Metagenomic Insights of the Root Colonizing Microbiome Associated with Symptomatic and Non-Symptomatic Bananas in Fusarium Wilt Infected Fields

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. General Characteristics of the Amplicon
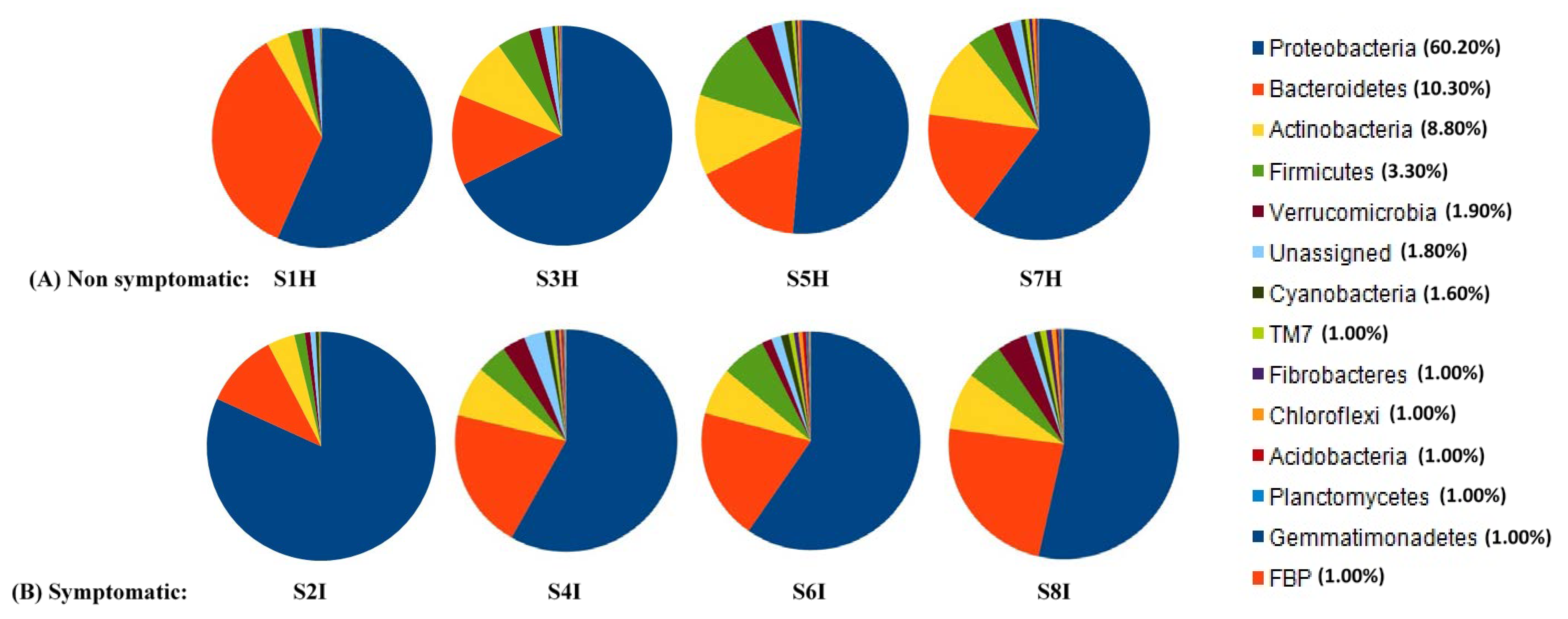
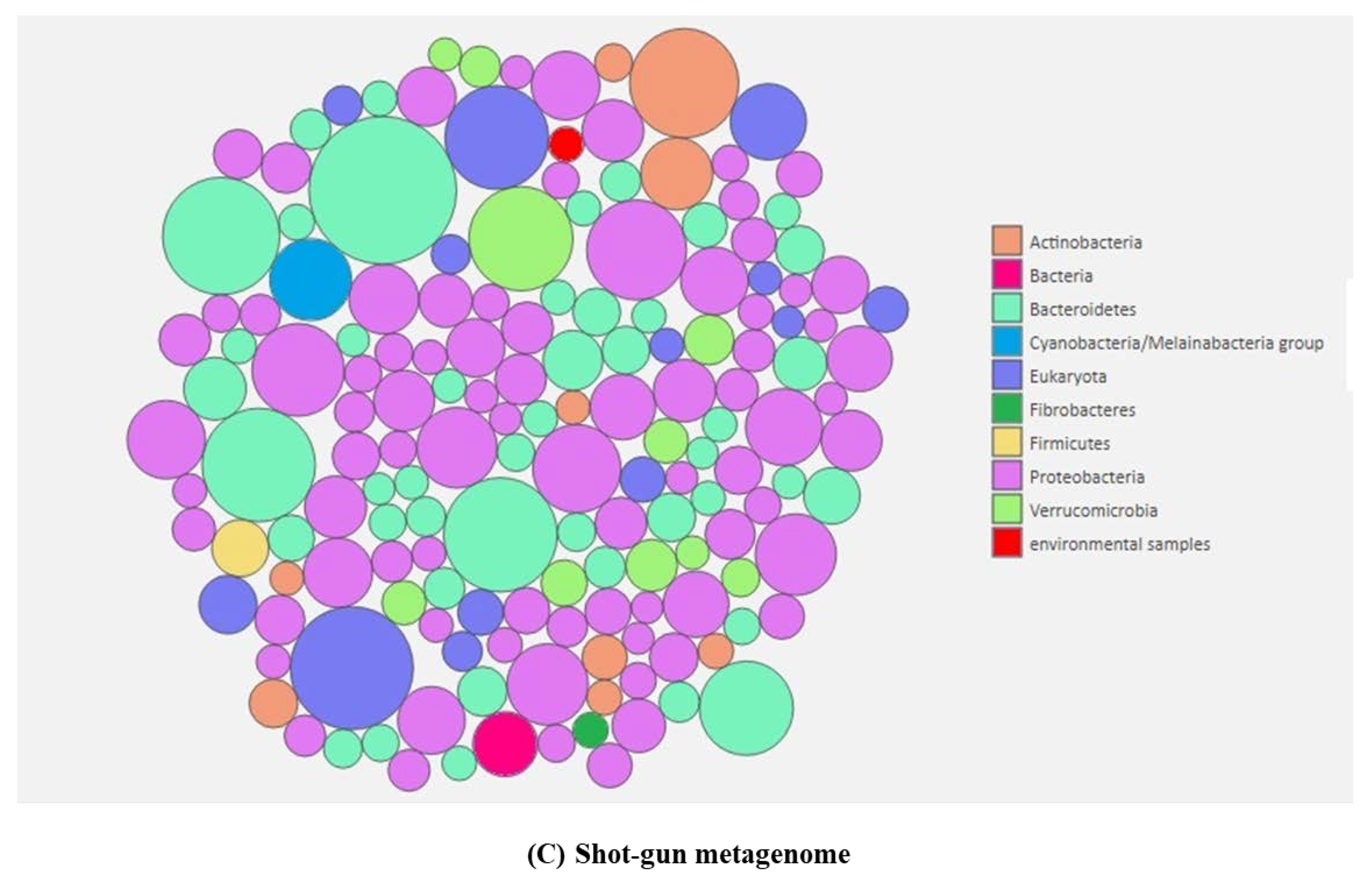
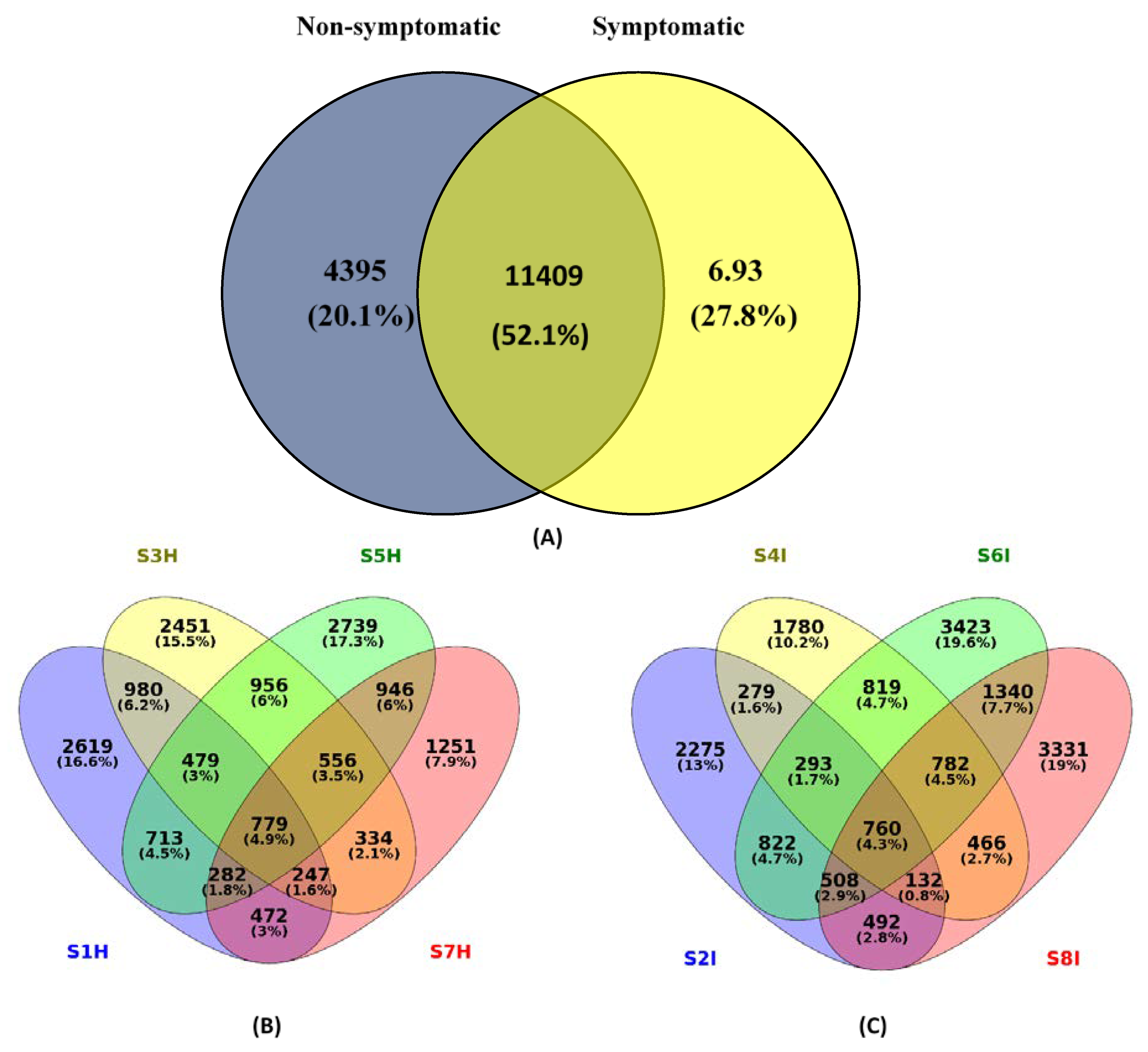
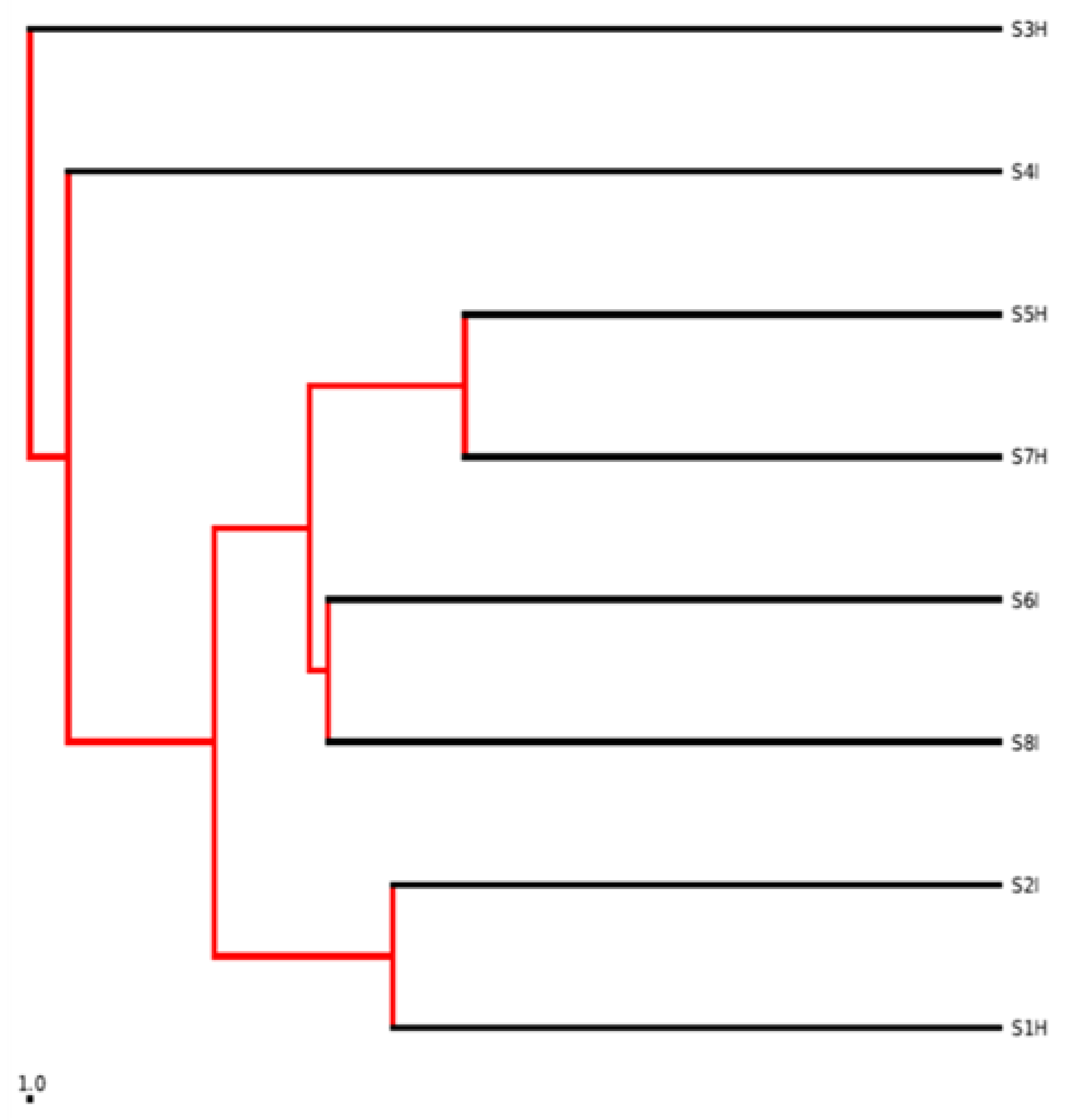
2.2. Root Associated Microbiome Composition and Structure in Symptomatic and Non-Symptomatic Banana
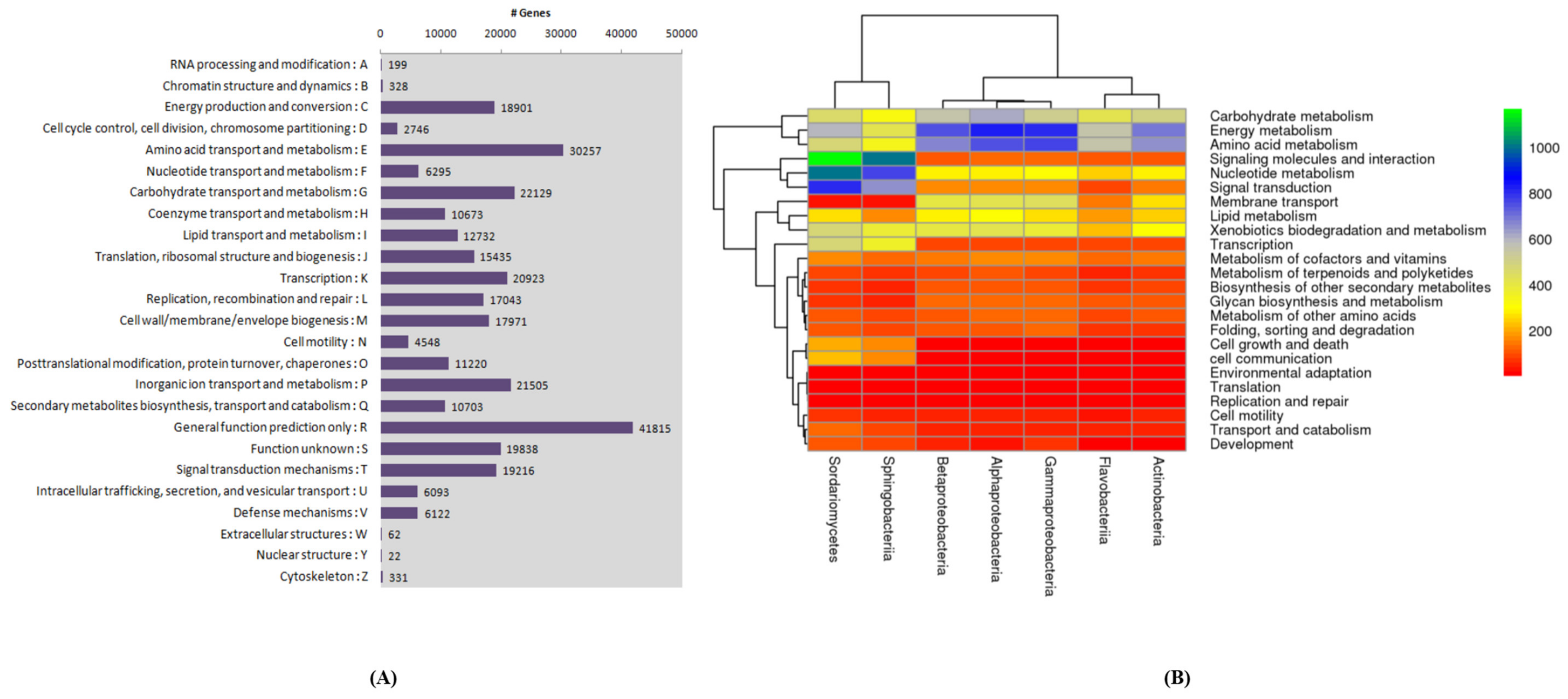
2.3. Growth Ameliorating Endophytes and Gene Function
3. Discussion
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. PCR Amplification of 16S rRNA Gene
4.3. Bioinformatic Analysis of 16s Results
4.4. Sequencing, Assembly and Binning of the Pathogenesis-Associated Metagenome
4.5. Metagenome Assembly and Gene Prediction
4.6. Taxonomic Classification
4.7. Functional Annotation
4.8. Annotation from COG Database and KEGG
4.9. Nucleotide Sequence Accession Numbers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Massart, S.; Perazzolli, M.; Höfte, M.; Pertot, I.; Jijakli, M.H. Impact of the omic technologies for understanding the modes of action of biological control agents against plant pathogens. Biocontrol 2015, 60, 725–746. [Google Scholar] [CrossRef]
- Chutulo, E.C.; Chalannavar, R.K. Endophytic mycoflora and their bioactive compounds from Azadirachta indica: A comprehensive review. J. Fungi 2018, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, M.; Kumar, A.; Kaushal, R. Bacillus pumilus strain YSPMK11 as plant growth promoter and biocontrol agent against Sclerotinia sclerotiorum. 3 Biotech 2017, 7, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proença, D.N.; Francisco, R.; Kublik, S.; Schöler, A.; Vestergaard, G.; Schloter, M.; Morais, P.V. The microbiome of endophytic, wood colonizing bacteria from Pine trees as affected by Pine wilt disease. Sci. Rep. 2017, 7, 4205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, A.; Tan, H.; Cao, L.; Zhang, R. Engineering banana endosphere microbiome to improve Fusarium wilt resistance in banana. Microbiome 2019, 7, 74. [Google Scholar] [CrossRef]
- Zhou, D.; Jing, T.; Chen, Y.; Wang, F.; Qi, D.; Feng, R.; Xie, J.; Li, H. Deciphering microbial diversity associated with Fusarium wilt-diseased and disease-free banana rhizosphere soil. BMC Microbiol. 2019, 19, 161. [Google Scholar] [CrossRef] [Green Version]
- Perrier, X.; Jenny, C.; Bakry, F.; Karamura, D.; Kitavi, M.; Dubois, C.; Hervouet, C.; Philippson, G.; De Langhe, E. East African diploid and triploid bananas: A genetic complex transported from South-East Asia. Ann. Bot. 2019, 123, 19–36. [Google Scholar] [CrossRef] [Green Version]
- Onyango, M.; Karamura, D.; Keeley, S.; Manshardt, R.; Haymer, D. Morphological characterisation of East African AAB and AA dessert bananas (Musa spp.). ISHS Acta Hort. 2011, 897, 95–105. [Google Scholar] [CrossRef]
- Swennen, R.; Blomme, G.; van Asten, P.; Lepoint, P.; Karamura, E.B.; Njukwe, E.; Tinzaara, W.; Viljoen, A.; Karangwa, P.; Coyne, D.L.; et al. Mitigating the impact of biotic constraints to build resilient banana systems in Central and Eastern Africa. In Agroecological Intensification of Farming Systems in the East and Central African Highlands; Vanlauwe, B., van Asten, P., Blomme, G., Eds.; Routledge: Oxon, UK, 2013; pp. 85–104. [Google Scholar]
- Butler, D. Fungus threatens top banana. Nature 2013, 504, 195–196. [Google Scholar] [CrossRef] [Green Version]
- Dita, M.; Barquero, M.; Heck, D.; Mizubuti, E.S.G.; Staver, C.P. Fusarium wilt of banana: Current knowledge on epidemiology and research needs towards sustainable disease management. Front. Plant Sci. 2018, 9, 1468. [Google Scholar] [CrossRef] [Green Version]
- Karangwa, P.; Mostert, D.; Ndayihanzamaso, P.; Dubois, T.; Niere, B.; Felde, A.Z.; Schouten, A.; Blomme, G.; Beed, F.; Viljoen, A. Genetic Diversity of Fusarium oxysporum f. sp. cubense in East and Central Africa. Plant Disease 2018, 102, 552–560. [Google Scholar]
- Zhang, L.; Yuan, T.; Wang, Y.; Zhang, D.; Bai, T.; Xu, S.; Wang, Y.; Tang, W.; Zheng, S.-J. Identification and evaluation of resistance to Fusarium oxysporum f. sp. cubense tropical race 4 in Musa acuminata Pahang. Euphytica 2018, 214, 106. [Google Scholar]
- Zuo, C.; Deng, G.; Li, B.; Huo, H.; Li, C.; Hu, C.; Kuang, R.; Yang, Q.; Dong, T.; Sheng, Q.; et al. Germplasm screening of Musa spp. for resistance to Fusarium oxysporum f. sp. cubense tropical race 4 (Foc TR4). Eur. J. Plant Pathol. 2018, 151, 723–734. [Google Scholar]
- Kema, G.H.J. Panama Disease. Wageningen: Wageningen University and Research. Available online: https://fusariumwilt.org/ (accessed on 3 December 2018).
- Ploetz, R.C.; Pegg, K.G. Fusarium wilt. In Diseases of Banana, Abaca and Enset; Jones, D.R., Ed.; CABI Publishing: New York, NY, USA, 2000; pp. 143–172. [Google Scholar]
- Mallon, C.A.; Poly, F.; Le Roux, X.; Marring, I.; van Elsas, J.D.; Salles, J.F. Resource pulses can alleviate the biodiversity-invasion relationship in soil microbial communities. Ecology 2015, 96, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Rybakova, D.; Grube, M.; Koberl, M. The plant microbiome explored: Implications for experimental botany. J. Exp. Bot. 2016, 67, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Okubo, T.; Anda, M.; Nakashita, H.; Yasuda, M.; Sato, S.; Kaneko, T.; Tabata, S.; Eda, S.; Momiyama, A.; et al. Community- and genome-based views of plant-associated bacteria: Plant-bacterial interactions in soybean and rice. Plant Cell Physiol. 2010, 51, 1398–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhaimi, N.S.M.; Goh, S.Y.; Ajam, N.; Othman, R.Y.; Chan, K.G.; Thong, K.L. Diversity of microbiota associated with symptomatic and non-symptomatic bacterial wilt-diseased banana plants determined using 16S rRNA metagenome sequencing. World J. Microbiol. Biotechnol. 2017, 33, 168. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, L.; Aziz, W.N.W. Molecular Identification of Endophytic Fungi from Banana Leaves (Musa spp.). Trop. Life Sci. Res. 2018, 29, 201–211. [Google Scholar] [CrossRef]
- Aira, M.; Gomez-Brandon, M.; Lazcano, C.; Baath, E.; Dominguez, J. Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol. Biochem. 2010, 42, 2276–2281. [Google Scholar] [CrossRef]
- Liu, H.; Carvalhais, L.C.; Schenk, P.M.; Dennis, P.G. Effects of jasmonic acid signalling on the wheat microbiome differ between body sites. Sci. Rep. 2017, 7, 41766. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Singh, R.; Dubey, A.K. Diversity and applications of endophytic actinobacteria of plants in special and other ecological niches. Front. Microbiol. 2018, 9, 1767. [Google Scholar] [CrossRef] [PubMed]
- Kyselkova, M.; Kopecký, J.; Frapolli, M.; Défago, G.; Ságová-Marecková, M.; Grundmann, G.L.; Moënne-Loccoz, Y. Comparison of rhizobacterial community composition in soil suppressive or conducive to tobacco black root rot disease. ISME J. 2009, 3, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Xing, K.; Jiang, J.H.; Xu, L.H.; Li, W.J. Biodiversity, bioactive natural products and biotechnological potential of plant-associated endophytic actinobacteria. Appl. Microbiol. Bioethanol. 2011, 89, 457–473. [Google Scholar] [CrossRef]
- Tian, B.Y.; Yang, J.K.; Zhang, K.Q. Bacteria used in biological control of plant-parasitic nematodes: Populations, mechanisms of action, and future prospects. FEMS Microbiol. Ecol. 2007, 61, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, M.; Wani, S.P. Plant-growth-promoting rhizobacteria: Drought stress alleviators to ameliorate crop production in drylands. Ann. Microbiol. 2016, 66, 35–42. [Google Scholar] [CrossRef]
- Ferrara, F.I.D.; Zilda, M.O.; Gonzales, H.H.S.; Eny, I.S.F.; Heloiza, R.B. Endophytic and rhizospheric enterobacteria isolated from sugar cane have different potentials for producing plant growth-promoting substances. Plant Soil 2012, 353, 409–417. [Google Scholar] [CrossRef]
- Dita, M.; Schilly, A.; Vargas, J.; Chaves, N.; Guzman, M.; Sandoval, J.; Staver, C. Endophyte microbiome of banana roots reveals high diversity and potential for agricultural uses. In Proceedings of the 2014 International Conference on Research on Food Security, Natural Resource Management and Rural Development, Tropentag, Prague, Czech Republic, 17–19 September 2014. [Google Scholar]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root associated microbiome of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [Green Version]
- Lagaert, S.; Belien, T.; Volckaert, G. Plant cell walls: Protecting the barrier from degradation by microbial enzymes. Sem. Cell Dev. Biol. 2009, 20, 1064–1073. [Google Scholar] [CrossRef]
- Solomon, E.B.; Matthews, K.R. Use of fluorescent microspheres as tool to investigate bacterial interactions with growing plants. J. Food Prot. 2005, 68, 870–873. [Google Scholar] [CrossRef]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome: Global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef]
- Spaepen, S.; Vanderleyden, J. Auxin and plant-microbe interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a001438. [Google Scholar] [CrossRef] [Green Version]
- White, R.A.; Callister, S.J.; Moore, R.J.; Baker, E.S.; Jansson, J.K. The past, present and future of microbiome analyses. Nature Protocols 2016, 11, 2049–2053. [Google Scholar] [CrossRef]
- Rolli, E.; Marasco, R.; Vigani, G.; Ettoumi, B.; Mapelli, F.; Deangelis, M.L.; Gandolfi, C.; Casati, E.; Previtali, F.; Gerbino, R.; et al. Improved plant resistance to drought is promoted by the root- associated microbiome as a water stress-dependent trait. Environ. Microbiol. 2015, 17, 316–331. [Google Scholar] [CrossRef]
- Su, L.; Shen, Z.; Ruan, Y.; Tao, C.; Chao, Y.; Li, R.; Shen, Q. Isolation of antagonistic endophytes from banana roots against Meloidogyne javanica and their effects on soil nematode community. Front. Microbiol. 2017, 8, 2070. [Google Scholar] [CrossRef]
- Vu, T.; Hauschild, R.; Sikora, R.A. Fusarium oxysporum endophytes induced systemic resistance against Radopholus similis on banana. Nematology 2006, 8, 847–852. [Google Scholar] [CrossRef]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [Green Version]
- Francis, O.E.; Bendall, M.; Manimaran, S.; Hong, C.; Clement, N.L.; Castro-Nallar, E.; Snell, Q.; Schaalje, G.B.; Clement, M.J.; Crandall, K.A.; et al. Pathoscope: Species identification and strain attribution with unassembled sequencing data. Genome Res. 2013, 23, 1721–1729. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushal, M.; Mahuku, G.; Swennen, R. Metagenomic Insights of the Root Colonizing Microbiome Associated with Symptomatic and Non-Symptomatic Bananas in Fusarium Wilt Infected Fields. Plants 2020, 9, 263. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9020263
Kaushal M, Mahuku G, Swennen R. Metagenomic Insights of the Root Colonizing Microbiome Associated with Symptomatic and Non-Symptomatic Bananas in Fusarium Wilt Infected Fields. Plants. 2020; 9(2):263. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9020263
Chicago/Turabian StyleKaushal, Manoj, George Mahuku, and Rony Swennen. 2020. "Metagenomic Insights of the Root Colonizing Microbiome Associated with Symptomatic and Non-Symptomatic Bananas in Fusarium Wilt Infected Fields" Plants 9, no. 2: 263. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9020263