transmembrane transporters, which provides the BBB endothelium a cellular polarization. The presence of TJs such as occludin (OCLN), claudin-5 (CLN-5), and junctional adhesion molecules (JAMs) are important for the BBB. These interendothelial junctions form a diffusion barrier that selectively prevents most blood-borne and xenobiotic hydrophilic substances from entering the brain through paracellular routes, protecting it from any undesired systemic and external influences [2,22]. Occludin and claudins are associated with cytoplasmic scaffolding and regulatory proteins called zona occludens (ZO-1, ZO-2, ZO-3) and cingulin, which anchor the TJs to the actin cytoskeleton, provide signaling functions, and guide the TJs in membrane distribution. In addition to TJs, the interendothelial space also features junctional complexes, including the adherens junction (AJs) and proteins such as cadherins. VE-cadherin (also known as Cadherin-5 or CD144) plays a particularly important role in the maintenance of cell-cell adhesion, contact inhibition, cytoskeleton remodeling, intracellular signaling, BBB permeability (via the modulation of CLN-5 expression [23]), and angiogenesis) [24]. Therefore, the loss of the AJ can lead to a loss of BBB integrity and its increased permeability [23,25]. However, the function and properties of the barrier do not entirely depend on the presence and expression of claudins and/or OCLN but rather on the organization, distribution, and interactions of these TJs with their homotypic counterpart on the adjacent cells [26].
Considering the intrinsic properties of the TJs physical barrier, the transport of water-soluble nutrients (such as D-glucose, monocarboxylic acids, and essential amino acids) from peripheral circulation into the brain parenchyma depends upon specific carrier-mediated transport systems with which the BBB endothelium is enriched, as summarized below. Although BBB properties are a characteristic feature of the brain microvascular endothelium, they are not intrinsic properties of the BBB endothelium. Rather, they are primarily acquired through an endothelial interaction with the surrounding environment during a process known as barriergenesis. Cellular cues derived from mural cells, immune cells, glial cells, and neural cells, all of which are part of the neurovascular unit (NVU) [27] during the various phases of brain development, are essential for the differentiation of the vascular endothelium into a BBB phenotype.
3.1. Endothelial Cells (ECs)
The brain microvascular endothelium is distinct from other tissue ECs, allowing them to tightly control the passage of ions, molecules, and cells between the blood and the brain. ECs of the CNS microcapillaries are joined by tight junctions (TJs), that restrict the paracellular transport of polar solutes [28,29,30]. Compared with the peripheral vasculature, the BBB endothelium exhibits an extremely low transcytosis and vesicle-mediated transcellular passage of solutes [31]. In addition, these microvascular endothelial cells present a regional polarization of transport mechanisms (both qualitative and quantitative) between the luminal and the basolateral side of the cell membrane so as to better control the influx and efflux of substances to and from the brain [32,33]. Transcytosis is an apical-to-basolateral vesicular-dependent intracellular transport mechanism and one of the key features of BBB. Transcytosis in brain endothelial cells is partially responsible for transporting several large molecules, including fatty acids and transferrin, across the BBB [34,35]. Transcytosis activity is higher during the early development period in the brain endothelial cells; however, it becomes dormant during the course of BBB maturation. Therefore, the upregulation of transcytosis (increased intracellular vesicles in the brain endothelial cells) is considered an early and precise indicator of BBB disruption [36,37]. Although it was previously assumed that changes in tight junction proteins primarily facilitated BBB permeability, recent evidence obtained from mice and zebrafish suggests that transcytosis is equally important in the regulation of BBB integrity [34].
Efflux transporters are primarily localized to the luminal surface of the BBB endothelial cells. They include the P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance associate proteins (MRPs) such as MRP1, MRP3, MRP4, and MRP6. Through the use of ATP hydrolysis, these transporters manage to drive their potentially harmful amphipathic and hydrophobic substrates across the ECs membrane back into the bloodstream against a concentration gradient, thus preventing them from entering into the brain [38,39]. These transporters function in parallel with various enzymes such as cytochrome P450s, monoaminoxidase (MAO), cholinesterases, catechol O-methyl transferase (COMT), endopeptidases, aminopeptidases, etc., that further increase the ability of BBB to remove/neutralize potentially harmful substances [3,40,41,42,43]. In contrast, other specialized transporters (that are mostly ATP independent), belonging to the solute carrier (SLC) superfamily, mediate the movement of small anionic and cationic molecules as well as nucleosides and peptides across the BBB, but also remove waste products from the CNS into the blood [44]. A brief overview of these transporters is presented here, although the reader is referred elsewhere for a more detailed and comprehensive description [45].
Moreover, the BBB endothelium has a higher amount of mitochondria compared to other vascular ECs. This is crucial to produce the necessary for ATP to support the metabolic work capability of the BBB and provide the ion gradient required for some of the transport functions [46]. Additionally, BBB ECs express very low amounts of leukocyte adhesion molecules (LAMs including E- and P-selectins) required for leukocyte entrance into the CNS across the endothelium [47,48], thus limiting the movement of immune cells into the brain [49,50,51], which is a de facto an immune-privileged organ. However, an elevated expression of LAMs was observed in the setting of neuroinflammatory diseases [47,49,51,52]. For instance, the infiltration of T and B lymphocytes, neutrophils, and macrophages was observed at multiple sclerosis (MS) active injury sites. Moreover, the infiltration of neutrophils, macrophages, and lymphocytes was reported in stroke [27,53].
3.2. The Basement Membrane
The endothelial vascular tube is surrounded by the following two types of basement membranes (BM): the inner vascular basement membrane and the outer parenchymal basement membrane. The inner vascular BM is composed of an extracellular matrix that is secreted by endothelial cells and pericytes. On the contrary, the outer parenchymal BM is secreted by astrocytic processes. These basement membranes are mainly comprised of laminin, agrin, type IV collagens, nidogen, heparin sulfate proteoglycans, and other glycoproteins. The vascular and parenchymal BM possess different isoforms of laminin. The vascular BM is composed of laminin α4 and laminin α5, whereas the parenchymal BM is comprised of laminin α1 and laminin α2 [54,55]. These basement membranes perform a pivotal role in many signaling processes and act as an additional barrier prior to access to the neural tissue. It is well acknowledged that matrix metalloproteinase enzymes (MMPs) disrupt the basement membranes, resulting in BBB dysfunction at the onset of different neurological disorders [27].
3.3. Astrocytes
Astrocytes are the most abundant cells in CNS and are responsible for the operation of different physiological and biological functions, including, but not limited to, neural parenchyma compartmentalization, pH regulation, ionic homeostasis, neurotransmitter uptake, and processing and signal mediation from neurons to vasculatures [56]. Interestingly, the glial/neuron ratio increases dramatically with brain complexity and volume (3). At the level of arterioles and venules, astrocytes contribute to the cerebral blood flow regulation through the release of vasoactive substances, thus modulating the vascular tone. At the brain microvascular level, astrocytes promote the differentiation of cerebrovascular endothelial cells to a BBB phenotype and contribute to the modulation of mature BBB functions and activity such as the induction and maintenance of tight junctions [2,57,58]. In vitro studies have also revealed that markers related to the development of a functional BBB, such as transferrin receptor, P-glycoprotein, gamma-GTP, etc., are typically upregulated in endothelial cells when co-cultured with astrocytes [58,59].
Recently, studies have demonstrated the crucial role of astrocytic endfeet in brain metabolism [9]. The communication between the astrocytes and the underlying microcapillaries occurs via the astrocytic endfeet, which ensheath the vascular tubes. These endfeets contain various proteins, including dystroglycan, dystrophin, aquaporin 4 (Aqp4), and the dystroglycan–dystrophin complex, which connects the endfeet cytoskeleton to the basement membrane through its binding with agrin [60,61]. Aqp4 is coordinated by this connection, which assists in maintaining cerebral water and ion homeostasis [27,62] as well as neurotransmitter regulation [63]. In contrast, activated astrocytes can secrete pro-inflammatory cytokines acting as inflammatory modulators and neurotoxins, causing neuronal damage [64,65].
In vivo studies have shown that the BBB is already experiencing development before the astrocytes envelop the brain capillaries; it has therefore been concluded that while astrocytes may not perform a significant role in the initial setting of BBB development, they enact a crucial role in the modulation and maintenance of BBB integrity post-formation [27,66].
3.4. Mural Cells and Pericytes
The vascular smooth muscle cells surrounding the large vessels and pericytes are known as mural cells. The pericytes are essential components of the brain capillary located on the abluminal surface of the microvascular endothelial tube and are embedded in the vascular BM [67]. They share a BM with endothelial cells, displaying a direct synaptic-like peg-socket focal contact with endothelium through N-cadherin and connexins [68]. CNS vasculature contains the greatest number of pericytes compared to other tissues with an approximate endothelial and its pericyte ratio is between 1:1 and 3:1 while the ratio is 100:1 for the muscle [69].
Pericytes are associated with different functions, including but not limited to maintaining BBB integrity, angiogenesis, microvascular stability, the regulation of the capillary diameter and cerebral blood flow, the deposition of the extracellular matrix, the regulation of immune cell filtration, and the removal of toxic metabolites [68,70,71,72,73]. Additionally, pericytes are crucial for regulating the formation of BBB during its development (Barriergenesis) and maintaining its integrity and function during adult and senior stages [66,74]. Moreover, other studies have demonstrated that pericytes perform an important role in cerebral autoregulation by expressing receptors for vascular mediators such as, angiotensin I [75], catecholamine [76], vasopressin vasoactive intestinal peptides [77], endothelin-1 [78], and vasopressin [79]. Different molecular identifiers, including PDGFR-β, NG2, Anpep (CD13), desmin, Rgs5, Abcc9, Kcnj8, Dlk, and Zic1 Immune Cells have been used to distinguish pericytes [68,80,81,82], although none of these are perfect identifiers of this cell type. It is challenging to determine pericytes’ precise involvement and functional contribution due to the lack of specific classification parameters (to define the exact type of pericyte) and corresponding specific markers [27].
3.5. Immune Cells
The blood vessels in the CNS interact with different immune cells, both in the CNS and in the blood. Perivascular macrophages and microglial cells are the principal immune cells within the CNS. Macrophages are monocyte lineage cells derived from blood-borne progenitors and reside on the vascular tube’s abluminal side [83,84]. These cells can phagocyte cellular debris and act as the first line of innate immunity [27]. Multiple studies have demonstrated that these cells can cross the BBB, and 80% of them are replaced within three months [85,86,87].
Microglial cells reside in CNS parenchymal immune cells that are derived from progenitors in the yolk sac and enter the brain during embryonic development [88]. These cells regulate neuronal development, the innate immune response, and wound healing and act as antigen-presenting cells in adaptive immunity [89,90]. Additionally, the interaction of blood-borne immune cells (neutrophils, T cells, and macrophages) with CNS vessels is also assumed to be integral for the maintenance of BBB integrity as they are capable of increasing vascular permeability by releasing reactive oxygen species once they are activated in response to an injury, disease condition or infection [91,92]. Therefore, it is crucial to identify the underlying mechanism behind the activation of both immune cells and the BBB and the interaction between the two to better understand the mechanisms that cause BBB disruption across different neurological diseases [27].
4. Adrenergic System and BBB
Several studies performed during the 1990s demonstrated the influence of the adrenergic system on BBB permeability. An increased BBB permeability was observed due to α-adrenoceptor activation either by the intracerebroventricular administration of agonists [93] or through the electrical stimulation of the locus coeruleus [94]. Moreover, the permeability of the BBB was facilitated by the blockade of the β-adrenoceptor. As a result of the α-adrenoceptor blockade and β-adrenoceptor stimulation, decreased BBB permeability became evident in various studies [95,96]. In all of these studies, alterations in the BBB permeability were accompanied by changes in pinocytotic activity in brain microvessel endothelial cells; however, the morphology of TJs remained unchanged [97].
5. BBB Dysfunction in CNS Disorders
The functional alterations of structural and cellular components in the BBB are responsible for BBB disruptions. These alterations may include changes that occur in tight junction expression, their distribution, and the local microenvironment that could be conducive to the opening of TJs, transport systems, enzymes, and the disruption of the basement membrane, which may ultimately lead to serum components and immune cell infiltration into the brain parenchyma, disrupt the CNS homeostasis and damage the surrounding brain tissues. Several studies have demonstrated that the disruption of the BBB is related to the onset and progression of various neurological and cerebrovascular diseases, including stroke, traumatic brain injury, brain tumor, multiple sclerosis, Alzheimer’s and Parkinson’s disease, epilepsy, edema, glaucoma, and amyotrophic lateral sclerosis. However, whether the disease conditions result from BBB impairment or BBB disruption occurs due to the disease pathology is still somewhat in dispute (for instance, in epilepsy). However, barrier disruption is often observed and can contribute to and exacerbate the developing pathology (see Table 1) [98]. We provide a summary overview of major CNS disorders and the implication of BBB impairments below.
5.1. Stroke
Stroke is the leading cause of permanent disability and is associated with various comorbidities such as hypertension and hyperglycemia [119]. Around 86% of stroke incidents are ischemic and are a result of the interruption of the blood and oxygen supply to a particular brain region [90], leading to a series of interrelated pathophysiological cascades that include but are not limited to BBB impairments [120,121]. The BBB’s disruption appears to start immediately after vessel occlusion and continues after the stroke event for an extended period [122]. However, it is not yet clear if the BBB disruption is the cause or the consequence of the post-stroke injury [123]. It has been observed from different experimental studies that hypoxia-ischemia conditions can affect the BBB by disrupting the TJs and damaging endothelial cells, resulting in increased permeability [124]. The TJs ensure a low paracellular permeability, ultimately preventing the occurrence of unwanted ion fluxes and paracellular diffusion across the BBB [125]. However, during ischemic stroke, the degradation of TJs occurs in a multistep and time-dependent way, comprising several signaling mechanisms [126]. In healthy conditions, the stability of TJ is maintained by anchoring the tight and adherens junctions (AJs; e.g., cadherin) to the actin cytoskeleton through tight junction-associated proteins such as ZO-1, ZO-2 and ZO-3 that act as linkers. The actin-myosin cytoskeleton is distributed in the form of short filaments and diffuse monomers between the endothelial cells. However, when subjected to hypoxic stress, the actin filaments polymerize into linear stress fibers, and the actin-myosin cytoskeleton contracts via myosin light chain phosphorylation, resulting in an increased cytoskeletal tension, the deterioration of the junction seals, and an increased BBB permeability [127,128,129]. Decreased expression levels in TJ transmembrane proteins (occludin, claudins, zona occludens, and junction adhesion molecules) has also been observed in stroke brains [125,126].
Pericytes, which constitute a functional component of NVU, closely interact with the capillary and venule endothelial cells via paracrine signaling and physical contact [130]. Pericyte plays a critical role in BBB maintenance due to its contractile, inductive, structural, and regulatory properties [131,132]. During an ischemic stroke, blood vessel constriction and a loss of pericytes occur, leading to a decreased cerebral blood flow and the loss of BBB integrity. It has been reported that during the hypoxic phase of an ischemic stroke, pericytes migrate from their original microvascular location, thereby impairing the viability of the BBB [131]. Moreover, an in vivo study in mice stroke models reported that the pericyte-derived vascular endothelial growth factor (VEGF) promotes the loss of BBB integrity in favor of angiogenesis following a stroke event [133]. VEGF is a crucial proangiogenic factor that stimulates endothelial cell proliferation, and its migration has been found to produce beneficial effects when administered before or after a stroke occurrence [134,135,136]. However, if VEGF is administered during the post-stroke acute phase, it can promote a leakage in BBB and cause a cerebral hemorrhage, resulting in an increased infarct volume [136].
Astrocytes, another crucial component of the NVU, participate in the maintenance of the BBB. However, they can also promote BBB disruption during an ischemic stroke. Astrocytes perform a dual role, depending on the phase of ischemia. During the acute phase, astrocytes are activated and secrete proinflammatory cytokines, inhibiting axonal generation, thus producing harmful effects. In contrast, astrocytes can perform a protective role during the chronic phase by participating in neurite sprouting, synapse formation, neurotrophic factor secretion, and rebuilding the BBB [137,138].
These events may be mediated by the release of soluble factors, such as cytokines, vascular endothelial growth factor (VEGF), and nitric oxide (NO). Higher levels of pro-inflammatory cytokines, including IL-1β, and TNF-α have been reported in animal brains after focal and global ischemia [139] and in the CSF of stroke patients [140]. It has also been observed, in vitro, that ischemic conditions can induce the secretion of IL-8 and monocyte chemoattractant protein-1 (MCP-1) in astrocyte and endothelial cells [141]. Another study has demonstrated that human astrocytes release inflammatory mediators under hypoxia, resulting in the upregulation of IL-8, ICAM-1, E-selectin, IL-1 β, TNF-α, and of MCP-1 genes in human cerebrovascular endothelial cells. The elevated level of cytokines upregulates endothelial and neutrophil adhesion molecules, resulting in the transmigration of leukocytes across the endothelium and the BBB. The recruitment of leukocytes is characterized by increased phosphotyrosine staining, decreased TJs proteins, and the redistributed AJs protein (vinculin), indicating BBB disruption [142].
Additionally, Mark KS et al. reported that under hypoxic conditions, sucrose permeability across primary bovine brain microvessel endothelial cells increased by more than 2.5 folds along with elevated expression of actin and altered the distribution of OCLN, ZO-1, and ZO-2 proteins [143]. Collectively, these experimental findings suggest that hypoxia-ischemia conditions can trigger the disruption of TJs and the loss of BBB integrity through a cascade of events involving VEGF, cytokines, and NO.
5.2. Multiple Sclerosis (MS)
Multiple sclerosis is an autoimmune disease in which reactive T cells interact with the antigen presented by macrophages- or microglia-expressing HLA-DR2a and HLADR2b. It leads to the destruction of the myelin sheath and the underlying axons [144]. NO, and various cytokines (interferon-γ, TNF-α, and IL-3), secreted by activated macrophages, damage oligodendrocytes and interfere with myelination and myelin gene expression [145,146]. Moreover, elevated amounts of reactive oxygen species (ROS) have been detected in MS lesions, leading to brain damage and contributing to several mechanisms underlying the pathogenesis of MS lesions [147]. As radiographic and histopathological evidence suggests, BBB disruption is one of the initial critical steps in multiple sclerosis. Radiographic analyses displayed Gd enhancing lesions; markers of BBB disruption are related to the active inflammation in the lesions and are considered an important diagnostic marker of MS [148].
Additionally, histopathological studies have revealed the origination of the myelin breakdown around parenchymal blood vessels [149]. Since BBB disruption creates a gateway for the entrance of inflammatory infiltrates into the perivascular space, it is hypothesized that a loss of BBB integrity could be one of the initial critical events in the lesion formation. This hypothesis is supported by evidence, such as the deposition of fibrinogen (a marker of endothelial permeability) [150], followed by a high infiltration of T cells in the demyelinated foci. TJs abnormalities, including the loss of claudin-3 [151,152], were also observed in the relapsing-remitting and progressive stages of MS, suggesting the opening of paracellular routes [148]. Furthermore, the failure of upregulating AQP4 and the retraction of astrocytic end-feet from the glia limitans [148] and the degradation of the basement membrane protein laminin [123] were both observed in MS, further inducing the loss of BBB viability.
5.3. Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disorder characterized by the progressive loss of the upper and lower motor neurons (LMNs) at the spinal or bulbar level [153]. It has been reported, in different studies, that the disruption of the BBB is associated with the loss of motor neurons, neuroinflammation, and motor impairment [154,155,156]. Oxidative stress performs a crucial role in the degeneration and dysfunction of motor neurons and astrocytes [157]. ROS generation in motor neurons, which results from excitotoxic activation, can induce the oxidative damage of glutamate transport in the surrounding astrocytes, resulting in increased excitatory stress, thereby promoting an ALS development [158,159]. Aqp4 and inward rectifying potassium channels (Kir) are essential for maintaining functional BBB astrocyte lining. In the ALS model, the ability of astrocytes to maintain the homeostasis of the surrounding environment is disrupted, and this imbalanced homeostasis negatively impacts the BBB viability, promotes neuronal dysfunction, and ultimately, neuronal cell death [159,160]. Several in-vivo studies have demonstrated the breakdown of BBB in a SOD1-G93A animal model for ALS [161,162,163,164,165]. This transgenic mouse model expresses the human SOD1 corresponding to the G93A mutation under the control of the cistronic human SOD1 promotor. Mutations in this gene have been linked to the onset of familial ALS (or Lou Gehrig’s disease), whereby the animals develop paralysis in one or more limbs within a few weeks of age.
Moreover, BBB abnormalities were also discovered in a postmortem study of an ALS patient [166]. Miyazaki et al. reported that MMP-9 activation in ALS patients and ALS animal models resulted in BBB damage prior to motor neuron degeneration. Additionally, there was a lack of association between the PCAM-1-positive endothelium and GFAP-positive astrocyte foot processes in patients and in vivo [154]. A direct correlation was observed between the CSF homocysteine increment and BBB disruption in ALS patients [167]. Another study also reported the astrocytic downregulation of the morphogenic protein sonic hedgehog, which resulted in an interleukin-1β mediated disruption of the BBB [168]. The same study also reported that the IL-1β mediated the upregulation of CCL2, CCL20, and CXCL2, which are pro-inflammatory chemokines that can facilitate immune cell migration, leading to BBB disruption and neuroinflammation. The involvement of TARDBP (a gene encoding for a protein called transactive response DNA binding protein 43 kDa—TDP-43, which regulates gene transcription) and angiopoietin (ANG) genes mutation in poor BBB integrity and neuroinflammation in ALS patients has also been proposed in another study [169]. Endothelial damage and/or impaired endothelium repair have also been proposed as causative factors for ALS onset [170].
5.4. Traumatic Brain Injury (TBI)
Traumatic brain injury or TBI is caused by the impact of direct or indirect external mechanical force to the brain, for instance, motor vehicle accidents, falls, assaults, sports-related incidents, etc. [171]. Almost 2.5 million people in the US require emergency care each year, and more than 5.3 million people live with a long-term disability caused by TBI [172,173,174]. The pathophysiology of TBI can be divided into two stages: primary or immediate injury and secondary or delayed injury [175,176]. The primary trauma includes acute pathological changes such as a shearing injury, hematomas, and contusions. The secondary injuries include oxidative stress, inflammation, cerebral edema, excitotoxicity, altered vascular permeability, altered calcium homeostasis, and BBB disruption [171]. Among these abovementioned pathophysiological consequences, disruption of the BBB as mediated by inflammation plays a significant role in the progression of brain injury and long-term neurological deficits associated with TBI [177]. The disruption of BBB is one of the notable pathophysiological features of TBI as related to neuroinflammatory events, which may result in brain edema and cell death. It has been observed that during or post-TBI, astrocytes and microglia can rapidly respond to injury through increased levels of multiple biological effects, which may also affect BBB function [178]. BBB integrity and low paracellular permeability are maintained through the presence and binding of inter-endothelial TJs between adjacent cells. TBI can cause endothelial cells to become damaged by disrupting blood flow, altering tight junction protein expression and the basal membrane, thus disrupting BBB integrity and increasing the paracellular permeability of the barrier [171,179]. Studies have shown that BBB disruption after TBI triggers leukocyte recruitment, inflammatory cell migration, proinflammatory cytokines, and ROS release. The generation of ROS related to TBI can further damage the BBB and mechanical trauma by promoting lipid peroxidation, protein backbone fragmentation, and DNA damage if it remains unchecked [171]. BBB disruption after TBI also stimulates the activation of the coagulation cascade, leading to the formation of intravascular blood clots and subsequent ischemia [175].
Alteration of the BBB following TBI occurs via two steps; the first step occurs within 4–6 h of tissue injury, and the second step occurs three days post-injury, affecting the cortex and the ipsilateral hippocampus [180]. Habgood et al. reported the passage of small and large molecules inside the brain after TBI, indicating the breakdown of the BBB. However, the loss of BBB integrity was temporary, as evidenced by the restoration of the BBB restriction of large and small molecules within 4–5 h and five days post-injury, respectively [152]. However, the findings of this study contrast with those of another group of researchers, indicating that restoring the BBB may require a significantly longer period of up to several years [181]. A recent study has demonstrated several inflammatory mechanisms behind BBB breakdown in mild traumatic brain injury (mTBI or concussion, the most common type of TBI) and hypertension [182]. Oxidative stress has been identified as the main cause of BBB impairment in the sub-acute stages of blast-induced traumatic brain injury (bTBI). Moreover, MMP activation after bTBI can also lead to the oxidative stress-mediated loss of BBB integrity caused by NADPH oxidase [183]. VEGF, MMP, NO, glutamate, and endothelin-1 are specific promoters in the loss of BBB integrity that have been associated with post-TBI astrocyte activation [184]. CSF/serum albumin ratio [185], TJs proteins [186,187], S100β [188,189], and plasma-soluble prion protein (PrPc) [190,191] are, conversely, potential biomarkers that have been associated with BBB disruption.
As suggested by a recent study, chronic traumatic encephalopathy (CTE) is also a neurodegenerative disorder related to repeated mTBIs, underlying the association between CTE development and concussive injuries in athletes and military personnel. However, the underlying molecular pathobiology of CTE is not well understood [192]. Markedly discontinuous or the absence of CLN-5, ZO-1, and BBB-associated tight junction components were observed in the regions of the perivascular p-Tau deposition, alongside immunohistochemical evidence of a damaged BBB [192]. Moreover, BBB disruption in the regions of the perivascular p-τ deposit has been reported in CTE and schizophrenia-diagnosed professional boxers. This p-τ deposition resulted in the loss of CLN-5 and increased extravasation of endogenous blood components such as fibrinogen and IgG [193]. Furthermore, the correlation between caspase-3-cleaved tau accumulation and the upregulation of cleaved-caspase-3 following chronic TBI suggests the involvement of apoptosis and neuroinflammation in the delated disruption of BBB following TBI [194].
5.5. Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by memory impairment [195]. Amyloid β (Aβ) is one of the major constituents of the amyloid plaque found in brain regions of patients with Alzheimer’s disease. The impairment of the BBB has been correlated with the pathogenesis of AD, where elevated levels of Aβ deposition and accumulation negatively affect the barrier integrity [195]. However, various studies have also demonstrated that BBB dysfunction plays a crucial role in generating Aβ [195,196,197] by activating β-secretase and γ-secretase [196,198]. At the BBB level, the receptor for advanced glycation end products (RAGE) is considered the prominent transporter of beta-amyloid into the brain from the systemic circulation.
In contrast, the low-density lipoprotein receptor-related protein (LRP)-1 carries the beta-amyloid in the opposite direction and out of the brain. A study of elderly human control and the AD hippocampi revealed the upregulation of RAGE receptors at the microvascular level while LRP-1 expression was downregulated in AD compared to the controls. The opposite pattern of expression was observed at the neuronal level [199]. The results from this study strongly support the proposition that changes in the relative distribution of RAGE and LRP-1 receptors affecting the brain microvasculature and neurons are a prodromal feature of AD. This study also suggests that a significant proportion of the amyloid accumulating within the brain is likely to originate from systemic circulation.
Additionally, changes and dysfunction in the BBB structural components, including pericytes, glial cells, vascular endothelial cells, the basement membrane protein (argin), and TJs, have been associated with an increased risk of AD [199]. Additional studies have also demonstrated that AD is associated with a decreased level of the glucose transporter, GLUT-1 [200], and p-glycoprotein [201]. Other features of AD, such as neuroinflammation and oxidative stress, both promoting BBB dysfunction, can reinforce the pathogenic cycle, thereby associating BBB alteration with the onset and progression of the disease [202,203,204]. However, additional studies are necessary to better dissect the pathogenic cascade leading to the onset of AD and to determine whether and to what measure BBB dysfunction acts as a causative factor or a derived effect that further contributes to the progressive worsening of the disease. For more detailed and extensive information on the subject, we refer the readers to recently published literature [205,206].
5.6. Parkinson’s Disease (PD)
Parkinson’s disease (PD) is a cognitive disorder that causes movement dysfunction. It is associated with multiple pathologic characteristics, including the formation of certain proteinaceous inclusions inside neurons, known as Lewy Bodies, and the loss of dopaminergic neurons in the Substantia Nigra pars compacta [207]. Although PD is related to multiple gene mutations, they are not isolated factors in promoting the onset of PD. Furthermore, the disease is thought to be related to a range of polygenetic and environmental cues. For example, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a lipophilic compound and crosses the BBB easily, that has been found to induce symptoms and the pathology of PD in the case of in vivo models [208]. Furthermore, a reduced P-gp function at the BBB has been observed in PD patients suggesting that a dysfunctional BBB may act as a causative mechanism in the onset of the disease [209,210]. Additional observations have revealed systemic vascular inflammation in PD patients [211], which may also harm the BBB; however, evidence of increased BBB permeability in PD patients is still lacking.
5.7. Huntington’s Disease (HD)
Huntington’s disease (HD) is an autosomal dominant inherited invariably fatal disorder that results from the expansion of glutamine residues in the HTT gene encoding for a protein called huntingtin (htt) as a result of a mutation in CAG—a trinucleotide repeat that exceeds its usual range [212]. The protein function is not completely understood but has been implicated in axonal transport [213] as well as in the transcription of the brain-derived neurotrophic factor (BDNF) that is produced by cortical neurons and promotes the survival of striatal neurons in the brain [214]. This protein contains between 6–35 glutamine residues in its standard form, but in individuals affected by Huntington’s disease, it presents more than 36 glutamine residues. The mutant huntingtin protein is ubiquitously expressed, but only certain brain regions such as the hypothalamus are affected. The reason as to why only selected neurons are affected by the disease is still unknown. However, hypothalamic changes in HD impacting the regulation of metabolism, sleep, and emotional responses can be considered an early manifestation of the disease. HD is characterized by the loss of neuronal cells with typical phenotypic features in patients, such as a compromised cognitive function, personality disorder, and hyperkinetic movements over the progression of the disease [215]. BBB disruption has been observed in the in vivo model of HD. Post-mortem tissues from human patients also presented similar morphological changes [216,217]. However, the causative implication of BBB impairment in HD is not well understood. A major challenge in HD research is presented by the lack of an appropriate rodent model to reproduce the neurodegeneration and disease progression features observed in human subjects [218,219].
5.8. Brain Tumor
Brain tumors have been proved to negatively impact BBB integrity and permeability while promoting the formation of a blood-tumor barrier (BTB) that is highly heterogeneous and characterized by numerous distinct features, including non-uniform permeability and the active efflux of molecules [220,221]. Studies have shown that around 30% of brain tumors are metastatic and derive from lung cancer, breast cancer, and melanomas [222]. Different studies have demonstrated the breakdown of inter-endothelial TJs in gliomas and metastatic adenocarcinoma in humans [223]. The downregulated expression of TJ proteins, CLN-5, OCLN, and CLN-1, have been observed in the brain microvessels of patients with glioblastoma multiforme even though the expression of ZO-1 (a scaffold protein that cross-links and anchors the TJ strand proteins to the cell cytoskeleton), remained unaltered [217]. The explanation behind the loss of TJs in the microvessels of brain tumors remains elusive. Still, the role of VEGF and the cytokines produced by the tumor cells certainly play a significant role in the elevated BBB vascular permeability and the formation of cerebral edemas [22,224]. The latter has also been linked to the substantial upregulation of AQP4 in various brain tumors, including astrocytoma and metastatic adenocarcinoma, and has is correlated with the opening of the BBB [225]. Several in-vivo studies using brain edema models have shown that mice that lack AQP4 have better survival rates than wild-type mice. AQP4 upregulation was also reported in rat models of brain injury and ischemia [226,227]. Although the brain is highly impermeable to cancerous cells, preventing their passage into CNS, BBB disruption is likely to provide a gateway for metastatic tumor cells to enter the brain parenchyma.
It has been demonstrated that BBB/BTB structural integrity is heterogeneous with respect to metastatic lesions and tumor types [220]. For instance, the fenestration of BBB ECs differs between four molecular subtypes of medulloblastoma, which consequently affect the transcytosis of drugs across the BTB and their therapeutic efficacy [228]. Moreover, the extent of BBB properties and its function can vary among brain metastases across different subtypes of breast cancer. A higher level of GLUT1 and BCRP expression has been observed in human epidermal growth factor receptor 2 (HER2)-positive breast cancer brain metastases compared to other subtypes [229]. In addition, the heterogeneity of BTB permeability has been observed in several pre-clinical studies [220]. For instance, the center of the tumor displays a higher level of leakiness compared to the peritumoral region and the surrounding brain microenvironment. A higher distribution of liposomes containing doxorubicin within the tumor was found in the intracranial GBM8401 glioma model relative to the surrounding brain tissue [230].
5.9. Septic Encephalopathy
Sepsis-associated encephalopathy (SAE) is a poorly understood diffuse brain dysfunction that occurs frequently and is secondary to systemic inflammation without an overt CNS infection. SAE is frequently diagnosed in patients with a severe systemic infection (70% of cases) and those in critically ill conditions who recovered in intensive care units. The exact pathophysiology of SAE is unknown, but it appears to encompass a variety of pathogenic mechanisms, including endothelial dysfunction, reduced cerebral blood flow and oxygen extraction in the brain tissue, circulating inflammatory mediators, and cerebral edema. This cascade of events results in activation of microglial and brain endothelial cell, TJs downregulation, and increased leukocyte recruitment. Thus, the resulting neurovascular inflammation and the BBB dysfunction exacerbate SAE pathology, leading to neuronal degradation and cell death and aggravating sepsis-induced brain dysfunction [231]. BBB breakdown in septic encephalopathy was assessed through a rodent model, using multiple BBB permeability markers such as colloidal iron oxide [232], 14C amino acid [233], and 125I-albumin [234]. Increased pinocytosis, their detachment from microvessel walls, dark and shrunken neurons, and the swelling of astrocytes end-feet are pathogenic features associated with BBB disruption as presented in these animal models of septic encephalopathy [231,232]. The involvement of the adrenergic system during the inflammatory response to sepsis, including the suppression of β2 adrenoreceptor stimulation and stimulation of α1 adrenoreceptor, potentially initiate the inflammatory cascade leading to the loss of BBB integrity [235]. Further information regarding the pathogenic mechanisms of BBB dysfunction in sepsis can be found elsewhere [236,237].
5.10. Hepatic Encephalopathy (HE)
Hepatic encephalopathy (HE) is a complex and potentially reversible neuropsychiatric disorder resulting from acute or chronic liver failure and is characterized by drowsiness, confusion, asterixis, extrapyramidal hypertonia, convulsion, and coma [238,239]. HE results from the impaired ability of the liver to metabolize neurotoxins, particularly ammonia, leading to several psychiatric/neurological deficits [238]. Association between a disrupted BBB and HE pathogenesis has been reported in several studies. Intact BBB has been observed in HE [240]; however, positron emission tomography studies have demonstrated an increased permeability of the BBB surface area to ammonia [238]. Additionally, alterations in the expression of genes coding for endothelial nitric oxide synthase and tight junction proteins in rat brain at coma/edema stage of encephalopathy with hepatic devascularization have been reported [241]. Moreover, an increased level of neurotoxin ammonia has been reported to be associated with edema and an altered morphology of astrocytes to Alzheimer’s type II astrocytes in the basal ganglia of patients with HE. This neurotoxin has been found to cause a decreased expression level of TJ CLN-12 [242]. McClung reported that ammonia exposure causes increases in the effective pore size of the BBB under certain conditions [243]. These studies indicate the potential influence of BBB breakdown in HE pathogenesis.
5.11. HIV Encephalitis
The activation of astrocytes and macrophages has been associated with an infection of the human immunodeficiency virus (HIV) in the CNS. Different types of cytokines, chemokines, reactive oxygen species, and several neurotoxins released by activated astrocytes and macrophages alter neurotransmitter activities and disrupt cellular function, promoting neuronal dysfunction and leukoencephalopathy [244]. In addition to TNF-α, other factors, including NO, arachidonic acid, platelet-activating factor, and quinolinic acid, contribute to pathology. TNF-α, mainly released by HIV-infected macrophages, affects oligodendrocytes [245]. Even though the mechanism of virus entry into the CNS is still not clear, once the virus permeates the brain, it impairs the BBB integrity and facilitates a further viral load into the CNS. For instance, serum proteins were present in the brain parenchyma of patients with HIV-associated dementia [246].
Additionally, the presence of fragmented or reduced levels of TJs expression such as ZO-1 and OCLN was observed in the brains of deceased patients with HIV-1 encephalitis [247]. The extravasation of albumin and the overexpression of ICAM-1 and VCAM1 were also observed in the gp120 transgenic mice model of HIV. Circulating gp120 is thought to negatively affect the integrity of the BBB [248,249]. Similar studies have also reported the cytotoxic effect of gp120 on the ECs of the brain microvessels, which could be responsible for a higher expression of metalloproteinases and/or induced oxidative stress, thus impacting the BBB viability [250,251].
Despite treatment with antiretroviral therapy, HIV-1 associated dementia (HAD) and cognitive impairments have been observed in HIV patients [252,253]. An imbalance between the matrix metalloproteinase (MMPs) and the tissue inhibitors of metalloproteinase (TIMPs) have been identified as the prodromal factors responsible for BBB disruption and HAD pathogenesis in HIV-1 patients [254]. It has also been demonstrated that HIV-infected cells release viral proteins such as gp120, Tat, and Nef and inflammatory cytokines and chemokines, decreasing BBB integrity and viability [255].
5.12. Epilepsy
Several studies have reported on the association between BBB disruption and epilepsy. BBB disruption is a causative factor and/or consequence of epilepsy [256]. Studies have shown that seizure activity can cause a dysfunction of the BBB [257,258,259,260,261]. Contrastingly, ample evidence suggests that BBB disruption can result in epilepsy or aggravate the epileptic condition [262,263,264,265,266,267,268,269,270]. An opening of the BBB has been observed following a seizure, and is probably associated with acute hypertension [271,272,273]. The disruption of the BBB has also been observed after traumatic brain injury (TBI), status epilepticus (SE), and temporal lobe epilepsy (TLE). It has been reported that a widespread BBB leakage occurs within minutes of post-SE, which can last from several hours to days [262,274,275,276,277,278,279,280,281,282,283], suggesting the disruption of BBB to be a consequence of epilepsy or seizure [256].
Additionally, a leakage in BBB has been observed in epileptic patients with contrast-enhanced MRI [284,285,286]. An analysis of brain tissue collected from epileptic patients also demonstrated an elevated albumin level in the brain parenchyma, suggesting blood-to-brain extravasation of large molecules [287,288]. Furthermore, the downregulation of regional GLUT-1 and a decreased uptake and metabolism level have been observed from patient samples in different studies [287,289,290].
It has been observed that BBB disruption may be epileptogenic or may contribute to the occurs of seizures. Several studies have demonstrated that BBB permeability is most evident during the acute phase, occurring soon after SE, although it extends into the latent phase in experimental models [191,262,274,275,277]. Interestingly, intense BBB leakage has been identified during the acute and latent phases without spontaneous seizures. This result indicates that BBB disruption does not induce seizures immediately but possibly performs a significant role in epileptogenesis. Similarly, significant BBB disruption was detected directly after TBI; however, seizure activity was only observed at later stages [268,269,270]. BBB disruption partnered with osmotic shock may also result in seizures in patients [291]. Moreover, several diseases with a disrupted BBB, including stroke, TBI, infection, and inflammation, may result in epilepsy and seizures [262,292]. Patients with a GLUT-1 deficiency have also developed epilepsy, suggesting the BBB transporter as crucial to maintaining normal brain function [293,294].
Several experiments have reported the extension of BBB disruption into the chronic phase of epilepsy [191,262,274,275,277]. Quantitative measurement of BBB disruption has revealed the gradual reduction of BBB leakage during epileptogenesis in most brain regions; however, leakage can continue for weeks or months after the initial insult in the ventral brain regions in epileptic rats, although to a lesser extent than during the acute phase [262,277]. Moreover, the disruption of BBB has been observed in the resected brain tissue of patients with drug-resistant epilepsy, for which the loss of BBB integrity was detected through albumin immunohistochemistry [256]. A positive correlation has been observed between BBB leakage and the severity of a seizure in epileptic rats during the chronic phase [262]. It has been reported that the opening of the BBB by mannitol in chronic epileptic rats resulted in a stable and progressive increase in the seizure frequency [262]. Because these events do not occur instantaneously but over a more extended period, it is possible that gradual changes after BBB disruption may result in a lower seizure threshold but an increased seizure frequency. Similar results have been observed in several studies in patients with post-traumatic epilepsy. A long-lasting focal increase in BBB permeability has been reported and associated with abnormal EEG activity and a reduced cerebral blood flow [268,269,270]. Similarly, a greater level of active spiking is observed in the resected epileptogenic foci of the disrupted BBB (characterized by albumin extravasation) in comparison to albumin, which is less extravasated regions [289]. Collectively, these data indicate a relationship between the occurrence of seizures and BBB leakage, suggesting that BBB impairments can further propel epileptogenesis and the progression of (already established) epilepsy in an already diseased brain [256].
5.13. Schizophrenia
The disruption of the BBB has been related to forms of psychosis such as schizophrenia. A weak relationship has been observed between schizophrenia and the TJ protein, claudin 5. Around 30% of schizophrenic patients have 22q11 deletion syndrome (22q11DS) and are CLN-5 haploinsufficient. Furthermore, it has been demonstrated through in vivo studies that the adeno-associated virus-mediated inhibition of CLN-5 results in the BBB’s disruption and abnormal behavioral outcomes. Experimental in vitro and in vivo studies have revealed a dose-dependent upregulation of CLN-5 expression after treatment with antipsychotic medications. However, a discontinuous expression of CLN-5 was observed in schizophrenic patients relative to the age-matched controls when their post-mortem brain samples were analyzed [295].
Additionally, it has been reported that the overall severity of schizophrenia (OSOS) and single group negative symptoms are related to BBB disruption [296]. The correlation between deficit schizophrenia and leaky BBB has been studied. It has been concluded that deficit schizophrenia results from BBB dysfunction, secondary to the breakdown of paracellular and vascular pathways [297].
5.14. Meningitis
The disruption of the BBB may enhance the transportation of various compounds inside the brain by altering permeability and may cause meninges inflammation [298]. A recent study has reported that meningitic E. coli can induce PDGF-B and ICAM-1 for in vitro and in vivo models. An increment in the PDGF-B and ICAM-1 potentially contributes to breaking the BBB and neuroinflammation by downregulating TJ proteins and recruiting neutrophils or monocytes, respectively [299]. Besides, microglia perform a vital role in triggering neuroinflammation by releasing chemokines and cytokines, which ultimately results in the infiltration of white blood cells through a vascular endothelium in the BBB and thereby disrupts BBB integrity [300].
6. Biological Targets for Restoring BBB Viability
The neurovascular unit (NVU) consists of endothelial cells, astrocytes, and pericytes where tight junction (TJ) related proteins including CLN-5, OCLN, and ZO-1 are present in the cellular membrane of endothelial cells [301,302]. These tight junction-associated proteins ensure that the adjacent endothelial cells are tightly bound and control the movement of molecules between the intra- and the extra-vascular spaces of BBB [302]. Various kinds of brain damage can trigger different molecular pathways, which can ultimately disrupt the integrity of BBB [303,304,305]. Some of the prominent molecules related to BBB disruption include, but are not limited to, vascular endothelial growth factors (VEGFs), matrix metalloproteinases (MMPs), and endothelins (ETs) [306,307]. The interaction between microglia and astrocytes can negatively impact the integrity of the BBB and can promote neuroinflammation, which is of particular interest to this study. Microglia and astrocytes, which are part of the NVU, are activated by various brain insults within a pro-inflammatory phenotype (M1 and A1). Microglial cells, which are more sensitive to pathogens or damage, are the first to activate (into the pro-inflammatory M1 phenotype) and produce pro-inflammatory mediators, including tumor necrosis factor (TNF), interleukin 1 beta (IL-1β), and a reactive oxygen species (ROS) which then triggers the reactive astrocytes into the A1 pro-inflammatory phenotype. In this inflammatory active form, A1 astrocytes begin releasing various chemokines (which sustain a self-feedback loop of microglial activation), MMPs (which degrade the extracellular matrix), and VEGF-A, which directly impact BBB integrity by disrupting CLN-5 and OCLN TJs, thereby inducing he breakdown of BBB and immune cell infiltration [100,308] (see also Figure 3). Studies have also reported that the inhibition of these factors may restore BBB integrity (see Table 1). The potential benefits of targeting this factor for the restoration of BBB integrity is further discussed below:
6.1. VEGFs
VEGFs are a well-known group of angiogenic factors that include VEGF-A, B, C, and D. VEGFs promote the proliferation and migration of endothelial cells and enhance the permeability of newly formed BBB with the assistance of VEGF-specific receptors (VEGFR-1, VEGFR-2) [309,310]. Brain damage has been found to upregulate the expression of VEGF both in experimental animal studies [311,312,313] and in post-mortem patients with brain damage [314,315]. One of the mechanisms of VEGF-mediated BBB disruption is related to the downregulation of CLN-5 and OCLN expression, which is associated with VEGF upregulation in astrocytes [316]. Subsequent studies by the same group have also shown that the inhibition of astrocytic VEGF-A reduced the BBB breakdown, lymphocyte infiltration, and inflammatory damage in a mouse.
Additionally, treatment administered using cavtratin, a selective eNOS inhibitor, protected against neurologic deficits in an MS mouse model and reduced VEGF-A-induced BBB disruption [100]. Similarly, treatment administered using an anti-VEGF antibody restored the BBB, leading to improved BBB selective permeability, reduced cerebral edema, and reduced infarct volume after an ischemic stroke in mice [317]. Furthermore, treatment with the VEGF receptor 2 (VEGFR-2) inhibitor SU5416 and the knockdown of VEGFR-2 reduced BBB damage after an ischemic stroke in mice [99]. Other studies have also exhibited the potential of VEGF inhibition as a viable target for restoring the BBB [318,319].
6.2. Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are zinc-dependent enzymes that can disrupt BBB permeability by degrading components of the extracellular matrix (ECM). MMPs can also degrade the TJ and basal lamina proteins, ultimately disrupting the BBB and thus facilitating leukocyte infiltration, brain edema formation, and the occurrence of a hemorrhage [320]. Studies have identified the role of several MMPs, such as MMP-2, -3, -9, and -10, in BBB damage mediated through degradation of the basal lamina in brain microvessels [320,321]. In an experimental ischemic stroke and traumatic brain injury, the inhibition of MMP-2 and MMP-9 reduced BBB damage and improved the outcomes [118,322,323,324]. MMP inhibitors such as GM6001 reduce BBB disruption and brain edema [101]. Similarly, a different MMP inhibitor known as BB-1101 protected the BBB integrity and viability following an ischemic stroke [325]. The inhibition of MMPs has also been associated with a reduced expression of the intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). These cell adhesion molecules are part of the immunoglobulin (Ig) superfamily and mediate the adhesion of lymphocytes, monocytes, and other immune cells to the vascular endothelium. Therefore, the downregulation of these molecules in response to MMPs’ inhibition will negatively impact inflammatory cell migration across the BBB [326]. Additionally, the inhibition of MMP2/9 with SB-3CT after schizophrenia in mice promoted BBB recovery and accelerated their neurological recovery [118]. Moreover, a tetracycline antibiotic, minocycline, has also been demonstrated to inhibit MMP enzyme and MMP production [327,328] which may ultimately reduce BBB disruption.
6.3. Endothelins
Endothelins (ETs) are endogenous vasoconstrictive proteins and are responsible for several CNS physiological and pathological consequences. The overexpression of ET-1 has been associated with BBB breakdown and cognitive dysfunction after an ischemic stroke in mice [329,330]. Furthermore, the upregulation of ET-1 promoted the expression of MMP2 and downregulated that of the inter endothelial TJ OCLN after an ischemic stroke [329]. Similarly, increased levels of ET-1 after a hemorrhagic stroke was associated with BBB disruption and brain edema formation [331]. Endothelin receptors, ETA, and ETB, have also been associated with ET-mediated BBB damage, and ETA antagonist, S-0139, was also found to reduce post-stroke increased BBB permeability and edema formation in mice [102]. Similarly, an anti-ETB known as BQ788 was found to significantly improve BBB integrity in an experimental animal model of epilepsy by inhibiting MMP-9 and ZO-1 degradation [103,104].
6.4. Adherens Junctions
Furthermore, changes in adherens junction (ADs) proteins expression have been observed under different pathological conditions and associated with BBB disruption. For instance, decreased cadherin expression or loss of cadherin has been observed in stroke, TBI, and brain tumors, with BBB integrity loss. In contrast, the restoration of their expression resulted in tight junction repair and disease regression [332]. However, the options that are currently available to restore ADs expression are limited. The use of CD5-2, a miR-27a/VE blocker, has been shown to increase VE-cadherin expression [105]. In fact, CD5-2 significantly improved BBB integrity and reduced cerebral cavernous malformation lesions in mice [333]. Additionally, a bioactive sphingolipid known as sphingosine-1-phosphate (S1P) has been shown to preserve BBB integrity by stabilizing the cadherin at the endothelial cell-cell contact regions [106]. Due to the functional link between adherens junctions and tight junctions, the modulation of cadherin may impact the formation of inter-endothelial tight junctions.
6.5. Tight Junctions
A decreased expression of tight junction proteins such as CLN-5 and ZO-1 has been observed in different neurological disease conditions. In mouse models of depression, chronic treatment with anti-depressants resulted in an increased CLN-5 expression [334]. An experimental antisense oligonucleotide for miR-501-3p prevented ZO-1 downregulation and cognition impairments in mice [107]. Similarly, a treatment with MS-275 (Entinostat), a histone deacetylase 1 (HDAC1) inhibitor, improved the BBB claudin-5 expression and reduced depression-like syndrome in a mice model of stress [108]. Likewise, melatonin treatment has also been associated with the mitigation of tight junction dysfunction through AMP-activated protein kinase [109].
Additionally, studies have shown the downregulation of tight junction proteins (CLN-5, OCLN, and ZO-2) after traumatic brain injury in mice [335]. Recent experimental data have also shown that Nrf2 enhancers such as metformin and sulforaphane can mitigate the post-injury reduction of tight junction proteins [336,337]. Similarly, blast-induced traumatic brain injury in mice reduced the BBB expression of tight junction proteins, and the phenomenon was reversed with a post-injury administration of dexamethasone [338]. Another compound, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPOL), a membrane-permeable radical scavenger, has also been demonstrated to improve BBB disruption by enhancing the expression of tight junction proteins after ischemic injury in a rat model of transient focal ischemia and splanchnic artery occlusion and reperfusion [339,340].
6.6. Endothelium
The endothelium is the thin layer of specialized endothelial cells that lines the inner luminal surface of blood vessels, and it is the innermost part of BBB. This layer of endothelial cells controls the vascular tone, blood fluidity, and extravasation of blood components within the brain parenchyma [3]. An injury or the dysfunction of this endothelium of BBB results in neurovascular inflammation, oxidative stress, thrombosis, and ischemia; therefore, this represents an important therapeutic target for several neurovascular disorders which affect BBB [341]. The study has shown the protective role of superoxide dismutase (SOD) conjugated with antibody (Ab/SOD) in managing acute vascular inflammation [342]. Similarly, ICAM-1 targeted catalase resulted in a marked reduction in oxidative stress, restored BBB integrity, and improved neurological function [343]. Enlimomab, a murine antibody for human ICAM-1, presented a significant therapeutic effect in repairing BBB after ischemic stroke in animal studies (101). Additionally, in a rat model of middle cerebral artery occlusion (MCAO) and a model of an ischemic stroke, the systemic administration of SOD1-cl-nanozymes reduced brain tissue injury along with sensory motor function recovery in rats [344].
6.7. Cytokines
One of the prominent characteristics of compromised BBB is the upregulation of inflammatory cytokines, such as TNF-β, IL-1β, TNF-α, and IL-6 [334]. Inhibiting these inflammatory cytokines has been shown to protect BBB integrity and ameliorate CNS disorders [345,346,347]. Treatment with the TNF-α inhibitor etanercept improved BBB integrity in a mouse model of depression [110]. Similarly, the use of anti-IL-6 antibodies improved BBB integrity in ovine fetuses [111]. Additionally, natalizumab, a humanized monoclonal antibody against the cell adhesion molecule α4-integrin, has been able to inhibit BBB endothelial inflammation by blocking the interaction between α4 integrin on white blood cells that are involved in inflammation and the VCAM-1 expressed on the vascular endothelium, thereby preventing white blood cells from entering the brain and spinal cord tissue [112,113].
6.8. Oxidative Stress
A critical class of intracellular signaling molecules known as reactive oxygen species (ROS), if accumulated in excess, can cause oxidative stress and cell damage, eventually leading to cell death. Increased production of ROS and oxidative stress have been associated with several neurological disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, multiple sclerosis, and stroke [348,349]. The activation of two oxidative stress-inducing enzymes, NADPH oxidase 4 (NOX4) and NOX5 promote BBB breakdown, leading to the infiltration of inflammatory cells in the brain parenchyma [114,115]. In a cerebral ischemic mice stroke model, the selective inhibition of NOX4 by GKT136901 (a NOX-1/4 inhibitor with potential application in diabetic nephropathy, stroke, or neurodegeneration) and NOX5 by ML090 (a pan-NOX inhibitor) improved BBB integrity and reduced infarct volume [114,115]. Additionally, the nuclear factor erythroid 2 (NFE2)-related factor 2 (Nrf2) is an emerging regulatory factor that participates in the modulation of cellular oxidative stress through the transcription of an array of antioxidant and detoxifying genes [350]. A reduced level of Nrf2 has been associated with increased susceptibility to brain injury due to its effect on BBB integrity [351], given that this nuclear factor has also been shown to regulate the expression of BBB endothelial tight and adherens junction proteins such as ZO-1 and OCLN [352]. Consequently, compounds such as metformin and sulforaphane, which enhance Nrf2 expression and activity, have been shown to effectively protect and/or restore BBB integrity after brain injuries [336,337].
6.9. Actin-Myosin Cytoskeleton
Phosphorylation of the myosin light chain promotes the contraction of the actin-myosin cytoskeleton, leading to an increased cytoskeleton tension, junction protein disorganization, and the widening of paracellular space, ultimately impacting the BBB [127]. From a mechanistic perspective, both phosphorylation and tight junction protein internalization are mediated by the activation of RhoA/Rho-associated proteins kinase (ROCK). Phosphorylation of tight junction proteins OCLN and CLN-5 by ROCKs promotes the migration of monocytes across the BBB [353]. Additionally, ROCKs activation in the capillaries of a mice model of AD led to BBB disruption and increased microvascular permeability [354]. Hence, the inhibition of ROCK1/ROCK2 has been proven to counteract BBB disruption resulting from brain injuries, such as cerebral ischemia, experimental autoimmune encephalomyelitis, and intracerebral hemorrhages [355]. Even though there are selective pharmacological inhibitors for ROCK1/ROCK2 isoforms, non-isoform specific ROCKs inhibitors, fasudil and Y-27632 are the most widely explored. Treatment using fasudil reduced the disruption of BBB and cavernous cerebral malformation in a murine model of an ischemic stroke [116,117]. Similarly, the inhibition of ROCK with Y-27632 or an ischemic stroke significantly reduced the cerebral lesion and edema volumes and improved the functional outcomes [356]. In the same study, the treatment of brain microvascular endothelial cells exposed to oxygen-glucose deprivation (OGD), using Y-27632 prevented the loss of intercellular junctions. Additionally, Slx-2119 (KD025) and SR3677, ROCK2-specific inhibitors, have also been shown to protect the cerebrovascular integrity in cerebral ischemia and reduce the production of amyloid-β in a mouse model of Alzheimer’s disease, respectively [357,358].
7. Conclusions
The BBB is a vital cellular and biological barrier that maintains the CNS microenvironment’s homeostasis by controlling the movement of molecules into and out of the brain parenchyma. This barrier acts as a gatekeeper so as to protect the brain from toxins, chemicals, inflammation, and pathogens. Therefore, a disruption of the BBB can lead to the onset and progression of several cerebrovascular and neurological diseases, which can be of a chronic or acute type [359]. In this review, we have summarized the role and functions of the BBB, the neurological disorders that are directly or indirectly associated with BBB disruption, and discussed potential therapeutic targets for restoring the impaired BBB. Studies have shown the involvement of different signaling pathways, such as VEGF, TNF-α, MMP and ET, in promoting the disruption of the BBB. Various experimental and clinical studies have revealed that some small molecular factors bear the potential of becoming viable therapeutic targets of intervention (see Figure 4). Unfortunately, clinical treatments that are capable of effectively restoring the disrupted BBB do not yet exist.
Even though substantial progress has been made in identifying many of the putative mechanisms and key factors involved in BBB disruption, further well-controlled pre-clinical and clinical studies are required to validate these pathogenic mechanisms. Furthermore, it is critically important to assess the pros and cons of modulating the activity of these molecular targets to promote the protection and/or restoration of BBB functions and integrity.
Understanding the relationship between BBB disruption and the subsequent brain pathophysiological cues is crucial for developing more effective and specific therapeutic strategies. Today, there is a major lacuna in our understanding of the underlying mechanisms linking BBB dysfunction and neurological disorders. In several instances, whether the impairment of the BBB is a direct causative factor of the CNS disorder or, instead, a result of the neurological disease, which can later negatively impact the outcome, is also unclear. However, regardless of its potential role as a prodromal factor for the onset of CNS disorders or a collateral casualty, BBB disruption has been established as a key player in worsening the disease’s outcome. Hence, combinatorial therapies targeting both the BBB and the brain parenchyma are more desirable than treatments targeting each of them as singular issues.
Moreover, due to the inherited heterogeneity of the brain structure and function, it is also important to consider region-specific BBB damage and subsequent therapeutic targets. Novel technologies are becoming available to assist in designing and delivering more effective and selective therapeutic strategies. These advanced techniques, like transcriptomic and proteomic analysis, single-cell RNA sequencing, and other advanced technologies, can help to elucidate the mechanistic differences of BBB damage as related to specific regional structures of the brain. Such an approach would assist in providing the necessary assistance for the development of more effective treatments.
Author Contributions
S.R.A. and A.A.S. conceived the study and prepared the drafting of the manuscript. L.C. assisted with the drafting of the manuscript and preparation of the figures. L.C. also oversaw the entire project and provided funding support. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health/National Institute on Drug Abuse 2R01DA029121-01A1, 1R01DA049737-01, and the National Institutes of Health/ National Institute of Neurological Disorders and Stroke 1R01NS117906-01 to Luca Cucullo.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AD | Alzheimer’s Disease |
| ALS | Amielotropic Lateral Sclerosis |
| BBB | Blood-Brain Barrier |
| BMECs | Human Brain Microvascular Endothelial Cells |
| CLN-5 | Claudin-5 |
| CNS | Central Nervous System |
| GLUT-1 | Glucose Transporter |
| HDAC | Histone Deacetylases |
| HDAC i | HDAC inhibitor |
| HIV | Human Immunodeficiency Virus |
| ICAM1 | Intercellular Adhesion Molecule 1 |
| MLC | Myosin Light Chain |
| MMPs | Matrix Metalloproteases |
| MRP | Multidrug Resistance-Associated Protein |
| MS | Multiple Sclerosis |
| NOX | NADPH Oxidase |
| Nrf2 | Nuclear Factor Erythroid 2 (NFE2)-Related factor 2) |
| NVU | Neurovascular Unit |
| OCLN | Occludin |
| PD | Parkinson’s Disease |
| P-gp | P-glycoprotein |
| VEGF | Vascular Endothelial Growth Factor |
| ROCK | RhoA/Rho-Associated Proteins Kinase |
| ROS | Reactive Oxygen Species |
| VCAM1 | Vascular Cell Adhesion Protein 1 |
| VEGFR2 | Vascular Endothelial Growth Factor Receptor-2 |
| TJs | Tight Junctions |
| ZO-1 | Zona Occludens-1 |
| 3D | 3 Dimensional |
References
- Krizbai, I.; Fazakas, C.; Haskó, J.; Molnár, J.; Nyúl-Tóth, A.; Farkas, A.; Wilhelm, I. Molecular structure and function of biological barriers. Acta Biol. Szeged. 2015, 59, 39–50. [Google Scholar]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.; Ghersi-Egea, J.F.; Perrin, R.; Leininger, B.; Siest, G. Drug metabolizing enzymes in the brain and cerebral microvessels. Brain Res. Brain Res. Rev. 1991, 16, 65–82. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Bhalerao, A.; Sivandzade, F.; Archie, S.R.; Chowdhury, E.A.; Noorani, B.; Cucullo, L. In vitro modeling of the neurovascular unit: Advances in the field. Fluids Barriers CNS 2020, 17, 22. [Google Scholar] [CrossRef]
- Jeong, S.M.; Hahm, K.D.; Shin, J.W.; Leem, J.G.; Lee, C.; Han, S.M. Changes in magnesium concentration in the serum and cerebrospinal fluid of neuropathic rats. Acta Anaesthesiol. Scand. 2006, 50, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Nischwitz, V.; Berthele, A.; Michalke, B. Speciation analysis of selected metals and determination of their total contents in paired serum and cerebrospinal fluid samples: An approach to investigate the permeability of the human blood-cerebrospinal fluid-barrier. Anal. Chim. Acta 2008, 627, 258–269. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Stubbs, J.; Hughes, I.E.; Parker, P. The distribution of potassium, sodium, chloride and urea between lumbar cerebrospinal fluid and blood serum in human subjects. Clin. Sci. 1963, 25, 97–105. [Google Scholar]
- Hansen, A.J. Effect of anoxia on ion distribution in the brain. Physiol. Rev. 1985, 65, 101–148. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Wolburg-Buchholz, K.; Mack, A.; Fallier-Becker, P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist 2009, 15, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, J.; Dobrowolska, A.; Nierwińska, K.; Małecki, A. Physiology and pharmacological role of the blood-brain barrier. Pharmacol. Rep. PR 2008, 60, 600–622. [Google Scholar]
- Nadal, A.; Fuentes, E.; Pastor, J.; McNaughton, P.A. Plasma albumin is a potent trigger of calcium signals and DNA synthesis in astrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 1426–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingrich, M.B.; Junge, C.E.; Lyuboslavsky, P.; Traynelis, S.F. Potentiation of NMDA Receptor Function by the Serine Protease Thrombin. J. Neurosci. 2000, 20, 4582. [Google Scholar] [CrossRef] [PubMed]
- Gingrich, M.B.; Traynelis, S.F. Serine proteases and brain damage—Is there a link? Trends Neurosci. 2000, 23, 399–407. [Google Scholar] [CrossRef]
- Olsson, Y.; Klatzo, I.; Sourander, P.; Steinwall, O. Blood-brain barrier to albumin in embryonic new born and adult rats. Acta Neuropathol. 1968, 10, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Tauc, M.; Vignon, X.; Bouchaud, C. Evidence for the effectiveness of the blood—CSF barrier in the fetal rat choroid plexus. A freeze-fracture and peroxidase diffusion study. Tissue Cell 1984, 16, 65–74. [Google Scholar] [CrossRef]
- Saunders, N.R. Development of the Blood—Brain Barrier to Macromolecules. In Barriers and Fluids of the Eye and Brain; Segal, M.B., Ed.; Macmillan Education UK: London, UK, 1992; pp. 128–155. [Google Scholar]
- Moos, T.; Møllgård, K. Cerebrovascular permeability to azo dyes and plasma proteins in rodents of different ages. Neuropathol. Appl. Neurobiol. 1993, 19, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Ennis, S.R.; Beer, M.E.; Betz, A.L. Developmental changes in blood-brain barrier potassium permeability in the rat: Relation to brain growth. J. Physiol. 1995, 488, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Preston, J.E.; Al-Sarraf, H.; Segal, M.B. Permeability of the developing blood-brain barrier to 14C-mannitol using the rat in situ brain perfusion technique. Dev. Brain Res. 1995, 87, 69–76. [Google Scholar] [CrossRef]
- Saunders, N.R.; Knott, G.W.; Dziegielewska, K.M. Barriers in the Immature Brain. Cell. Mol. Neurobiol. 2000, 20, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- Harris, E.S.; Nelson, W.J. VE-cadherin: At the front, center, and sides of endothelial cell organization and function. Curr. Opin. Cell Biol. 2010, 22, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vasc. Pharm. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Hamm, S.; Dehouck, B.; Kraus, J.; Wolburg-Buchholz, K.; Wolburg, H.; Risau, W.; Cecchelli, R.; Engelhardt, B.; Dehouck, M.P. Astrocyte mediated modulation of blood-brain barrier permeability does not correlate with a loss of tight junction proteins from the cellular contacts. Cell Tissue Res. 2004, 315, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Brightman, M.W.; Reese, T.S. Junctions between intimately apposed cell membranes in the vertebrate brain. J. Cell Biol. 1969, 40, 648–677. [Google Scholar] [CrossRef] [PubMed]
- Westergaard, E.; Brightman, M.W. Transport of proteins across normal cerebral arterioles. J. Comp. Neurol. 1973, 152, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Coomber, B.L.; Stewart, P.A. Morphometric analysis of CNS microvascular endothelium. Microvasc. Res. 1985, 30, 99–115. [Google Scholar] [CrossRef]
- Betz, A.L.; Goldstein, G.W. Polarity of the blood-brain barrier: Neutral amino acid transport into isolated brain capillaries. Science 1978, 202, 225–227. [Google Scholar] [CrossRef]
- Betz, A.L.; Firth, J.A.; Goldstein, G.W. Polarity of the blood-brain barrier: Distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res. 1980, 192, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Bien-Ly, N.; Yu, Y.J.; Bumbaca, D.; Elstrott, J.; Boswell, C.A.; Zhang, Y.; Luk, W.; Lu, Y.; Dennis, M.S.; Weimer, R.M. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J. Exp. Med. 2014, 211, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Dege, T.; Kunze, R.; Ernst, A.-S.; Lorenz, H.; Böhler, L.-I.; Korff, T.; Marti, H.H.; Heiland, S.; Bendszus, M. Early Blood–Brain barrier disruption in ischemic stroke initiates Multifocally around Capillaries/Venules. Stroke 2018, 49, 1479–1487. [Google Scholar] [CrossRef]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscher, W.; Potschka, H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, W. ABC transporters and drug efflux at the blood-brain barrier. Rev. Neurosci. 2010, 21, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Gonzalez-Martinez, J.; Hossain, M.; Cucullo, L.; Fazio, V.; Janigro, D.; Marchi, N. Pattern of P450 expression at the human blood-brain barrier: Roles of epileptic condition and laminar flow. Epilepsia 2010, 51, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.P.; Gehlhaus, M.; Knoth, R.; Volk, B. Expression and function of cytochrome p450 in brain drug metabolism. Curr. Drug Metab. 2007, 8, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Marchi, N.; Desai, N.K.; Puvenna, V.; Hossain, M.; Gonzalez-Martinez, J.; Alexopoulos, A.V.; Janigro, D. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia 2011, 52, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, C.; Hossain, M.; Solanki, J.; Najm, I.M.; Marchi, N.; Janigro, D. Overexpression of pregnane X and glucocorticoid receptors and the regulation of cytochrome P450 in human epileptic brain endothelial cells. Epilepsia 2017, 58, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittapalli, R.K.; Manda, V.K.; Adkins, C.E.; Geldenhuys, W.J.; Lockman, P.R. Exploiting nutrient transporters at the blood-brain barrier to improve brain distribution of small molecules. Ther. Deliv. 2010, 1, 775–784. [Google Scholar] [CrossRef]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [Green Version]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Upadhyay, U.M.; Tamargo, R.J. Inflammation in stroke and focal cerebral ischemia. Surg. Neurol. 2006, 66, 232–245. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Henninger, D.D.; Panés, J.; Eppihimer, M.; Russell, J.; Gerritsen, M.; Anderson, D.C.; Granger, D.N. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J. Immunol. 1997, 158, 1825–1832. [Google Scholar] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B. Immune cell entry into the central nervous system: Involvement of adhesion molecules and chemokines. J. Neurol. Sci. 2008, 274, 23–26. [Google Scholar] [CrossRef]
- Planas, A.M. Role of Immune Cells Migrating to the Ischemic Brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef]
- Wu, C.; Ivars, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 2009, 15, 519–527. [Google Scholar] [CrossRef]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J. Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 2006, 209, 2304–2311. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef]
- Cucullo, L.; McAllister, M.S.; Kight, K.; Krizanac-Bengez, L.; Marroni, M.; Mayberg, M.R.; Stanness, K.A.; Janigro, D. A new dynamic in vitro model for the multidimensional study of astrocyte-endothelial cell interactions at the blood-brain barrier. Brain Res. 2002, 951, 243–254. [Google Scholar] [CrossRef]
- Noell, S.; Wolburg-Buchholz, K.; Mack, A.F.; Beedle, A.M.; Satz, J.S.; Campbell, K.P.; Wolburg, H.; Fallier-Becker, P. Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. Eur. J. Neurosci. 2011, 33, 2179–2186. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Fallier-Becker, P.; Noell, S.; Mack, A.F. Structure and functions of aquaporin-4-based orthogonal arrays of particles. Int. Rev. Cell Mol. Biol. 2011, 287, 1–41. [Google Scholar] [CrossRef]
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef]
- Simard, M.; Arcuino, G.; Takano, T.; Liu, Q.S.; Nedergaard, M. Signaling at the gliovascular interface. J. Neurosci. 2003, 23, 9254–9262. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. Int. J. Mol. Sci. 2021, 22, 4280. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Sims, D.E. The pericyte—A review. Tissue Cell 1986, 18, 153–174. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Shepro, D.; Morel, N.M. Pericyte physiology. FASEB J. 1993, 7, 1031–1038. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [Green Version]
- Healy, D.P.; Wilk, S. Localization of immunoreactive glutamyl aminopeptidase in rat brain. II. Distribution and correlation with angiotensin II. Brain Res. 1993, 606, 295–303. [Google Scholar] [CrossRef]
- Elfont, R.M.; Sundaresan, P.R.; Sladek, C.D. Adrenergic receptors on cerebral microvessels: Pericyte contribution. Am. J. Physiol. 1989, 256, R224–R230. [Google Scholar] [CrossRef] [PubMed]
- Benagiano, V.; Virgintino, D.; Maiorano, E.; Rizzi, A.; Palombo, S.; Roncali, L.; Ambrosi, G. VIP-like immunoreactivity within neurons and perivascular neuronal processes of the human cerebral cortex. Eur. J. Histochem. 1996, 40, 53–56. [Google Scholar] [PubMed]
- Dehouck, M.P.; Vigne, P.; Torpier, G.; Breittmayer, J.P.; Cecchelli, R.; Frelin, C. Endothelin-1 as a mediator of endothelial cell-pericyte interactions in bovine brain capillaries. J. Cereb. Blood Flow. Metab. 1997, 17, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zwieten, E.J.; Ravid, R.; Swaab, D.F.; Van de Woude, T. Immunocytochemically stained vasopressin binding sites on blood vessels in the rat brain. Brain Res. 1988, 474, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Smyth, L.C.D.; Rustenhoven, J.; Scotter, E.L.; Schweder, P.; Faull, R.L.M.; Park, T.I.H.; Dragunow, M. Markers for human brain pericytes and smooth muscle cells. J. Chem. Neuroanat. 2018, 92, 48–60. [Google Scholar] [CrossRef]
- Alarcon-Martinez, L.; Yilmaz-Ozcan, S.; Yemisci, M.; Schallek, J.; Kılıç, K.; Can, A.; Di Polo, A.; Dalkara, T. Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. Elife 2018, 7. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, W.F.; Kimura, H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 1988, 239, 290–292. [Google Scholar] [CrossRef]
- Polfliet, M.M.; Zwijnenburg, P.J.; van Furth, A.M.; van der Poll, T.; Döpp, E.A.; Renardel de Lavalette, C.; van Kesteren-Hendrikx, E.M.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. Meningeal and perivascular macrophages of the central nervous system play a protective role during bacterial meningitis. J. Immunol. 2001, 167, 4644–4650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, E.R.; Sung, J.H.; Manivel, J.C.; Chenggis, M.L.; Blazar, B.R.; Krivit, W. Male donor-derived cells in the brains of female sex-mismatched bone marrow transplant recipients: A Y-chromosome specific in situ hybridization study. J. Neuropathol. Exp. Neurol. 1993, 52, 460–470. [Google Scholar] [CrossRef]
- Vass, K.; Hickey, W.F.; Schmidt, R.E.; Lassmann, H. Bone marrow-derived elements in the peripheral nervous system. An immunohistochemical and ultrastructural investigation in chimeric rats. Lab. Investig. 1993, 69, 275–282. [Google Scholar] [PubMed]
- Williams, K.; Alvarez, X.; Lackner, A.A. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia 2001, 36, 156–164. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Conde, J.R.; Fendrick, S.E.; Flanary, B.E.; Mariani, C.L. Role of microglia in the central nervous system’s immune response. Neurol. Res. 2005, 27, 685–691. [Google Scholar] [CrossRef]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ghorpade, A.; Rasmussen, J.; Limoges, J.; Liu, X.J.; Stins, M.; Fiala, M.; Way, D.; Kim, K.S.; Witte, M.H.; et al. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999, 155, 1599–1611. [Google Scholar] [CrossRef] [Green Version]
- Hudson, L.C.; Bragg, D.C.; Tompkins, M.B.; Meeker, R.B. Astrocytes and microglia differentially regulate trafficking of lymphocyte subsets across brain endothelial cells. Brain Res. 2005, 1058, 148–160. [Google Scholar] [CrossRef]
- Sarmento, A.; Borges, N.; Azevedo, I. Adrenergic influences on the control of blood-brain barrier permeability. Naunyn. Schmiedebergs Arch. Pharm. 1991, 343, 633–637. [Google Scholar] [CrossRef]
- Sarmento, A.; Borges, N.; Lima, D. Influence of electrical stimulation of locus coeruleus on the rat blood-brain barrier permeability to sodium fluorescein. Acta Neurochir. 1994, 127, 215–219. [Google Scholar] [CrossRef]
- Borges, N.; Sarmento, A.; Azevedo, I. Dynamics of experimental vasogenic brain oedema in the rat: Changes induced by adrenergic drugs. J. Auton. Pharm. 1999, 19, 209–217. [Google Scholar] [CrossRef]
- Borges, N.; Shi, F.; Azevedo, I.; Audus, K.L. Changes in brain microvessel endothelial cell monolayer permeability induced by adrenergic drugs. Eur. J. Pharm. 1994, 269, 243–248. [Google Scholar] [CrossRef]
- Sarmento, A.; Borges, N.; Azevedo, I. Adrenergic Mechanisms and Blood-Brain Barrier Permeability. Crit. Care Med. 2005, 33, 1474. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharm. 2006, 1, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Reeson, P.; Tennant, K.A.; Gerrow, K.; Wang, J.; Weiser Novak, S.; Thompson, K.; Lockhart, K.L.; Holmes, A.; Nahirney, P.C.; Brown, C.E. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J. Neurosci. 2015, 35, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [Green Version]
- Shigemori, Y.; Katayama, Y.; Mori, T.; Maeda, T.; Kawamata, T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir. Suppl. 2006, 96, 130–133. [Google Scholar] [CrossRef]
- Matsuo, Y.; Mihara, S.; Ninomiya, M.; Fujimoto, M. Protective effect of endothelin type A receptor antagonist on brain edema and injury after transient middle cerebral artery occlusion in rats. Stroke 2001, 32, 2143–2148. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/ endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ko, A.R.; Hyun, H.W.; Kang, T.C. ETB receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1 protein degradation following status epilepticus. Neuroscience 2015, 304, 355–367. [Google Scholar] [CrossRef]
- Zhao, Y.; Ting, K.K.; Li, J.; Cogger, V.C.; Chen, J.; Johansson-Percival, A.; Ngiow, S.F.; Holst, J.; Grau, G.; Goel, S.; et al. Targeting Vascular Endothelial-Cadherin in Tumor-Associated Blood Vessels Promotes T-cell-Mediated Immunotherapy. Cancer Res. 2017, 77, 4434–4447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, K.; Cabochette, P.; Diéguez-Hurtado, R.; Wiesner, C.; Wakayama, Y.; Grassme, K.S.; Hubert, M.; Guenther, S.; Belting, H.G.; Affolter, M.; et al. Wnt/β-catenin signaling regulates VE-cadherin-mediated anastomosis of brain capillaries by counteracting S1pr1 signaling. Nat. Commun. 2018, 9, 4860. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Sun, P.; Zhang, X.; Hamblin, M.H.; Yin, K.J. Endothelium-targeted deletion of the miR-15a/16-1 cluster ameliorates blood-brain barrier dysfunction in ischemic stroke. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Dudek, K.A.; Dion-Albert, L.; Lebel, M.; LeClair, K.; Labrecque, S.; Tuck, E.; Ferrer Perez, C.; Golden, S.A.; Tamminga, C.; Turecki, G.; et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. Proc. Natl. Acad. Sci. USA 2020, 117, 3326–3336. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xue, G.X.; Liu, W.C.; Shu, H.; Wang, M.; Sun, Y.; Liu, X.; Sun, Y.E.; Liu, C.F.; Liu, J.; et al. Melatonin alleviates lipopolysaccharide-compromised integrity of blood-brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell 2017, 16, 414–421. [Google Scholar] [CrossRef]
- Cheng, Y.; Desse, S.; Martinez, A.; Worthen, R.J.; Jope, R.S.; Beurel, E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav. Immun. 2018, 69, 556–567. [Google Scholar] [CrossRef]
- Zhang, J.; Sadowska, G.B.; Chen, X.; Park, S.Y.; Kim, J.E.; Bodge, C.A.; Cummings, E.; Lim, Y.P.; Makeyev, O.; Besio, W.G.; et al. Anti-IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus. FASEB J. 2015, 29, 1739–1753. [Google Scholar] [CrossRef] [Green Version]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, S.P.; Simonsen, H.J.; Varatharaj, A.; Galea, I.; Frederiksen, J.L.; Larsson, H.B.W. Permeability of the blood-brain barrier predicts no evidence of disease activity at 2 years after natalizumab or fingolimod treatment in relapsing-remitting multiple sclerosis. Ann. Neurol. 2018, 83, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Casas, A.I.; Geuss, E.; Kleikers, P.W.M.; Mencl, S.; Herrmann, A.M.; Buendia, I.; Egea, J.; Meuth, S.G.; Lopez, M.G.; Kleinschnitz, C.; et al. NOX4-dependent neuronal autotoxicity and BBB breakdown explain the superior sensitivity of the brain to ischemic damage. Proc. Natl. Acad. Sci. USA 2017, 114, 12315–12320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, A.I.; Kleikers, P.W.M.; Geuss, E.; Langhauser, F.; Adler, T.; Busch, D.H.; Gailus-Durner, V.; de Angelis, M.H.; Egea, J.; Lopez, M.G.; et al. Calcium-dependent blood-brain barrier breakdown by NOX5 limits postreperfusion benefit in stroke. J. Clin. Investig. 2019, 129, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Zhang, Y.; Chen, H.; Li, J. Rho kinase: A new target for treatment of cerebral ischemia/reperfusion injury. Neural Regen Res. 2013, 8, 1180–1189. [Google Scholar] [CrossRef]
- Shenkar, R.; Shi, C.; Austin, C.; Moore, T.; Lightle, R.; Cao, Y.; Zhang, L.; Wu, M.; Zeineddine, H.A.; Girard, R.; et al. RhoA Kinase Inhibition with Fasudil Versus Simvastatin in Murine Models of Cerebral Cavernous Malformations. Stroke 2017, 48, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tsirka, S.E. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain 2005, 128, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- van der Worp, H.B.; Raaijmakers, T.W.; Kappelle, L.J. Early complications of ischemic stroke. Curr. Treat. Options Neurol. 2008, 10, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Sarvari, S.; Moakedi, F.; Hone, E.; Simpkins, J.W.; Ren, X. Mechanisms in blood-brain barrier opening and metabolism-challenged cerebrovascular ischemia with emphasis on ischemic stroke. Metab. Brain Dis. 2020, 35, 851–868. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, L.; An, C.; Wang, R.; Yang, L.; Yu, W.; Li, P.; Gao, Y. The blood brain barrier in cerebral ischemic injury—Disruption and repair. Brain Hemorrhages 2020, 1, 34–53. [Google Scholar] [CrossRef]
- Balkaya, M.; Kim, I.-d.; Shakil, F.; Cho, S. CD36 deficiency reduces chronic BBB dysfunction and scar formation and improves activity, hedonic and memory deficits in ischemic stroke. J. Cereb. Blood Flow Metab. 2020, 41, 486–501. [Google Scholar] [CrossRef]
- Krueger, M.; Bechmann, I.; Immig, K.; Reichenbach, A.; Härtig, W.; Michalski, D. Blood—Brain Barrier Breakdown Involves Four Distinct Stages of Vascular Damage in Various Models of Experimental Focal Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2014, 35, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.C.; Davis, T.P. Calcium Modulation of Adherens and Tight Junction Function. Stroke 2002, 33, 1706–1711. [Google Scholar] [CrossRef]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, F.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef]
- Hicks, K.; O’Neil, R.G.; Dubinsky, W.S.; Brown, R.C. TRPC-mediated actin-myosin contraction is critical for BBB disruption following hypoxic stress. Am. J. Physiol. Cell Physiol. 2010, 298, C1583–C1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkat, P.; Chopp, M.; Chen, J. Blood–Brain Barrier Disruption, Vascular Impairment, and Ischemia/Reperfusion Damage in Diabetic Stroke. J. Am. Heart Assoc. 2017, 6, e005819. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.Y.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair After Ischemic Stroke. Transl. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Agalliu, D.; Yu, C.; Fisher, M. The Role of Pericytes in Blood-Brain Barrier Function and Stroke. Curr. Pharm. Des. 2012, 18, 3653–3662. [Google Scholar] [CrossRef] [PubMed]
- Wildman, S. Sympathetic nerve-derived ATP regulates renal medullary vasa recta diameter via pericyte cells: A role for regulating medullary blood flow? Front. Physiol. 2013, 4, 307. [Google Scholar]
- Bai, Y.; Zhu, X.; Chao, J.; Zhang, Y.; Qian, C.; Li, P.; Liu, D.; Han, B.; Zhao, L.; Zhang, J.; et al. Pericytes Contribute to the Disruption of the Cerebral Endothelial Barrier via Increasing VEGF Expression: Implications for Stroke. PLoS ONE 2015, 10, e0124362. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular Endothelial Growth Factor and Angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Zechariah, A.; ElAli, A.; Doeppner, T.R.; Jin, F.; Hasan, M.R.; Helfrich, I.; Mies, G.; Hermann, D.M. Vascular Endothelial Growth Factor Promotes Pericyte Coverage of Brain Capillaries, Improves Cerebral Blood Flow During Subsequent Focal Cerebral Ischemia, and Preserves the Metabolic Penumbra. Stroke 2013, 44, 1690–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.v.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Ståhlberg, A.; Aprico, K.; Larsson, K.; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, Z.; Xin, H.; Chopp, M. The role of astrocytes in mediating exogenous cell-based restorative therapy for stroke. Glia 2014, 62, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Feuerstein, G.Z.; Liu, T.; Barone, F.C. Cytokines, inflammation, and brain injury: Role of tumor necrosis factor-alpha. Cereb. Brain Metab. Rev. 1994, 6, 341–360. [Google Scholar]
- Tarkowski, E.; Rosengren, L.; Blomstrand, C.; Wikkelsö, C.; Jensen, C.; Ekholm, S.; Tarkowski, A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin. Exp. Immunol. 1997, 110, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Smith, C.; Shapiro, A.; Monette, R.; Hutchison, J.; Stanimirovic, D. Increased expression of bioactive chemokines in human cerebromicrovascular endothelial cells and astrocytes subjected to simulated ischemia in vitro. J. Neuroimmunol. 1999, 101, 148–160. [Google Scholar] [CrossRef]
- Bolton, S.J.; Anthony, D.C.; Perry, V.H. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 1998, 86, 1245–1257. [Google Scholar] [CrossRef]
- Mark, K.S.; Davis, T.P. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1485–H1494. [Google Scholar] [CrossRef] [Green Version]
- Wekerle, H.; Hohlfeld, R. Molecular mimicry in multiple sclerosis. N. Engl. J. Med. 2003, 349, 185–186. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Sheng, W.S.; Peterson, P.K. Tumor necrosis factor-alpha production by human fetal microglial cells: Regulation by other cytokines. Dev. Neurosci. 1995, 17, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.E.; Ignarro, L.J.; Sherman, M.P.; Melinek, J.; Lane, T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 1993, 151, 2132–2141. [Google Scholar]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef]
- Spencer, J.I.; Bell, J.S.; DeLuca, G.C. Vascular pathology in multiple sclerosis: Reframing pathogenesis around the blood-brain barrier. J. Neurol. Neurosurg. Psychiatry 2018, 89, 42–52. [Google Scholar] [CrossRef]
- Prineas, J.W.; Parratt, J.D. Oligodendrocytes and the early multiple sclerosis lesion. Ann. Neurol. 2012, 72, 18–31. [Google Scholar] [CrossRef]
- Vos, C.M.; Geurts, J.J.; Montagne, L.; van Haastert, E.S.; Bö, L.; van der Valk, P.; Barkhof, F.; de Vries, H.E. Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol. Dis. 2005, 20, 953–960. [Google Scholar] [CrossRef]
- McQuaid, S.; Cunnea, P.; McMahon, J.; Fitzgerald, U. The effects of blood–brain barrier disruption on glial cell function in multiple sclerosis. Biochem. Soc. Trans. 2009, 37, 329–331. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 89, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ilieva, H.; Hallagan, L.; Bell, R.; Singh, I.; Paquette, N.; Thiyagarajan, M.; Deane, R.; Fernandez, J.A.; Lane, S.; et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J. Clin. Investig. 2009, 119, 3437–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4356386. [Google Scholar] [CrossRef]
- Dal-Cim, T.; Poluceno, G.G.; Lanznaster, D.; de Oliveira, K.A.; Nedel, C.B.; Tasca, C.I. Guanosine prevents oxidative damage and glutamate uptake impairment induced by oxygen/glucose deprivation in cortical astrocyte cultures: Involvement of A1 and A2A adenosine receptors and PI3K, MEK, and PKC pathways. Purinergic Signal. 2019, 15, 465–476. [Google Scholar] [CrossRef]
- Martorana, F.; Foti, M.; Virtuoso, A.; Gaglio, D.; Aprea, F.; Latronico, T.; Rossano, R.; Riccio, P.; Papa, M.; Alberghina, L.; et al. Differential Modulation of NF-κB in Neurons and Astrocytes Underlies Neuroprotection and Antigliosis Activity of Natural Antioxidant Molecules. Oxid. Med. Cell. Longev. 2019, 2019, 8056904. [Google Scholar] [CrossRef] [Green Version]
- Bataveljić, D.; Nikolić, L.; Milosević, M.; Todorović, N.; Andjus, P.R. Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1G93A rat model. Glia 2012, 60, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, C.; Mitrecic, D.; Demetter, P.; De Decker, R.; Authelet, M.; Boom, A.; Pochet, R. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009, 1301, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Andjus, P.R.; Bataveljić, D.; Vanhoutte, G.; Mitrecic, D.; Pizzolante, F.; Djogo, N.; Nicaise, C.; Gankam Kengne, F.; Gangitano, C.; Michetti, F.; et al. In vivo morphological changes in animal models of amyotrophic lateral sclerosis and Alzheimer’s-like disease: MRI approach. Anat. Rec. 2009, 292, 1882–1892. [Google Scholar] [CrossRef]
- Bataveljić, D.; Stamenković, S.; Bačić, G.; Andjus, P.R. Imaging cellular markers of neuroinflammation in the brain of the rat model of amyotrophic lateral sclerosis. Acta Physiol. Hung. 2011, 98, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE 2007, 2, e1205. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Lin, W.; Zheng, M.; Liu, Q.; He, B.; Luo, C.; Lu, X.; Pei, Z.; Su, H.; Yao, X. Alterations in AQP4 expression and polarization in the course of motor neuron degeneration in SOD1G93A mice. Mol. Med. Rep. 2017, 16, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, X.; Li, X.; Wang, H.; Wang, T. Elevated cerebrospinal fluid homocysteine is associated with blood-brain barrier disruption in amyotrophic lateral sclerosis patients. Neurol. Sci. 2020, 41, 1865–1872. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticozzi, N.; Tiloca, C.; Mencacci, N.E.; Morelli, C.; Doretti, A.; Rusconi, D.; Colombrita, C.; Sangalli, D.; Verde, F.; Finelli, P.; et al. Oligoclonal bands in the cerebrospinal fluid of amyotrophic lateral sclerosis patients with disease-associated mutations. J. Neurol. 2013, 260, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Garbuzova-Davis, S.; Woods, R.L., 3rd; Louis, M.K.; Zesiewicz, T.A.; Kuzmin-Nichols, N.; Sullivan, K.L.; Miller, A.M.; Hernandez-Ontiveros, D.G.; Sanberg, P.R. Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS ONE 2010, 5, e10614. [Google Scholar] [CrossRef]
- Sivandzade, F.; Alqahtani, F.; Cucullo, L. Traumatic Brain Injury and Blood–Brain Barrier (BBB): Underlying Pathophysiological Mechanisms and the Influence of Cigarette Smoking as a Premorbid Condition. Int. J. Mol. Sci. 2020, 21, 2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Tremblaye, P.B.; O’Neil, D.A.; LaPorte, M.J.; Cheng, J.P.; Beitchman, J.A.; Thomas, T.C.; Bondi, C.O.; Kline, A.E. Elucidating opportunities and pitfalls in the treatment of experimental traumatic brain injury to optimize and facilitate clinical translation. Neurosci. Biobehav. Rev. 2018, 85, 160–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Büki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Durazzo, T.C.; Abadjian, L.; Kincaid, A.; Bilovsky-Muniz, T.; Boreta, L.; Gauger, G.E. The Influence of Chronic Cigarette Smoking on Neurocognitive Recovery after Mild Traumatic Brain Injury. J. Neurotrauma 2013, 30, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, L.; Wilson, C.; Grant, G. Frontiers in Neuroscience. Blood–Brain Barrier Pathophysiology following Traumatic Brain Injury. In Translational Research in Traumatic Brain Injury; Laskowitz, D., Grant, G., Eds.; CRC Press/Taylor and Francis Group, LLC.: Boca Raton, FL, USA, 2016. [Google Scholar]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wang, H.L.; Pu, H.J.; Shi, Y.J.; Zhang, J.; Zhang, W.T.; Wang, G.H.; Hu, X.M.; Leak, R.K.; Chen, J.; et al. Ethyl pyruvate protects against blood-brain barrier damage and improves long-term neurological outcomes in a rat model of traumatic brain injury. CNS Neurosci. 2015, 21, 374–384. [Google Scholar] [CrossRef]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood–Brain Barrier Pathophysiology in Traumatic Brain Injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef] [Green Version]
- Alves, J.L. Blood–brain barrier and traumatic brain injury. J. Neurosci. Res. 2014, 92, 141–147. [Google Scholar] [CrossRef]
- Başkaya, M.K.; Rao, A.M.; Doğan, A.; Donaldson, D.; Dempsey, R.J. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 1997, 226, 33–36. [Google Scholar] [CrossRef]
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years After Traumatic Brain Injury in Humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Szarka, N.; Toth, L.; Czigler, A.; Kellermayer, Z.; Ungvari, Z.; Amrein, K.; Czeiter, E.; Bali, Z.K.; Tadepalli, S.A.; Wahr, M.; et al. Single Mild Traumatic Brain Injury Induces Persistent Disruption of the Blood-Brain Barrier, Neuroinflammation and Cognitive Decline in Hypertensive Rats. Int. J. Mol. Sci. 2019, 20, 3223. [Google Scholar] [CrossRef] [Green Version]
- Kuriakose, M.; Younger, D.; Ravula, A.R.; Alay, E.; Rama Rao, K.V.; Chandra, N. Synergistic Role of Oxidative Stress and Blood-Brain Barrier Permeability as Injury Mechanisms in the Acute Pathophysiology of Blast-induced Neurotrauma. Sci. Rep. 2019, 9, 7717. [Google Scholar] [CrossRef] [Green Version]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagos, F.T.; Empey, P.E.; Wang, P.; Ma, X.; Poloyac, S.M.; Bayir, H.; Kochanek, P.M.; Bell, M.J.; Clark, R.S.B. Exploratory Application of Neuropharmacometabolomics in Severe Childhood Traumatic Brain Injury. Crit. Care Med. 2018, 46, 1471–1479. [Google Scholar] [CrossRef]
- Higashida, T.; Kreipke, C.W.; Rafols, J.A.; Peng, C.; Schafer, S.; Schafer, P.; Ding, J.Y.; Dornbos, D., 3rd; Li, X.; Guthikonda, M.; et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J. Neurosurg. 2011, 114, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, R.; Yu, K.; Weatherwax, T.; Zheng, H.; Liu, W.; Liu, K.J. Blood Occludin Level as a Potential Biomarker for Early Blood Brain Barrier Damage Following Ischemic Stroke. Sci. Rep. 2017, 7, 40331. [Google Scholar] [CrossRef] [PubMed]
- Zongo, D.; Ribéreau-Gayon, R.; Masson, F.; Laborey, M.; Contrand, B.; Salmi, L.R.; Montaudon, D.; Beaudeux, J.L.; Meurin, A.; Dousset, V.; et al. S100-B protein as a screening tool for the early assessment of minor head injury. Ann. Emerg. Med. 2012, 59, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Han, L.; Guo, S.; Wang, L.; Xiong, Z.; Chen, Z.; Chen, W.; Liang, J. Serum τ protein as a potential biomarker in the assessment of traumatic brain injury. Exp. Ther. Med. 2016, 11, 1147–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, N.; Akonasu, H.; Shishkin, R.; Taghibiglou, C. Plasma soluble prion protein, a potential biomarker for sport-related concussions: A pilot study. PLoS ONE 2015, 10, e0117286. [Google Scholar] [CrossRef]
- Rigau, V.; Morin, M.; Rousset, M.C.; de Bock, F.; Lebrun, A.; Coubes, P.; Picot, M.C.; Baldy-Moulinier, M.; Bockaert, J.; Crespel, A.; et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain 2007, 130, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Doherty, C.P.; O’Keefe, E.; Wallace, E.; Loftus, T.; Keaney, J.; Kealy, J.; Humphries, M.M.; Molloy, M.G.; Meaney, J.F.; Farrell, M.; et al. Blood–Brain Barrier Dysfunction as a Hallmark Pathology in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2016, 75, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, M.; Aherne, S.; O’Riordan, S.; O’Keeffe, E.; Greene, C.; Campbell, M. Blood-brain barrier dysfunction in a boxer with chronic traumatic encephalopathy and schizophrenia. Clin. Neuropathol. 2019, 38, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, O.Y.; Glushakov, A.O.; Borlongan, C.V.; Valadka, A.B.; Hayes, R.L.; Glushakov, A.V. Role of Caspase-3-Mediated Apoptosis in Chronic Caspase-3-Cleaved Tau Accumulation and Blood-Brain Barrier Damage in the Corpus Callosum after Traumatic Brain Injury in Rats. J. Neurotrauma 2018, 35, 157–173. [Google Scholar] [CrossRef]
- Cai, Z.; Qiao, P.-F.; Wan, C.-Q.; Cai, M.; Zhou, N.-K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Kho, D.; Graham, E.S.; Nicholson, L.F.B.; O’Carroll, S.J. Statins Inhibit Fibrillary β-Amyloid Induced Inflammation in a Model of the Human Blood Brain Barrier. PLoS ONE 2016, 11, e0157483. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, F.; Du, Y.-F.; Long, Y.; Reed, M.N.; Hu, M.; Suppiramaniam, V.; Hong, H.; Tang, S.-S. Targeted inhibition of RAGE reduces amyloid-β influx across the blood-brain barrier and improves cognitive deficits in db/db mice. Neuropharmacology 2018, 131, 143–153. [Google Scholar] [CrossRef]
- Atwal, J.K.; Chen, Y.; Chiu, C.; Mortensen, D.L.; Meilandt, W.J.; Liu, Y.; Heise, C.E.; Hoyte, K.; Luk, W.; Lu, Y.; et al. A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci. Transl. Med. 2011, 3, 84ra43. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. The blood-brain barrier and cerebrovascular pathology in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1999, 893, 113–125. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfield, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Ratka, A. Oxidative stress and β-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 2011, 13, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martinez-Gomez, P.A.; Campa-Cordoba, B.B.; Apatiga-Perez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garces-Ramirez, L.; Luna-Munoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. [Google Scholar] [CrossRef] [PubMed]
- Apatiga-Perez, R.; Soto-Rojas, L.O.; Campa-Cordoba, B.B.; Luna-Viramontes, N.I.; Cuevas, E.; Villanueva-Fierro, I.; Ontiveros-Torres, M.A.; Bravo-Munoz, M.; Flores-Rodriguez, P.; Garces-Ramirez, L.; et al. Neurovascular dysfunction and vascular amyloid accumulation as early events in Alzheimer’s disease. Metab. Brain Dis. 2021. [Google Scholar] [CrossRef]
- Jagmag, S.A.; Tripathi, N.; Shukla, S.D.; Maiti, S.; Khurana, S. Evaluation of Models of Parkinson’s Disease. Front. Neurosci. 2015, 9, 503. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, M.; Møller, A.; Jensen, L.H.; Pedersen, A.; Harajehi, J.B.; Pakkenberg, H. MPTP-induced Parkinsonism in minipigs: A behavioral, biochemical, and histological study. Neurotoxicol. Teratol. 1999, 21, 169–175. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.M.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.H.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood–brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Yu, C.C.; Chen, H.L.; Chen, M.H.; Lu, C.H.; Tsai, N.W.; Huang, C.C.; Chang, Y.Y.; Li, S.H.; Chen, Y.S.; Chiang, P.L.; et al. Vascular Inflammation Is a Risk Factor Associated with Brain Atrophy and Disease Severity in Parkinson’s Disease: A Case-Control Study. Oxid. Med. Cell Longev. 2020, 2020, 2591248. [Google Scholar] [CrossRef]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A Worldwide Study of the Huntington’s Disease Mutation: The Sensitivity and Specificity of Measuring CAG Repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic signaling: New functions of huntingtin and axonal transport in neurological disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M.; Sasaki, H.; Tsukita, S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J. Cell Biol. 1999, 147, 891–903. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furies, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef]
- Neuhaus, J.; Risau, W.; Wolburg, H. Induction of blood-brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann. N. Y. Acad. Sci. 1991, 633, 578–580. [Google Scholar] [CrossRef]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Groothuis, D.R.; Vriesendorp, F.J.; Kupfer, B.; Warnke, P.C.; Lapin, G.D.; Kuruvilla, A.; Vick, N.A.; Mikhael, M.A.; Patlak, C.S. Quantitative measurements of capillary transport in human brain tumors by computed tomography. Ann. Neurol. 1991, 30, 581–588. [Google Scholar] [CrossRef]
- Norden, A.D.; Wen, P.Y.; Kesari, S. Brain metastases. Curr. Opin. Neurol. 2005, 18, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Long, D.M. Capillary ultrastructure and the blood-brain barrier in human malignant brain tumors. J. Neurosurg. 1970, 32, 127–144. [Google Scholar] [CrossRef] [PubMed]
- De Vries, H.E.; Blom-Roosemalen, M.C.; van Oosten, M.; de Boer, A.G.; van Berkel, T.J.; Breimer, D.D.; Kuiper, J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Krishna, S.; Bell, B.A. Aquaporin-4 expression is increased in oedematous human brain tumours. J. Neurol. Neurosurg. Psychiatry 2002, 72, 262–265. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, M.; Yamashita, T.; Kumura, E.; Tamatani, M.; Kobayashi, A.; Yokawa, T.; Maruno, M.; Kato, A.; Ohnishi, T.; Kohmura, E.; et al. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Brain Res. Mol. Brain Res. 2000, 78, 131–137. [Google Scholar] [CrossRef]
- Vizuete, M.L.; Venero, J.L.; Vargas, C.; Ilundáin, A.A.; Echevarría, M.; Machado, A.; Cano, J. Differential upregulation of aquaporin-4 mRNA expression in reactive astrocytes after brain injury: Potential role in brain edema. Neurobiol. Dis. 1999, 6, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Yonemori, K.; Tsuta, K.; Ono, M.; Shimizu, C.; Hirakawa, A.; Hasegawa, T.; Hatanaka, Y.; Narita, Y.; Shibui, S.; Fujiwara, Y. Disruption of the blood brain barrier by brain metastases of triple-negative and basal-type breast cancer but not HER2/neu-positive breast cancer. Cancer 2010, 116, 302–308. [Google Scholar] [CrossRef]
- Yang, F.Y.; Wong, T.T.; Teng, M.C.; Liu, R.S.; Lu, M.; Liang, H.F.; Wei, M.C. Focused ultrasound and interleukin-4 receptor-targeted liposomal doxorubicin for enhanced targeted drug delivery and antitumor effect in glioblastoma multiforme. J. Control. Release 2012, 160, 652–658. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Davies, D.C.; Moss, R.F.; Tighe, D.; Bennett, E.D. Pathophysiology of septic encephalopathy: A review. Crit. Care Med. 2000, 28, 3019–3024. [Google Scholar] [CrossRef] [PubMed]
- Clawson, C.C.; Hartmann, J.F.; Vernier, R.L. Electron microscopy of the effect of gram-negative endotoxin on the blood-brain barrier. J. Comp. Neurol. 1966, 127, 183–198. [Google Scholar] [CrossRef]
- Jeppsson, B.; Freund, H.R.; Gimmon, Z.; James, J.H.; von Meyenfeldt, M.F.; Fischer, J.E. Blood-brain barrier derangement in sepsis: Cause of septic encephalopathy? Am. J. Surg. 1981, 141, 136–142. [Google Scholar] [CrossRef]
- Deng, X.; Wang, X.; Andersson, R. Endothelial barrier resistance in multiple organs after septic and nonseptic challenges in the rat. J. Appl. Physiol. 1995, 78, 2052–2061. [Google Scholar] [CrossRef]
- Tighe, D.; Moss, R.; Bennett, D. Cell surface adrenergic receptor stimulation modifies the endothelial response to SIRS. Systemic Inflammatory Response Syndrome. New Horiz. 1996, 4, 426–442. [Google Scholar] [PubMed]
- Nwafor, D.C.; Brichacek, A.L.; Mohammad, A.S.; Griffith, J.; Lucke-Wold, B.P.; Benkovic, S.A.; Geldenhuys, W.J.; Lockman, P.R.; Brown, C.M. Targeting the Blood-Brain Barrier to Prevent Sepsis-Associated Cognitive Impairment. J. Cent. Nerv. Syst. Dis. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Hernandes, M.S. Sepsis-Associated Encephalopathy and Blood-Brain Barrier Dysfunction. Inflammation 2021. [Google Scholar] [CrossRef]
- Dhanda, S.; Sandhir, R. Blood-Brain Barrier Permeability Is Exacerbated in Experimental Model of Hepatic Encephalopathy via MMP-9 Activation and Downregulation of Tight Junction Proteins. Mol. Neurobiol. 2018, 55, 3642–3659. [Google Scholar] [CrossRef] [PubMed]
- Bismuth, M.; Funakoshi, N.; Cadranel, J.-F.; Blanc, P. Hepatic encephalopathy: From pathophysiology to therapeutic management. Eur. J. Gastroenterol. Hepatol. 2011, 23, 8–22. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.; Shawcross, D.; Damink, S.W.O.; Jalan, R. Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab. Brain Dis. 2007, 22, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Sawara, K.; Desjardins, P.; Chatauret, N.; Kato, A.; Suzuki, K.; Butterworth, R.F. Alterations in expression of genes coding for proteins of the neurovascular unit in ischemic liver failure. Neurochem. Int. 2009, 55, 119–123. [Google Scholar] [CrossRef]
- Bélanger, M.; Asashima, T.; Ohtsuki, S.; Yamaguchi, H.; Ito, S.; Terasaki, T. Hyperammonemia induces transport of taurine and creatine and suppresses claudin-12 gene expression in brain capillary endothelial cells in vitro. Neurochem. Int. 2007, 50, 95–101. [Google Scholar] [CrossRef]
- McClung, H.J.; Sloan, H.R.; Powers, P.; Merola, A.J.; Murray, R.; Kerzner, B.; Pollack, J.D. Early changes in the permeability of the blood-brain barrier produced by toxins associated with liver failure. Pediatr. Res. 1990, 28, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharer, L.R. Pathology of HIV-1 infection of the central nervous system. A review. J. Neuropathol. Exp. Neurol. 1992, 51, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T.; McArthur, J.C.; Narayan, O. The neurobiology of human immunodeficiency virus infections. FASEB J. 1988, 2, 2970–2981. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K.; Cash, K.S. Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: Immunohistochemical localization of serum proteins in postmortem brain. Ann. Neurol. 1992, 32, 658–666. [Google Scholar] [CrossRef]
- Dallasta, L.M.; Pisarov, L.A.; Esplen, J.E.; Werley, J.V.; Moses, A.V.; Nelson, J.A.; Achim, C.L. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 1999, 155, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Toneatto, S.; Finco, O.; van der Putten, H.; Abrignani, S.; Annunziata, P. Evidence of blood-brain barrier alteration and activation in HIV-1 gp120 transgenic mice. Aids 1999, 13, 2343–2348. [Google Scholar] [CrossRef]
- Cioni, C.; Annunziata, P. Circulating gp120 alters the blood-brain barrier permeability in HIV-1 gp120 transgenic mice. Neurosci. Lett. 2002, 330, 299–301. [Google Scholar] [CrossRef]
- Huang, M.B.; Hunter, M.; Bond, V.C. Effect of extracellular human immunodeficiency virus type 1 glycoprotein 120 on primary human vascular endothelial cell cultures. AIDS Res. Hum. Retrovir. 1999, 15, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Louboutin, J.-P.; Strayer, D.S. Blood-Brain Barrier Abnormalities Caused by HIV-1 gp120: Mechanistic and Therapeutic Implications. Sci. World J. 2012, 2012, 482575. [Google Scholar] [CrossRef]
- Sacktor, N.; Skolasky, R.L.; Seaberg, E.; Munro, C.; Becker, J.T.; Martin, E.; Ragin, A.; Levine, A.; Miller, E. Prevalence of HIV-associated neurocognitive disorders in the Multicenter AIDS Cohort Study. Neurology 2016, 86, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Jessen Krut, J.; Mellberg, T.; Price, R.W.; Hagberg, L.; Fuchs, D.; Rosengren, L.; Nilsson, S.; Zetterberg, H.; Gisslén, M. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS ONE 2014, 9, e88591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Shepherd, N.; Lan, J.; Li, W.; Rane, S.; Gupta, S.K.; Zhang, S.; Dong, J.; Yu, Q. MMPs/TIMPs imbalances in the peripheral blood and cerebrospinal fluid are associated with the pathogenesis of HIV-1-associated neurocognitive disorders. Brain Behav. Immun. 2017, 65, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Strazza, M.; Pirrone, V.; Wigdahl, B.; Nonnemacher, M.R. Breaking down the barrier: The effects of HIV-1 on the blood-brain barrier. Brain Res. 2011, 1399, 96–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet, E.A.; Aronica, E.; Gorter, J.A. Blood–brain barrier dysfunction, seizures and epilepsy. Semin. Cell Dev. Biol. 2015, 38, 26–34. [Google Scholar] [CrossRef]
- Friedman, A. Blood–brain barrier dysfunction, status epilepticus, seizures, and epilepsy: A puzzle of a chicken and egg? Epilepsia 2011, 52, 19–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFβ signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Marchi, N.; Granata, T.; Ghosh, C.; Janigro, D. Blood–brain barrier dysfunction and epilepsy: Pathophysiologic role and therapeutic approaches. Epilepsia 2012, 53, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, E.; da Costa Araujo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J. Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Seiffert, E.; Dreier, J.P.; Ivens, S.; Bechmann, I.; Tomkins, O.; Heinemann, U.; Friedman, A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef] [Green Version]
- Ivens, S.; Kaufer, D.; Flores, L.P.; Bechmann, I.; Zumsteg, D.; Tomkins, O.; Seiffert, E.; Heinemann, U.; Friedman, A. TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007, 130, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Tomkins, O.; Friedman, O.; Ivens, S.; Reiffurth, C.; Major, S.; Dreier, J.; Heinemann, U.; Friedman, A. Blood–brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol. Dis. 2007, 25, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Marchi, N.; Oby, E.; Batra, A.; Uva, L.; De Curtis, M.; Hernandez, N.; Van Boxel-Dezaire, A.; Najm, I.; Janigro, D. In vivo and in vitro effects of pilocarpine: Relevance to ictogenesis. Epilepsia 2007, 48, 1934–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, N.; Fan, Q.; Ghosh, C.; Fazio, V.; Bertolini, F.; Betto, G.; Batra, A.; Carlton, E.; Najm, I.; Granata, T. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol. Dis. 2009, 33, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, A.; Golan, H.; Melamed, I.; Pascual-Marqui, R.; Friedman, A. Focal cortical dysfunction and blood–brain barrier disruption in patients with postconcussion syndrome. J. Clin. Neurophysiol. 2005, 22, 1–9. [Google Scholar] [CrossRef]
- Tomkins, O.; Shelef, I.; Kaizerman, I.; Eliushin, A.; Afawi, Z.; Misk, A.; Gidon, M.; Cohen, A.; Zumsteg, D.; Friedman, A. Blood–brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 2008, 79, 774–777. [Google Scholar] [CrossRef] [Green Version]
- Tomkins, O.; Feintuch, A.; Benifla, M.; Cohen, A.; Friedman, A.; Shelef, I. Blood-brain barrier breakdown following traumatic brain injury: A possible role in posttraumatic epilepsy. Cardiovasc. Psychiatry Neurol. 2011, 2011, 765923. [Google Scholar] [CrossRef] [Green Version]
- Bolwig, T.G.; Hertz, M.M.; Westergaard, E. Acute hypertension causing blood-brain barrier breakdown during epileptic seizures. Acta Neurol. Scand. 1977, 56, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Nilsson, B. The pathophysiology of the blood-brain barrier dysfunction induced by severe hypercapnia and by epileptic brain activity. Acta Neuropathol. 1977, 38, 153–158. [Google Scholar] [CrossRef]
- Petito, C.K.; Schaefer, J.A.; Plum, F. Ultrastructural characteristics of the brain and blood-brain barrier in experimental seizures. Brain Res. 1977, 127, 251–267. [Google Scholar] [CrossRef]
- Ravizza, T.; Gagliardi, B.; Noé, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Ndode-Ekane, X.; Hayward, N.; Gröhn, O.; Pitkänen, A. Vascular changes in epilepsy: Functional consequences and association with network plasticity in pilocarpine-induced experimental epilepsy. Neuroscience 2010, 166, 312–332. [Google Scholar] [CrossRef] [PubMed]
- Leroy, C.; Roch, C.; Koning, E.; Namer, I.J.; Nehlig, A. In the lithium–pilocarpine model of epilepsy, brain lesions are not linked to changes in blood–brain barrier permeability: An autoradiographic study in adult and developing rats. Exp. Neurol. 2003, 182, 361–372. [Google Scholar] [CrossRef]
- Van Vliet, E.; Otte, W.; Gorter, J.; Dijkhuizen, R.; Wadman, W. Longitudinal assessment of blood–brain barrier leakage during epileptogenesis in rats. A quantitative MRI study. Neurobiol. Dis. 2014, 63, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zucker, D.K.; Wooten, G.F.; Lothman, E.W. Blood-brain barrier changes with kainic acid-induced limbic seizures. Exp. Neurol. 1983, 79, 422–433. [Google Scholar] [CrossRef]
- Ruth, R. Increased cerebrovascular permeability to protein during systemic kainic acid seizures. Epilepsia 1984, 25, 259–268. [Google Scholar] [CrossRef]
- Lassmann, H.; Petsche, U.; Kitz, K.; Baran, H.; Sperk, G.; Seitelberger, F.; Hornykiewicz, O. The role of brain edema in epileptic brain damage induced by systemic kainic acid injection. Neuroscience 1984, 13, 691–704. [Google Scholar] [CrossRef]
- Marchi, N.; Johnson, A.J.; Puvenna, V.; Johnson, H.L.; Tierney, W.; Ghosh, C.; Cucullo, L.; Fabene, P.F.; Janigro, D. Modulation of peripheral cytotoxic cells and ictogenesis in a model of seizures. Epilepsia 2011, 52, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.-M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef] [Green Version]
- Michalak, Z.; Sano, T.; Engel, T.; Miller-Delaney, S.F.; Lerner-Natoli, M.; Henshall, D.C. Spatio-temporally restricted blood–brain barrier disruption after intra-amygdala kainic acid-induced status epilepticus in mice. Epilepsy Res. 2013, 103, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.W.; Merchut, M.; Fine, M.; Azar-Kia, B. Complex partial seizure-induced transient MR enhancement. J. Comput. Assist. Tomogr. 1992, 16, 814–816. [Google Scholar] [CrossRef]
- Alvarez, V.; Maeder, P.; Rossetti, A.O. Postictal blood-brain barrier breakdown on contrast-enhanced MRI. Epilepsy Behav. 2010, 17, 302–303. [Google Scholar] [CrossRef]
- Rüber, T.; David, B.; Lüchters, G.; Nass, R.D.; Friedman, A.; Surges, R.; Stöcker, T.; Weber, B.; Deichmann, R.; Schlaug, G.; et al. Evidence for peri-ictal blood-brain barrier dysfunction in patients with epilepsy. Brain 2018, 141, 2952–2965. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Gee, M.N.; Swartz, B.E.; Mandelkern, M.A.; Blahd, W.H.; Landaw, E.M.; Delgado-Escueta, A.V. Dynamic [18F]fluorodeoxyglucose positron emission tomography and hypometabolic zones in seizures: Reduced capillary influx. Ann. Neurol. 1998, 43, 801–808. [Google Scholar] [CrossRef]
- Mihály, A.; Bozóky, B. Immunohistochemical localization of extravasated serum albumin in the hippocampus of human subjects with partial and generalized epilepsies and epileptiform convulsions. Acta Neuropathol. 1984, 65, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Hyman, S.; Cornford, M.E.; Landaw, E.M.; Delgado-Escueta, A.V. Interictal seizure resections show two configurations of endothelial Glut1 glucose transporter in the human blood-brain barrier. J. Cereb. Blood Flow Metab. 1998, 18, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Janigro, D. Blood-brain barrier, ion homeostatis and epilepsy: Possible implications towards the understanding of ketogenic diet mechanisms. Epilepsy Res. 1999, 37, 223–232. [Google Scholar] [CrossRef]
- Marchi, N.; Angelov, L.; Masaryk, T.; Fazio, V.; Granata, T.; Hernandez, N.; Hallene, K.; Diglaw, T.; Franic, L.; Najm, I.; et al. Seizure-promoting effect of blood-brain barrier disruption. Epilepsia 2007, 48, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Oby, E.; Janigro, D. The blood-brain barrier and epilepsy. Epilepsia 2006, 47, 1761–1774. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Trifiletti, R.R.; Jacobson, R.I.; Ronen, G.M.; Behmand, R.A.; Harik, S.I. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N. Engl. J. Med. 1991, 325, 703–709. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Leary, L.; Wang, D. Glucose transporter 1 deficiency syndrome and other glycolytic defects. J. Child. Neurol. 2002, 17 (Suppl. S3), 3S15-23; discussion 13S24-15. [Google Scholar]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef]
- Maes, M.; Vojdani, A.; Geffard, M.; Moreira, E.G.; Barbosa, D.S.; Michelin, A.P.; Semeão, L.O.; Sirivichayakul, S.; Kanchanatawan, B. Schizophrenia phenomenology comprises a bifactorial general severity and a single-group factor, which are differently associated with neurotoxic immune and immune-regulatory pathways. Biomol. Concepts 2019, 10, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Sirivichayakul, S.; Kanchanatawan, B.; Vodjani, A. Breakdown of the Paracellular Tight and Adherens Junctions in the Gut and Blood Brain Barrier and Damage to the Vascular Barrier in Patients with Deficit Schizophrenia. Neurotox. Res. 2019, 36, 306–322. [Google Scholar] [CrossRef]
- Raza, M.W.; Shad, A.; Pedler, S.J.; Karamat, K.A. Penetration and activity of antibiotics in brain abscess. J. Coll. Physicians Surg. Pak. 2005, 15, 165–167. [Google Scholar] [PubMed]
- Yang, R.C.; Qu, X.Y.; Xiao, S.Y.; Li, L.; Xu, B.J.; Fu, J.Y.; Lv, Y.J.; Amjad, N.; Tan, C.; Kim, K.S.; et al. Meningitic Escherichia coli-induced upregulation of PDGF-B and ICAM-1 aggravates blood-brain barrier disruption and neuroinflammatory response. J. Neuroinflamm. 2019, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- Thorsdottir, S.; Henriques-Normark, B.; Iovino, F. The Role of Microglia in Bacterial Meningitis: Inflammatory Response, Experimental Models and New Neuroprotective Therapeutic Strategies. Front. Microbiol. 2019, 10, 576. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Prakash, R.; Carmichael, S.T. Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr. Opin. Neurol. 2015, 28, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef]
- Nag, S.; Manias, J.L.; Stewart, D.J. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009, 118, 197–217. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nagai, N.; Umemura, K. A Review of the Mechanisms of Blood-Brain Barrier Permeability by Tissue-Type Plasminogen Activator Treatment for Cerebral Ischemia. Front. Cell Neurosci. 2016, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Roy, H.; Bhardwaj, S.; Ylä-Herttuala, S. Biology of vascular endothelial growth factors. FEBS Lett. 2006, 580, 2879–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xia, R.; Jiang, Y.; Wang, L.; Gao, F. Vascular endothelial growth factors enhance the permeability of the mouse blood-brain barrier. PLoS ONE 2014, 9, e86407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jośko, J. Cerebral angiogenesis and expression of VEGF after subarachnoid hemorrhage (SAH) in rats. Brain Res. 2003, 981, 58–69. [Google Scholar] [CrossRef]
- Lee, C.; Agoston, D.V. Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J. Neurotrauma 2010, 27, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Sköld, M.K.; von Gertten, C.; Sandberg-Nordqvist, A.C.; Mathiesen, T.; Holmin, S. VEGF and VEGF receptor expression after experimental brain contusion in rat. J. Neurotrauma 2005, 22, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, R.; Ago, T.; Kamouchi, M.; Kuroda, J.; Kuwashiro, T.; Hata, J.; Sugimori, H.; Fukuda, K.; Gotoh, S.; Makihara, N.; et al. Clinical significance of plasma VEGF value in ischemic stroke—Research for biomarkers in ischemic stroke (REBIOS) study. BMC Neurol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, P.M.; Jackson, E.K.; Wisniewski, S.R.; Clark, R.S.; Adelson, P.D.; Kochanek, P.M. Vascular endothelial growth factor is increased in cerebrospinal fluid after traumatic brain injury in infants and children. Neurosurgery 2004, 54, 605–611. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, R.; Nakase, H.; Tamaki, R.; Sakaki, T. Vascular endothelial growth factor antagonist reduces brain edema formation and venous infarction. Stroke 2005, 36, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, M.; Igarashi, H.; Kawamura, K.; Takahashi, T.; Kakita, A.; Takahashi, H.; Nakada, T.; Nishizawa, M.; Shimohata, T. Inhibition of VEGF signaling pathway attenuates hemorrhage after tPA treatment. J. Cereb. Blood Flow Metab. 2011, 31, 1461–1474. [Google Scholar] [CrossRef]
- Lange, C.; Storkebaum, E.; de Almodóvar, C.R.; Dewerchin, M.; Carmeliet, P. Vascular endothelial growth factor: A neurovascular target in neurological diseases. Nat. Rev. Neurol. 2016, 12, 439–454. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A.; Tepper, D.; Leonard, A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front. Neurol. 2013, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Guo, S.; Lok, J.; Navaratna, D.; Whalen, M.J.; Kim, K.W.; Lo, E.H. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr. Pharm. Des. 2012, 18, 3645–3648. [Google Scholar] [CrossRef]
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 2006, 12, 441–445. [Google Scholar] [CrossRef]
- Sifringer, M.; Stefovska, V.; Zentner, I.; Hansen, B.; Stepulak, A.; Knaute, C.; Marzahn, J.; Ikonomidou, C. The role of matrix metalloproteinases in infant traumatic brain injury. Neurobiol. Dis. 2007, 25, 526–535. [Google Scholar] [CrossRef]
- Trujillo, K.A.; Akil, H. Opioid and non-opioid behavioral actions of dynorphin A and the dynorphin analogue DAKLI. NIDA Res. Monogr. 1990, 105, 397–398. [Google Scholar]
- Sood, R.R.; Taheri, S.; Candelario-Jalil, E.; Estrada, E.Y.; Rosenberg, G.A. Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J. Cereb. Blood Flow Metab. 2008, 28, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Jin, S.M.; Kim, H.J.; Lee, Y.C. Matrix metalloproteinase inhibitor regulates inflammatory cell migration by reducing ICAM-1 and VCAM-1 expression in a murine model of toluene diisocyanate-induced asthma. J. Allergy Clin. Immunol. 2003, 111, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Golub, L.M.; Ramamurthy, N.; McNamara, T.F.; Gomes, B.; Wolff, M.; Casino, A.; Kapoor, A.; Zambon, J.; Ciancio, S.; Schneir, M.; et al. Tetracyclines inhibit tissue collagenase activity. J. Periodontal. Res. 1984, 19, 651–655. [Google Scholar] [CrossRef]
- Brundula, V.; Rewcastle, N.B.; Metz, L.M.; Bernard, C.C.; Yong, V.W. Targeting leukocyte MMPs and transmigration: Minocycline as a potential therapy for multiple sclerosis. Brain 2002, 125, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.W.; Chung, S.S.; Chung, S.K. Endothelial endothelin-1 over-expression using receptor tyrosine kinase tie-1 promoter leads to more severe vascular permeability and blood brain barrier breakdown after transient middle cerebral artery occlusion. Brain Res. 2009, 1266, 121–129. [Google Scholar] [CrossRef]
- Zhang, X.; Yeung, P.K.; McAlonan, G.M.; Chung, S.S.; Chung, S.K. Transgenic mice over-expressing endothelial endothelin-1 show cognitive deficit with blood-brain barrier breakdown after transient ischemia with long-term reperfusion. Neurobiol. Learn. Mem. 2013, 101, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Yeung, P.K.; Shen, J.; Chung, S.S.; Chung, S.K. Targeted over-expression of endothelin-1 in astrocytes leads to more severe brain damage and vasospasm after subarachnoid hemorrhage. BMC Neurosci. 2013, 14, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zheng, M.; Shimoni, O.; Banks, W.A.; Bush, A.I.; Gamble, J.R.; Shi, B. Development of Novel Therapeutics Targeting the Blood–Brain Barrier: From Barrier to Carrier. Adv. Sci. 2021, 8, 2101090. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Choi, J.; Ting, K.K.; Coleman, P.; Chen, J.; Cogger, V.C.; Wan, L.; Shi, Z.; Moller, T.; et al. Targeting miR-27a/VE-cadherin interactions rescues cerebral cavernous malformations in mice. PLoS Biol. 2020, 18, e3000734. [Google Scholar] [CrossRef]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B.; et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivandzade, F.; Alqahtani, F.; Sifat, A.; Cucullo, L. The cerebrovascular and neurological impact of chronic smoking on post-traumatic brain injury outcome and recovery: An in vivo study. J. Neuroinflamm. 2020, 17, 133. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Förster, C.; Waschke, J.; Burek, M.; Leers, J.; Drenckhahn, D. Glucocorticoid effects on mouse microvascular endothelial barrier permeability are brain specific. J. Physiol. 2006, 573, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; McDonald, M.C.; Mazzon, E.; Filipe, H.M.; Costantino, G.; Caputi, A.P.; Thiemermann, C. Beneficial effects of tempol, a membrane-permeable radical scavenger, in a rodent model of splanchnic artery occlusion and reperfusion. Shock 2000, 14, 150–156. [Google Scholar] [CrossRef]
- Rak, R.; Chao, D.L.; Pluta, R.M.; Mitchell, J.B.; Oldfield, E.H.; Watson, J.C. Neuroprotection by the stable nitroxide Tempol during reperfusion in a rat model of transient focal ischemia. J. Neurosurg. 2000, 92, 646–651. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Contreras, O.A.; Greineder, C.F.; Kiseleva, R.Y.; Parhiz, H.; Walsh, L.R.; Zuluaga-Ramirez, V.; Myerson, J.W.; Hood, E.D.; Villa, C.H.; Tombacz, I.; et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2020, 117, 3405. [Google Scholar] [CrossRef]
- Shuvaev, V.V.; Han, J.; Tliba, S.; Arguiri, E.; Christofidou-Solomidou, M.; Ramirez, S.H.; Dykstra, H.; Persidsky, Y.; Atochin, D.N.; Huang, P.L.; et al. Anti-inflammatory effect of targeted delivery of SOD to endothelium: Mechanism, synergism with NO donors and protective effects in vitro and in vivo. PLoS ONE 2013, 8, e77002. [Google Scholar] [CrossRef] [PubMed]
- Lutton, E.M.; Razmpour, R.; Andrews, A.M.; Cannella, L.A.; Son, Y.J.; Shuvaev, V.V.; Muzykantov, V.R.; Ramirez, S.H. Acute administration of catalase targeted to ICAM-1 attenuates neuropathology in experimental traumatic brain injury. Sci. Rep. 2017, 7, 3846. [Google Scholar] [CrossRef] [Green Version]
- Manickam, D.S.; Brynskikh, A.M.; Kopanic, J.L.; Sorgen, P.L.; Klyachko, N.L.; Batrakova, E.V.; Bronich, T.K.; Kabanov, A.V. Well-defined cross-linked antioxidant nanozymes for treatment of ischemic brain injury. J. Control. Release 2012, 162, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Senatorov, V.V., Jr.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Dilena, R.; Mauri, E.; Aronica, E.; Bernasconi, P.; Bana, C.; Cappelletti, C.; Carrabba, G.; Ferrero, S.; Giorda, R.; Guez, S.; et al. Therapeutic effect of Anakinra in the relapsing chronic phase of febrile infection-related epilepsy syndrome. Epilepsia Open 2019, 4, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Kenney-Jung, D.L.; Vezzani, A.; Kahoud, R.J.; LaFrance-Corey, R.G.; Ho, M.L.; Muskardin, T.W.; Wirrell, E.C.; Howe, C.L.; Payne, E.T. Febrile infection-related epilepsy syndrome treated with anakinra. Ann. Neurol. 2016, 80, 939–945. [Google Scholar] [CrossRef]
- Prasad, S.; Liles, T.; Cucullo, L. Chapter 44—Brain, Nrf2, and Tobacco: Mechanisms and Countermechanisms Underlying Oxidative-Stress-Mediated Cerebrovascular Effects of Cigarette Smoking. In Neuroscience of Nicotine; Preedy, V.R., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 355–363. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro. 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajja, R.K.; Green, K.N.; Cucullo, L. Altered Nrf2 Signaling Mediates Hypoglycemia-Induced Blood–Brain Barrier Endothelial Dysfunction In Vitro. PLoS ONE 2015, 10, e0122358. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Ramirez, S.H.; Sato, S.; Kiyota, T.; Cerny, R.L.; Kaibuchi, K.; Persidsky, Y.; Ikezu, T. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am. J. Pathol. 2008, 172, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Park, J.C.; Baik, S.H.; Han, S.H.; Cho, H.J.; Choi, H.; Kim, H.J.; Choi, H.; Lee, W.; Kim, D.K.; Mook-Jung, I. Annexin A1 restores Aβ(1-42) -induced blood-brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell 2017, 16, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; McLaurin, J. Rho-associated protein kinases as therapeutic targets for both vascular and parenchymal pathologies in Alzheimer’s disease. J. Neurochem. 2018, 144, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.L.; Srivastava, K.; Sprigg, N.; Bath, P.M.; Bayraktutan, U. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J. Neurochem. 2014, 129, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Zheng, Y.; von Bornstadt, D.; Wei, Y.; Balcioglu, A.; Daneshmand, A.; Yalcin, N.; Yu, E.; Herisson, F.; Atalay, Y.B.; et al. Selective ROCK2 Inhibition In Focal Cerebral Ischemia. Ann. Clin. Transl. Neurol. 2014, 1, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herskowitz, J.H.; Feng, Y.; Mattheyses, A.L.; Hales, C.M.; Higginbotham, L.A.; Duong, D.M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Seyfried, N.T.; et al. Pharmacologic inhibition of ROCK2 suppresses amyloid-β production in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 19086–19098. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R. The blood-brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration summarizing some of the major neurological disorders associated with impairment of the BBB. Note that in some cases, there is a clear, direct causative association where impairment of the BBB is the major prodromal factor for the onset and/or progression of the CNS disorder (e.g., post-ischemic brain injury). In other instances, whether the BBB damage is the causative factor or a derivate effect of the brain disorder further impacting the disease at a later stage is less clear (e.g., Epilepsy).
Figure 1.
Schematic illustration summarizing some of the major neurological disorders associated with impairment of the BBB. Note that in some cases, there is a clear, direct causative association where impairment of the BBB is the major prodromal factor for the onset and/or progression of the CNS disorder (e.g., post-ischemic brain injury). In other instances, whether the BBB damage is the causative factor or a derivate effect of the brain disorder further impacting the disease at a later stage is less clear (e.g., Epilepsy).

Figure 2.
Schematic illustration of the BBB anatomy. A cross-section of a brain microcapillary segment depicting the innermost luminal compartment composed of a uniform layer of a tightly packed endothelial cell (EC) surrounded by an additional envelope of pericytes (embedded within the basal membrane and astrocytic foot processes which tightly ensheaths the brain microcapillary. The movement of substances across the BBB endothelium is controlled by a multimodal barrier that includes tight junctions (gating barrier to paracellular diffusion of polar molecules); efflux transporters (P-gp, MRPs, BCRP, etc.) with high affinity for lipophilic substances such as cytochrome P450 enzymes, MAO, etc. (metabolic/enzymatic barrier).
Figure 2.
Schematic illustration of the BBB anatomy. A cross-section of a brain microcapillary segment depicting the innermost luminal compartment composed of a uniform layer of a tightly packed endothelial cell (EC) surrounded by an additional envelope of pericytes (embedded within the basal membrane and astrocytic foot processes which tightly ensheaths the brain microcapillary. The movement of substances across the BBB endothelium is controlled by a multimodal barrier that includes tight junctions (gating barrier to paracellular diffusion of polar molecules); efflux transporters (P-gp, MRPs, BCRP, etc.) with high affinity for lipophilic substances such as cytochrome P450 enzymes, MAO, etc. (metabolic/enzymatic barrier).

Figure 3.
Schematic illustration summarizing the effects of brain damage on BBB integrity. The production and activation of MMPs, VEGFs, and ETs are upregulated in various brain cells following brain damage. These factors can then impair the viability and integrity of the BBB by negatively impacting the tight junctions between adjacent endothelial cells.
Figure 3.
Schematic illustration summarizing the effects of brain damage on BBB integrity. The production and activation of MMPs, VEGFs, and ETs are upregulated in various brain cells following brain damage. These factors can then impair the viability and integrity of the BBB by negatively impacting the tight junctions between adjacent endothelial cells.

Figure 4.
Structure of blood-brain barrier (BBB) and potential therapeutic targets to restore the BBB viability. Targeting angiogenesis, oxidative stress, cytoskeleton reorganization, and inflammation can effectively protect and potentially restore the viability of the BBB. This includes MMPs and angiogenesis inhibitors (e.g., antibodies targeting VEGF/VEGFR), oxidative stress inhibitors/reducers (such as NOX inhibitors and Nrf2 enhancers) that are promoters of cytoskeleton reorganization (e.g., ROCK inhibitors). Blocking inflammation by targeting immune cells can prevent their recruitment, thus protecting the BBB.
Figure 4.
Structure of blood-brain barrier (BBB) and potential therapeutic targets to restore the BBB viability. Targeting angiogenesis, oxidative stress, cytoskeleton reorganization, and inflammation can effectively protect and potentially restore the viability of the BBB. This includes MMPs and angiogenesis inhibitors (e.g., antibodies targeting VEGF/VEGFR), oxidative stress inhibitors/reducers (such as NOX inhibitors and Nrf2 enhancers) that are promoters of cytoskeleton reorganization (e.g., ROCK inhibitors). Blocking inflammation by targeting immune cells can prevent their recruitment, thus protecting the BBB.


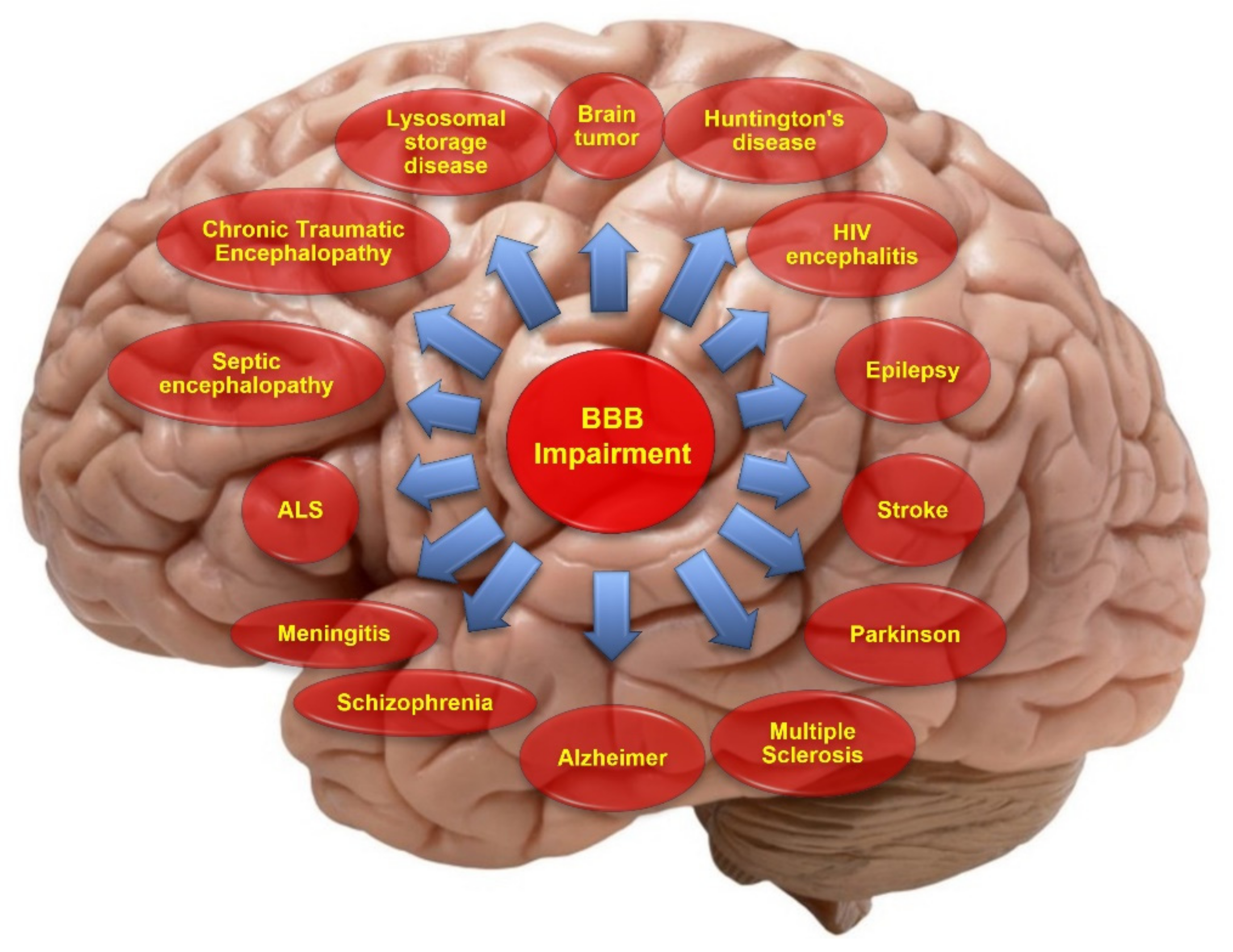{kind=link}
{kind=link}
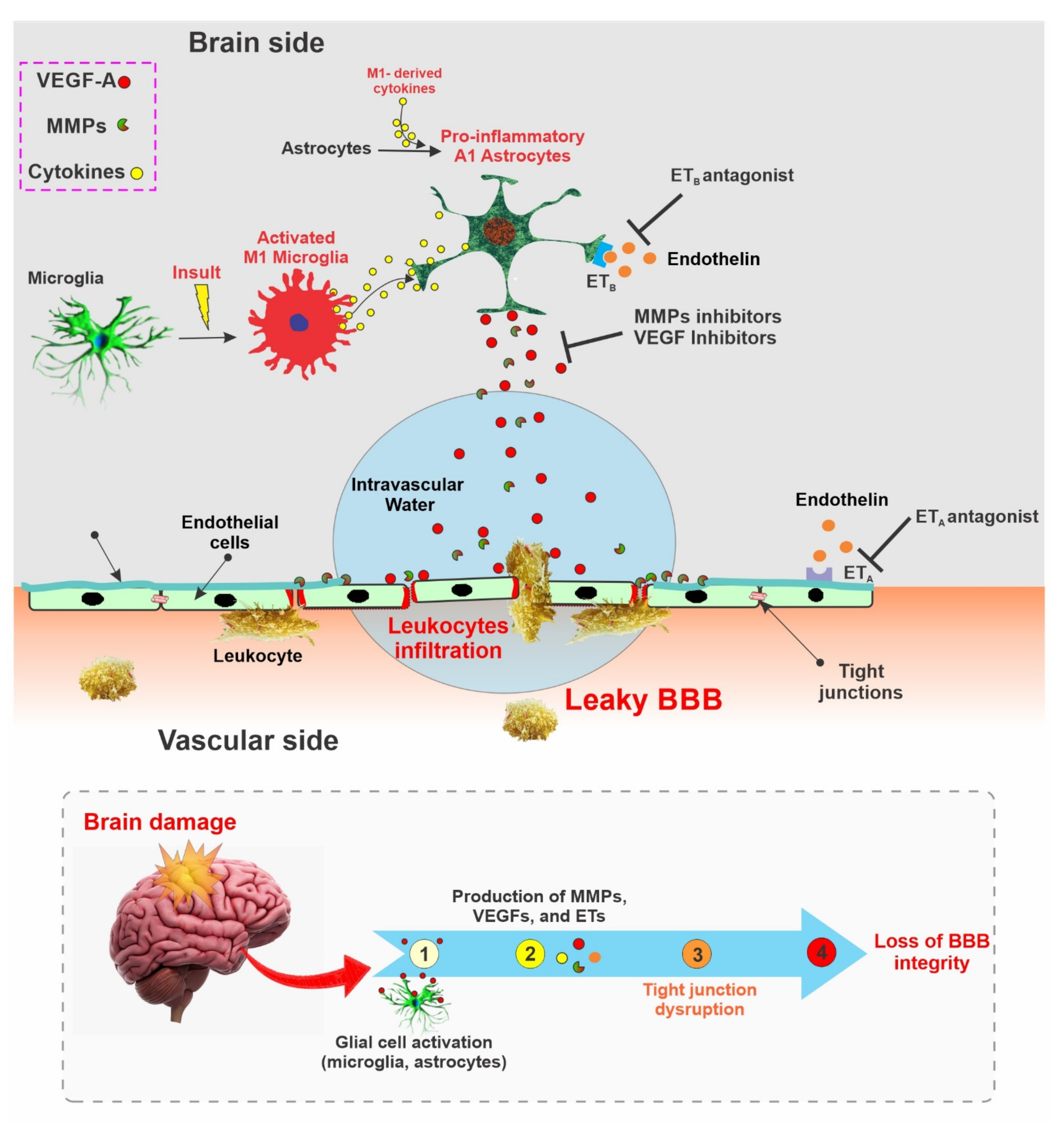{kind=link}
{kind=link}
Table 1.
Blood-Brain Barrier disruption associated with different disease conditions and potential therapeutic targets.
Table 1.
Blood-Brain Barrier disruption associated with different disease conditions and potential therapeutic targets.
| Pathophysiology of BBB Disruption | Associated Diseases | Therapeutic Targets | Ref. |
|---|---|---|---|
| Upregulation of VEGF and Activation of VEGFR2 | ALS, AD, PD, epilepsy, ischemic stroke | anti-VEGF antibody and VEGFR 2 inhibitor (e.g., SU5416) | [99] |
| eNOS activation | Stroke and TBI | selective eNOS inhibitor (e.g., cavtratin) | [100] |
| Upregulation of MMPs | Schizophrenia, Stroke, and TBI | Inhibition of MMPs (e.g., GM6001) | [101] |
| Activation of endothelin receptors, ETA, ETB | Stroke and epilepsy | Inhibition of ETA and ETB (e.g., S-0139, BQ788) | [102,103,104] |
| Downregulation of VE-Cadherine | MS, Stroke | Inhibition of miR-27a/VE by CD5-2; antibody against ICAM (Enlimomab) | [105] |
| Disorganization of adherens junctions | MS, Stroke | Stabilize the adherens junctions using sphingosine-1-phosphate (S1P) | [106] |
| Reduced expression of TJ proteins | Depression, Stroke, Stress, | Induction of TJs protein expression. (e.g., antisense oligonucleotide for miR-501-3p, HDAC1 inhibitor, MS-275 -Entinostat) | [107,108] |
| Imbalance of AMP-activated protein kinase pathway | Stroke | Activation of AMP-activated protein kinase pathway by melatonin | [109] |
| Activation or upregulation of inflammatory cytokines e.g., TNF-β, IL-1β, TNF-α and IL-6 | Stroke, TBI, Cognitive impairments, Seizures, MS | Inhibition of inflammatory cytokines (e.g., etanercept, anti-IL-6 antibody, and natalizumab) | [110,111,112,113] |
| Oxidative stress induction by activation of NOX4 and NOX5 | Stroke | selective inhibition of NOX4 or NOX5 (GKT136901 and ML090) | [114,115] |
| Contraction of actin-myosin cytoskeleton via myosin light chain Phosphorylation | Stroke | Inhibition of RhoA/Rho-associated proteins kinase (ROCK). e.g., fasudil | [116,117] |
| Upregulation of MMPs | Stroke, TBI and Schizophrenia | Inhibition of MMP2/9 (e.g., SB-3CT) | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111779
AMA Style
Archie SR, Al Shoyaib A, Cucullo L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics. 2021; 13(11):1779. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111779
Chicago/Turabian StyleArchie, Sabrina Rahman, Abdullah Al Shoyaib, and Luca Cucullo. 2021. "Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview" Pharmaceutics 13, no. 11: 1779. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111779
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.