
Escherichia coli Shiga Toxins and Gut Microbiota Interactions


1
Environmental Diseases Research Center, Korea Research Institute of Bioscience and Biotechnology, 125 Gwahak-ro, Daejeon 34141, Korea
2
Department of Biomolecular Science, KRIBB School of Bioscience, Korea University of Science and Technology (UST), 127 Gajeong-ro, Yuseong-gu, Daejeon 34113, Korea
*
Authors to whom correspondence should be addressed.
Toxins 2021, 13(6), 416; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060416
Submission received: 2 April 2021
/
Revised: 3 June 2021
/
Accepted: 8 June 2021
/
Published: 11 June 2021
(This article belongs to the Special Issue Toxins: The New Frontier for Understanding the Barrier between Commensalism and Pathogenicity?)
Abstract
:Escherichia coli (EHEC) and Shigella dysenteriae serotype 1 are enterohemorrhagic bacteria that induce hemorrhagic colitis. This, in turn, may result in potentially lethal complications, such as hemolytic uremic syndrome (HUS), which is characterized by thrombocytopenia, acute renal failure, and neurological abnormalities. Both species of bacteria produce Shiga toxins (Stxs), a phage-encoded exotoxin inhibiting protein synthesis in host cells that are primarily responsible for bacterial virulence. Although most studies have focused on the pathogenic roles of Stxs as harmful substances capable of inducing cell death and as proinflammatory factors that sensitize the host target organs to damage, less is known about the interface between the commensalism of bacterial communities and the pathogenicity of the toxins. The gut contains more species of bacteria than any other organ, providing pathogenic bacteria that colonize the gut with a greater number of opportunities to encounter other bacterial species. Notably, the presence in the intestines of pathogenic EHEC producing Stxs associated with severe illness may have compounding effects on the diversity of the indigenous bacteria and bacterial communities in the gut. The present review focuses on studies describing the roles of Stxs in the complex interactions between pathogenic Shiga toxin-producing E. coli, the resident microbiome, and host tissues. The determination of these interactions may provide insights into the unresolved issues regarding these pathogens.
Keywords:
Shiga toxins; Shiga toxin types 1 and 2; Shiga-toxin-producing Escherichia coli (STEC); commensal microbes; bacterial toxins; gut microbiota; hemolytic uremic syndrome (HUS)Key Contribution: The review provides an overview comprehensively describing the current understanding of the roles of Stxs and STEC at their interfaces with commensal microbiota in the gut, aiming at focusing on interactions with human microbiota.
1. Introduction
Numerous supportive public health measures have led people to erroneously believe that epidemics of many bacterial infectious diseases are no longer a serious health risk. However, Shiga-toxin-producing Escherichia coli (STEC) still poses a threat to public health. Shiga toxin (Stx) is prototypically synthesized by the bacterium Shigella dysenteriae serotype 1, with genetically and structurally related toxin variants produced by certain serotypes of E. coli, including enterohaemorrhagic strains of E. coli (EHEC) [1]. Bacillary dysentery due to infection by Stx-producing bacteria, characterized by acute infectious diarrhea, primarily affects children aged <5 years [2]. Endemic bacillary dysentery occurs globally, including in portions of Africa, Southeast Asia, and the Indian subcontinent, with an estimated 2–7 per 1000 children per year requiring clinical care and 164,300 deaths per year attributable to shigellosis [3]. By contrast, STEC-associated illnesses in young children are more prevalent in developed countries, in which residents consume higher levels of beef and beef products [4,5]. Major outbreaks of diarrheal diseases caused by EHEC may be due to the ingestion of foods such as uncooked meat, unpasteurized milk, water contaminated with these bacteria, and by the contamination of foods used in the preparation of fast-food [6]. Perhaps the largest outbreak of hemorrhagic colitis was caused by an O157 infection in and around Sakai City, Japan, in 1996, which resulted in approximately 1000 hospitalizations among 7000 infected cases [7]. Many non-O157 STEC serotypes have been increasingly reported, and a massive outbreak caused by the hybrid STEC/enteroaggregative E. coli (EAEC) O104:H4 strain occurred in northern Germany from May to June 2011 [8]. More recently, 439 outbreaks and 5 deaths caused by EHEC-contaminated romaine lettuce were reported in multiple states of the United States in 2018 [9].
Stx-producing bacteria have received substantial attention as emergent pathogens due to the dangerous toxins they produce. These exotoxins are the principal virulence factors associated with the pathogenesis of bloody diarrheal diseases, bacillary dysentery, and hemorrhagic colitis progressing to acute renal failure in infected patients, primarily in children. This phenomenon, collectively referred to as hemolytic uremic syndrome (HUS), is the leading cause of pediatric acute renal failure in many countries, including countries in the European Union and the United States [10,11]. Both Stxs and the inflammatory innate immune cells activated by these toxins contribute to the pathogenesis of HUS by rendering blood vessels in the colon, kidney, and central nervous system (CNS) more sensitive to the detrimental action of Stxs. Studies in animals found that treatment with purified Stxs induces intestinal and renal epithelial and endothelial cells to express neutrophil and monocyte chemoattractants. This, in turn, induces the infiltration of peripheral blood mononuclear cells (PBMCs) into the lamina propria and kidneys [12,13]. These findings suggest that the infiltration of inflammatory cells into sites of toxin-induced damage causes the localized expression of cytokines, which in turn facilitate vascular damage via immunopathological reactions.
Studies on Stxs-induced host signaling pathways have indicated that these toxins, which act as multifunctional bacterial proteins, promote ribotoxic stress, apoptosis, endoplasmic reticulum (ER) stress, inflammatory responses, and autophagy in host cells [14]. In addition to the toxigenic and immunopathological potential of Stxs in patients, these toxins interact with multiple cell types in vitro and are responsible for the pathogenic characteristics of HUS in animal models. Although the Stxs-mediated pathogenesis of HUS is not fully understood, comprehensive knowledge of the role of Stxs in altering the composition (also referred to as ‘dysbiosis’) of the intestinal microbiota in a host infected with EHEC compared with a healthy control must be used to help identify the host factors or the commensal microbial-derived products that exacerbate tissue damage or protect against intoxication caused by toxin-producing bacteria. Studies have evaluated the precise correlations between Stx-mediated pathogenesis and intestinal indigenous commensal microbes. This review summarizes the current understanding of the roles of E. coli Stxs and STEC at their interfaces with commensal microbiota in the gut, mainly focusing on interactions with human microbiota.
2. Toxins
Stxs produced by bacteria, including EHEC and Shigella dysenteriae serotype 1, act as primary virulence factors. Each ribosome-inactivating holotoxin possess an AB5 molecular configuration, as confirmed by high-resolution structural analysis, consisting of a large monomeric 32 kDa A subunit and small homo-pentameric 7.7 kDa B subunits [15] (Figure 1). The enzymatic subunit A of the holotoxin is associated with cytotoxic activity, whereas subunit B binds to host receptors on cell surfaces. Stxs bind with high affinity mainly to the glycolipid Gal (α1→4)Gal (β1→4)Glc (β1→1) ceramide (globotriaosylceramide or Gb3), or to a lesser extent, GalNAcβ1-3Galα1-4Galβ1-4GlcβCer (globotetraosylceramide or Gb4) or Galβ1-3GalNAcβ1-3Galαl-4Galβ1-4Glcβl-lCer (globopentaosylceramide or Gb5), on the host cell surface, which act as the toxin receptors [16,17]. In particular, the B-subunits mediate the specific binding of toxins to target cell receptors and their receptor-mediated uptake into target cells [18]. Of note, as a Gb3-independent Stx-binding receptor, Brigotti et al. demonstrated that the Toll-like receptor 4 (TLR4), a well-characterized pattern recognition receptor for lipopolysaccharide, binds to the toxin on the surface of human neutrophils, which are lacking Gb3, without the internalization or intracellular processing of the toxin [19]. Many different cell types, including human renal proximal tubular epithelial cells, human brain microvascular endothelial cells, and primary human monocyte-derived macrophages, express functional Gb3 on their membrane and therefore are target cells for the binding of Stxs (reviewed in [20]). Following the binding of Stxs to their receptors on host cell surfaces, these toxins may be endocytosed via clathrin-coated pits into early endosomes. Alternatively, Stxs may cluster at the plasma membrane, followed by their uptake by a clathrin-independent mechanism [21,22]. Otherwise, Stxs are unable to enter the cell cytosol directly. A recent, extensively updated review of Stxs-toxin receptor interactions on host cell surfaces provided a concise summary and new insight into receptor analog-mediated therapeutic approaches against bacterial verotoxins and Stxs-mediated cytotoxicity [23]. Following its binding to Gb3 on the cell surface, the holotoxin is internalized via receptor-mediated endocytosis and trafficked intracellularly to early endosomes, the trans-Golgi network through the Golgi apparatus, and the lumen of the ER by a process called retrograde transport to deliver Stxs. The precise mechanisms underlying the retrograde transport of Stxs remain largely unexplored, although it occurs in a KDEL-receptor-independent manner [24]. During the intracellular processing of the toxin, subunit A is cleaved by the host protease furin into two fragments: A1, the catalytically active fragment with N-glycosidase activity; and A2, which remains covalently associated through a disulfide bond and is essential for the assembly of the AB5 configuration [15]. Following the reduction of the bond, the A1 fragment retrotranslocates across the ER membrane into the cytosol by utilizing host cellular endoplasmic reticulum-associated protein degradation (ERAD) machinery. The integral membrane Sec61 complex translocon core unit may be involved in the translocation of the A1 subunit. The A1 subunit, which has RNA N-glycosidase activity, inactivates the 60 S subunit of host cell ribosomes by cleaving the N-glycosidic bond at a single specific adenine residue in 28S rRNA (in rats, A4324), leading to the inhibition of the EF-1–dependent aminoacyl-tRNA binding and ultimately preventing aminoacyl-tRNA from binding to the ribosome. This holotoxin subsequently enters the host cell cytosol, leading to multiple cellular responses, including the inhibition of protein synthesis, apoptotic cell death, ER stress, autophagy, and inflammation [25,26,27] (Figure 2). As an alternative way of transporting Stxs into host cells, Stx-containing microvesicles, derived from the plasma membrane of host blood cells in the circulation, reach the kidneys and the toxin is transferred to the renal target cells, including the glomerular endothelial cells, where it is released from the microvesicles and elicits cytotoxic effects by reaching the ribosomes and inhibiting protein synthesis [28]. During STEC-associated HUS, Stxs directly activate complements by triggering a cascade of signaling events and delaying the cofactor activation of surface adhesion factor H (FH) bound to the toxin, leading to the complement-mediated hemolysis with the release of microvesicles from the fragmented red blood cells [29,30]. Buelli et al. reviewed the etiology of complement activation in various experimental models and HUS patients [31]. However, due to the complex pathophysiological cascade of events that ultimately leads to the clinical symptoms of Shigatoxemia, there are no satisfactory therapeutic interventions or regimens for treating children infected with EHEC.
Two major structural types of Stxs have been identified to date: Shiga toxin type 1 (Stx1) and Shiga toxin type 2 (Stx2). Each of these is further subdivided into subtypes, Stx1a, Stx1c, and Stx1d; and Stx2a, Stx2b, Stx2c, Stx2d, Stx2e, Stx2f, Stx2g (reviewed in [32,33]), Stx2h [34], and Stx2i [35,36]. Each subtype consists of a number of variants, which are released by Stx subtype-specific STEC strains. The overall amino acid identity of Stx1 and Stx2 is only 56%, although residues involved in enzymatic activity and binding to cells are more highly conserved. By contrast, variations within the Stx1 and Stx2 categories are much lower, with members having 84–99% amino acid identity. Because the Stx2 variants differ from one another to a greater extent than the Stx1 variants, they have different levels of toxicity in animal models of disease and have different receptor preferences [37]. More importantly, the Stx1a and Stx2a subtypes most commonly cause severe pathogenesis in humans.
The multiple functions of Stxs have been extensively investigated, both in vitro and in vivo, by our group and others. Over the course of these studies, we have devised methods of interrupting Stx-induced host injury signaling mechanisms activated by apoptosis via ER stress, autophagy, or pro-inflammatory cytokine/chemokine production. However, the precise pathogenic potential roles of EHEC-Stxs and other commensal microbiota have not yet been completely investigated. These interactions may be a crucial step in determining the yet unknown mechanisms by which the toxin encounters host microbiota and crosses the mucosal layer.
3. Crosstalk with Gut Microbiota in Intestinal Pathology upon STEC Infection
Approximately 1014 bacteria, consisting of 500 to 1000 species, are present in the gastrointestinal (GI) tract of a human adult. This population of very diverse bacteria, called the gut microbiota, includes protective bacteria, as well as bacteria that could potentially be harmful to, but maintain a symbiotic relationship with, the host. Cross-regulation between the host and the gut microbiota maintains a homeostatic balance of bacteria, keeping the GI tract healthy and preventing an overgrowth of potentially pathogenic bacteria [38,39]. The Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria groups make up the largest share of the normal gut microbiota [40]. Dysbiosis refers to an imbalance in the qualitative and quantitative composition and metabolic activity of intestinal microbiota and may be associated with a variety of disorders including inflammatory bowel disease (IBD), allergies, diabetes, obesity, and multiple sclerosis [41]. Dysbiosis may increase the risk of various gut bacterial infections and has also been linked to STEC infection [42]. Changes in gut microbiota according to diet are well-known [43,44], and Zumbrun et al. reported that dietary choice modulates susceptibility to STEC infection. Mice that were fed a high fiber diet (HFD) exhibited a decrease in native Escherichia species and increased colonization of STEC, weight loss, and mortality compared with mice that were fed a low fiber diet (LFD) [45]. A colitis murine model generated using dextran sulfate sodium similarly had an increased risk of STEC infection [46]. These reports indicate that the robustness of the gut microbiota plays an important role in the defense against STEC infection (Figure 2). Several case reports demonstrated that STEC infection and disease are more likely to occur in children than in adults [47,48]. The gut microbiota of children, which is less mature than that of adults, has a low diversity and number of communities [49]. This is likely one reason for the high rate of STEC infections in children because STEC may colonize individuals with weak/non-healthy microbiota.
During intestinal infection, STEC can reach the ileum and colon and cause disease through survival and colonization [50,51]. STEC colonization is a type of attaching and effacing (A/E) lesion that relies on the type three secretion system (T3SS) to transfer effector proteins to host cells and on the apoptosis of enterocytes located on the apical surface of the intestines. STEC attachment is accompanied by the localized destruction of microvilli [52,53]. Bacteria closely attach to intestinal cells via T3SS and a series of effector proteins encoded in the locus of enterocyte effacement (LEE). Various effector proteins help to sustain the presence of bacteria in the intestinal tract, as well as playing a pivotal role in the toxic effects of these bacteria [54]. After colonization, Stxs produced by STEC pass through intestinal epithelial cells (IECs) into the bloodstream, allowing them to reach target organs, including the kidneys, brain, and eyes, and causing diseases such as HUS [55].
STEC-associated immune responses of the host can include the overstimulation of proinflammatory cytokine production, immune cell activation, and complement activation by Stxs, resulting in primary tissue injury [56,57,58,59]. A continuum of events can result in the development of HUS. Stxs may bind to IECs and mediate the translocation of toxin molecules to and through the basolateral membrane. STEC strains and non-pathogenic commensal E. coli showed differential inflammatory responses using an in vitro IEC infection model system [60]. Although no single transcriptional or cytokine response pattern was reported to be characteristic of the early stages of STEC infection, STEC strains and commensal E. coli differed significantly in the expression of genes involved in amino acid biosynthesis and in uptake and respiration. These three classes of hypothetical genes were found in a fairly high percentage of other STEC pathotypes [60].
Although STEC infection can occur in a variety of animals, cattle are the main reservoir [61]. STEC colonizes the recto-anal junction (RAJ) in cattle but is asymptomatic [62,63]. This is because the Gb3 expression is lower than in humans, and there are reports that even when Gb3 is expressed in the kidneys and brain, Stxs cannot bind to blood vessels in the bovine GI tract [64]. To colonize the RAJ, STEC uses an acid resistance (AR) system. Glutamate decarboxylase GadA and GadB consume protons by converting glutamate into gamma-amino butyric acid (GABA), which helps to protect cells against acidic stress in the GI tract [65,66]. The LuxR homologue SdiA detects acyl-homoserine lactone (AHL) produced by other bacteria and induces a quorum-sensing (QS) system [67]. STEC detects AHL through SdiA in the rumen, inhibits LEE expression, and activates the GadA/B-mediated AR system to survive in an acidic environment. STEC migrates to the RAJ, and LEE is expressed in the absence of AHL, allowing for the colonization of the GI compartment [66]. E. coli O157:H7 in feces is defined as a super-shedder (SS) at a level higher than 104 CFU per gram [68]. Several studies have compared changes in the microbiota between non-shedders (NS) and SS. STEC infection alters the abundance and diversity of some gut microbiota in cattle [69,70]. A paper reviewing data on changes of the microbial count in cattle due to a recent STEC infection classified and organized the microbial differences [71]. The composition of the gut microbiota has been reported to be clinically altered. Lower numbers of Bifidobacteriales and Clostridiales were found in the feces of patients infected with STEC O26:H11 than in the healthy subjects [72]. Bifidobacteria are involved in the NF-κB and SOCS signaling pathways in IEC lines by downregulating the mRNA levels of inflammatory cytokines in response to stimulation with intact bacterial cells or bacterial cell wall components such as LPS [73,74]. Moreover, Bifidobacteria have been reported to have protective efficacy in mice infected with EHEC O157:H7 [75]. Many studies have demonstrated that Clostridium species are probiotics that control the intestinal inflammatory response caused by LPS, suggesting that they have preventive and therapeutic effects on EHEC infection [76,77,78]. Taken together, these findings suggest that STEC infection through intestinal colonization may affect the microbial composition and abundance, which may increase the risk of infection.
In addition to its role in the effects of STEC, Stx is a virulence factor that alone has the potential to affect intestinal tissue damage and microbiota. Clinically, STEC virulence genes, including the stx2a and eae genes that encode Stxs, have been detected in the feces of STEC-infected patients through meta-genomic analysis [72]. This suggests that Stx not only works by migrating to the lamina propria but can also act in the intestinal lumen. Purified Stx proteins induce ribotoxic stress, apoptosis, and inflammatory responses in a variety of cells. Stxs released after STEC colonization bind to intestinal epithelium but not to normal intestinal cells that do not express Gb3 [79,80]. The human IEC lines Caco-2 and HEp-2 cells, which express Gb3, were sensitive to Stxs, as shown by the induction of apoptosis through ER stress [81,82]. Although fully differentiated T84 cells do not express Gb3 and are resistant to Stx [82], the long-term exposure of T84 cells to Stx2 during in vitro organ culture (IVOC) induces internalization and damage [83]. Dysbiosis due to EHEC colonization of the intestine is also affected by Stxs. For example, Stx2 may be involved in increasing the colonization capacity of EHECs by increasing the expression of necleolin in HEp-2 cells [84]. Moreover, a Stx2 neutralizing antibody has been reported to protect mice against weight loss and death by reducing EHEC colonization [85]. These studies suggest that this toxin is the single substance involved in promoting bacterial intestinal infections and exacerbating disease. In addition, Stx1 was found to modulate the expression of galetin-3, which is associated with sodium absorption in the intestine and may contribute to diarrhea [86]. The Stx-induced inflammatory responses of IECs may enhance colonization by EHECs [55,87]. Compared with homologous mutations without the Stx gene in rabbit colon epithelium, wild-type EHEC modulates more diverse transcriptome responses and regulates cytokine gene expression [88].
Stxs stimulate the release of pro-inflammatory cytokines by various host cell types, including those in the endothelium [89]. Serum concentrations of IL-8, MCP-1, and G-CSF have been reported to be higher in pediatric patients with HUS than in controls, with these concentrations associated with disease severity [90,91,92]. E. coli O157:H7 enteritis was associated with the production of GRO-α, MIP-1β, and MCP-1 in blood, regardless of the occurrence of HC or HUS [93]. STEC infection-associated alterations in the expression of cytokines and chemokines at specific cellular levels is important for understanding the disease. A study assessing the effects of infection of IECs with STECs (97-3250: STEC O26: H11, 4865/96: STEC O145: H28) and HS (commensal E. coli O9:H4) on the expression of cytokine mRNA and protein found that 97-3250 promoted greater polymorphonuclear leukocytes (PMN) penetration than 4865/96 or HS by upregulating the expression of various chemokines, including CXCL8/IL-8, and by enhancing PMN chemotaxis. Moreover, of the strains tested, 97-3250 had the greatest effect on gene expression [60]. The presence of polymorphonucleocytes (PMNs) in the stool is considered a risk indicator for the development of HUS. Several invasive pathogens, including STEC, cause fever and inflammatory diarrhea, which is characterized by a high level of PMNs in stools [94]. Increases in the numbers of macrophages and leukocytes, as well as neutrophils, are associated with disease development [90,95].
4. Effects of Probiotics on STEC and Stxs in the Gut
E. coli is mainly found in the intestinal cecum and colon of mammals and resides in the mucosal layer, from which it moves into the intestinal lumen and is excreted into the feces. Pathogenic E. coli-mediated diseases, such as food poisoning, intestinal tissue damage, and particularly bloody diarrhea in young children, are major concerns in public health and medical expense-related economic problems worldwide. More importantly, certain strains of STEC in the gut may cause severe extraintestinal or extrarenal illnesses in humans. Certain Gb3-expressing cell types in the gut, such as Paneth cells, may serve as portals for ingress of Stxs. Several epidemiological studies have shown that infection with STEC isolates expressing Stx2a is more pathogenic than infection with strains producing Stx1a or Stx1a+Stx2a [4,33,36]. Infection with strains producing Stx2a may lead to extraintestinal complications, although the cause of the latter is likely to be multi-factorial and can include the constitutive regulation of stx gene expression; antibiotic usage during the prodromal diarrheal phase, inducing the phage-mediated lytic cycle; the presence or absence of additional E. coli virulence factors; and variations in host responses to toxins, which all contribute to the outcome of infection. Moreover, stx genes are carried in the genomes of temperate phages [96,97], located in the late gene region downstream of the late promoters and upstream of the lysis cassette, highly expressed upon activation of phage-mediated lytic cycle and the toxin subunits assembled in the periplasm are secreted by the lytic cycle [98]. Therefore, antibiotics are not recommended for patients with HUS because they activate the phage-mediated lytic cycle in STEC [99] that lyses bacterial host cells to release Stxs and free phage particles that can infect other bacteria and transduce stx genes [100,101,102,103,104,105]. Given that antibiotic therapy is not indicated for these reasons, besides numerous therapeutic approaches utilizing Stxs-specific neutralizing antibodies, toxin receptor analogs, and vaccinations [106], studies have attempted to increase the population of intestinal microbiota that can inhibit STEC colonization and/or Stx expression (Table 1, Figure 3), thereby limiting HUS development.
4.1. Inhibitory Effects of Microbiota on STEC
4.1.1. Inhibition of E. coli O157:H7 Growth
Bacteroides strains, which are abundant in human gut microbiota, are the most studied commensal bacteria against E. coli O157:H7. For instance, intestinal E. coli O157:H7 colonization was significantly lower in gnotobiotic mice pre-colonized with Bacteroides fragilis (B. fragilis) [107]. Due to the reduced colonization of E. coli O157:H7 in the intestines of mice pre-colonized with B. fragilis, the translocation of E. coli O157:H7 to other organs such as the kidneys, heart, liver, and spleen was significantly lower than in mice with non-commensal bacteria, leading to increased survival outcomes against E. coli O157:H7 [107]. Similar to Bacteroides strains, Lactobacilli such as Lactobacillus reuteri (L. reuteri) also have inhibitory effects against E. coli O157:H7 colonization independent of the antimicrobial compound reuterin, suggesting that L. reuteri has a direct role in protecting against EHEC [108]. The colonization of E. coli O157:H7 was decreased, and subsequently, the necrosis of the kidneys and weight loss were significantly ameliorated in L. reuteri-fed mice [108]. Cattle that were fed Lactobacillus acidophilus (L. acidophilus) also showed the inhibition of E. coli O157:H7 colonization in the feces [109].
In addition to the direct role of microbiota against the colonization of E. coli O157:H7, indirect roles such as the secretion of molecules, modulation of pH, and competition for nutrients have been reported to suppress the growth of EHEC. For instance, intestinal N-acetylglucosamine (NAG), which is derived from the degradation of commensals such as Bacteroides thetaiotaomicron (B. thetaiotaomicron) by mucin, inhibits the colonization of BALB/c mouse intestines by E. coli O157:H7 and represses the expression of T3SS-encoding genes [110]. Reuterin, which was converted from glycerol by L. reuteri, completely suppressed the growth of E. coli O157:H7 incubated in bovine rumen fluid [111]. L. acidophilus strain La-5 was also found to control the transcription of E. coli O157:H7 genes associated with colonization by secreting molecules that act as QS signal inhibitors or interact directly with regulators of bacterial gene transcription [112]. Hydrogen peroxide produced by Lactobacillus lactis [113] and lactic acid produced by Lactobacillus strains [136] were found to significantly reduce the colonization of E. coli O157:H7, the latter by altering pH [114]. Likewise, yoghurt containing Bifidobacterium bifidum (B. bifidum) was found to inhibit colonization by E. coli O157:H7, followed by a reduction in pH [115]. Mice administrated E. coli O157:H7 and the acetic acid-producing Bifidobacterium breve (B. breve) show significantly reduced amounts of E. coli O157:H7 in the feces, leading to higher survival rates and body weight than mice administrated E. coli O157:H7 alone, suggesting that the acetate secreted by B. breve lowered the pH in the intestines and thus inhibited disease pathogenesis [116]. Butyrate-producing Clostridium butyricum (C. butyricum) inhibits the growth of E. coli O157:H7 and reduces its lethality in gnotobiotic mice [78]. Another novel butyrate-producing bacteria, Anaerostipes butyraticus, was found in low-E. coli O157:H7-shedding calves and cattle, suggesting that butyrate-producing bacteria in the GI tract can be used to treat E. coli O157:H7 infection [137]. Similarly, other butyrate-producing bacteria, such as strains of Porphyromonadaceae [138], Lachnospiraceae [139], Ruminococcaceae [140], and Clostridium sartagoforme [141], increased with age in cattle, suggesting a relationship between the attachment/shedding of E. coli O157:H7 and butyrate-producing bacteria [137]. Competition for nutrients by probiotics was reported to inhibit the colonization of E. coli O157:H7. Enterobacter asburiae reduced E. coli O157:H7 survival 20- to 30-fold on lettuce by competition for carbon and nitrogen substrates [117].
4.1.2. Regulation of the Host Immune System
Commensal microbiota not only inhibit E. coli O157:H7 growth but also regulate host immune responses to reduce the pathogenesis of HUS. For example, mice infected with E. coli O157:H7 and fed Lactobacillus rhamnosus (L. rhamnosus) HN001 show increased intestinal anti-E. coli IgA responses [120]. In addition, blood leukocyte activity was higher in L. rhamnosus HN001-fed mice than in controls, leading to the decreased translocation of E. coli O157:H7 and associated lethality [120]. Similarly, infant rabbits administered Lactobacillus casei (L. casei) and infected with E. coli O157:H7 show increased levels of IgAs against Stx1, Stx2, and E. coli O157:H7 in the intestines [118]. L. casei-fed infant rabbits show decreased diarrhea, damage of the intestinal mucus, and colonization of E. coli O157:H7 independent of pH and fermented products such as lactic acid, suggesting that L. casei enhances local immune responses to E. coli O157:H7 [118]. Similar to Lactobacilli, Bifidobacteria also enhance the host immune response to E. coli O157:H7. The proportions of phagocytically active cells in the blood and peritoneum were significantly higher in Bifidobacterium lactis (B. lactis)-fed mice infected with E. coli O157:H7 than in controls [121]. In addition, the level of intestinal IgA against E. coli O157:H7 was higher in B. lactis-fed mice than in controls, which may help to inhibit the translocation of E. coli O157:H7 [121]. The levels of serum IgG and IgM, intestinal IgA antibodies against E. coli O157:H7, were also higher and the levels of fecal E. coli O157:H7 and intestinal injuries were lower in Bifidobacterium thermacidophilum-fed mice than in control mice, leading to decreased lethality [119].
4.1.3. Reduction of Stx Production, Gene Expression, and Stx Phage Particle Release
B. thetaiotaomicron produces a soluble factor with a molecular weight <3 kDa that inhibits the SOS response of E. coli O157:H7 mediated by RecA and consequently inhibits Stx2 phage particle release and Stx2 synthesis independent of pH [122]. B. thetaiotaomicron also inhibited the production of Stx2 by E. coli O157:H7 via uptake of vitamin B12 [123]. Interestingly, mutated B. thetaiotaomicron, which does not express an outer membrane receptor for vitamin B12, did not significantly inhibit the production of Stx2 by E. coli O157:H7, suggesting that vitamin B12 is essential for activating the LEE operon of the latter [123]. B. thetaiotaomicron also inhibits Stx phage production in E. coli O153:H25, which may reduce the production of Stxs [124]. Moreover, the reduction of the pH due to the production of organic acids by probiotics such as Pediococcus pentosaceus, L. rhamnosus GG, and Bifidobacterium thermophilum reduces the gene expression of Stx2 in E. coli O157:H7 [125]. B. fragilis-fed gnotobiotic mice also have a decreased level of Stxs in their feces, leading to reduced lethality [107]. B. breve, which reduces lethality in mice infected with E. coli O157:H7, inhibits Stx production due to a reduced intestinal pH and a higher concentration of acetic acid [116]. Bifidobacterium infantis and Bifidobacterium longum also reduce production of Stxs in the intestines of gnotobiotic mice [75]. C. butyricum, which reduces lethality induced by E. coli O157:H7 in gnotobiotic mice, decreases the fecal level of Stxs [78]. L. casei-fed infant rabbits demonstrated decreased level of Stxs in the large intestines due to anti-Stx1 and -Stx2 IgA antibodies in the colon [118].
4.1.4. Reduction of A/E Lesions
Interestingly, it is reported that Lactobacillus strains reduce the pathogenesis of E. coli O157:H7 by reducing A/E lesions. For instance, the cell-free spent medium (CFSM) of L. acidophilus decreased attachment of E. coli O157:H7 to epithelial cells in vitro such as HeLa and HEp-2 cells, suggesting that the CFSM may block QS mechanisms in EHEC [126]. The biologically active fraction of the CFSM of L. acidophilus also reduces attachment of E. coli O157:H7 to the intestinal epithelium of ICR mice and subsequently decreases body weight loss [126]. Similar to the CFSM of Lactobacilli, the surface-layer protein extracts of Lactobacillus helveticus (L. helveticus) decreased the A/E lesions of E. coli O157:H7 and preserved the barrier function of the monolayers of HEp-2 and T84 cells in vitro [127]. In addition, L. rhamnosus inhibits the adhesion of E. coli O157:H7 to Hep-2 and T84 cells by adhering to these cells, thereby reducing the A/E lesions of E. coli O157:H7 [128].
4.2. Enhancing Effects of Microbiota on STEC
4.2.1. Enhancement of Stxs Production
It is reported that the incubation of nonpathogenic phage-susceptible E. coli with toxin-encoding phages resulted in up to a 40-fold greater production of toxin when compared with lysogens alone, suggesting that nonpathogenic phage-susceptible commensal E. coli may play a role in the pathogenesis of HUS [129]. Similarly, intestinal Stx production was upregulated in CD-1 mice colonized with nonpathogenic phage-susceptible E. coli after infection with E. coli O157:H7 [130]. Moreover, the DNase colicins (E8/9) produced by some strains of nonpathogenic E. coli were found to increase Stx2 production from 8- to 64-fold compared with controls via the activation of an SOS response causing damage to E. coli O157:H7 DNA [131].
4.2.2. Increased Expression of E. coli O157:H7 Virulence Genes
In addition to commensal strains of E. coli, strains of other probiotic species have been found to enhance the pathogenesis of E. coli O157:H7. Bacteroides, the abundant type of bacteria in human intestines [142], was found to enhance the virulence of E. coli O157:H7 as well as disease progression, although the inhibitory effects of Bacteroides against E. coli O157:H7 are well-known (Section 4.1). For instance, B. thetaiotaomicron increased the expression of E. coli O157:H7 virulence genes such as ler, the master LEE regulator, by regulating the transcription factor Cra, which is responsive to fluctuations in sugar concentrations [132]. Moreover, the in vitro addition of succinate, a major by-product of Bacteroides [143], increased the expression of the LEE-encoded protein EspA in E. coli O157:H7 but not in the cra mutant, suggesting an interplay between succinate and Cra [132]. Due to the increased expression of E. coli O157:H7 virulence genes, treatment of B. thetaiotaomicron-reconstituted C3H/HeJ mice with antibiotics resulted in significant weight loss following infection of Citrobacter rodentium, a natural mouse pathogen homologous to E. coli O157:H7 [132].
4.2.3. Increased Colonization of E. coli O157:H7
Along with the secreted molecules such as succinate, the proteases secreted by Bacteroides were found to enhance the processing of the T3SS of E. coli O157:H7, increasing effector translocations and A/E lesion formation on host cells, which enhances the colonization of E. coli O157:H7 [133]. Furthermore, the fucose produced by B. thetaiotaomicron via multiple fucosidases that cleave fucose from host glycans in the intestines also enhances the colonization of E. coli O157:H7 [134]. A two-component signal transduction system named FusKR in E. coli O157:H7, in which FusK is the histidine sensor kinase and FusR is the response regulator, senses fucose and controls the expression of E. coli O157:H7 virulence genes, leading to robust colonization of E. coli O157:H7 [134].
4.2.4. Increased Motility of E. coli O157:H7
Interestingly, a multi-omics study using an organ-on-a-chip microfluidic culture found that four human microbiome metabolites, 4-methyl benzoic acid, 3,4-dimethylbenzoic acid, hexanoic acid, and heptanoic acid, induced the expression of E. coli O157:H7 flagellin, which contributes to the pathogenesis of HUS [135]. These metabolites, however, did not alter colonization by E. coli O157:H7 or the concentration of Stx1, suggesting that metabolites derived from the human microbiome induce the pathogenesis of HUS, not by colonization and toxin production but by enhancing bacterial motility [135].
4.2.5. Increased Expression of Toxin Receptors on Host Cells
Mice fed a HFD containing 10% guar gum showed elevated levels of butyrate, which is derived from fiber by their microbiota. In contrast to the ability of butyrate to inhibit the pathogenesis of E. coli O157:H7, the high concentrations of butyrate in HFD-fed mice were found to enhance E. coli O157:H7 colonization and lethality, as well as weight loss [45]. Elevated concentrations of butyrate produced by normal gut microbiota in HFD-mice also enhanced the expression of Gb3, the receptor of Stxs, suggesting that normal gut microbiota may indirectly enhance the pathogenesis of HUS [45].
5. Conclusions and Future Perspectives
Attempts to identify Stxs-induced risk factors in host cellular responses have revealed that these toxins have a wide range of novel properties that are associated with pathogenesis. Although studies have described Stx-induced signaling pathways that are associated with tissue damage, inflammation, and complement activation resulting from the immunopathological responses to these bacterial toxins, many details on the interfaces between STEC and commensal microbiota (Figure 3) remain to be determined. In particular, no coherent mechanism to date has defined the targets for intervention in HUS disease progression that might explain the dynamic immune modulation, or the mediators of inflammation associated with Stxs interacting with commensal gut microbiota. Additional studies are needed to better understand the intricate pathophysiology involving Stxs-associated bacterial communities in the gut. The precise mechanism by which changes of intestinal microbiota in response to EHEC Stxs may enhance host defense or exacerbate multi-organ damage warrants further investigation. These studies may help to identify potential targets for the disruption of innate immune responses or the protection of primary organs from damage induced by Stx1a and Stx2a.
Author Contributions
Written and revised by K.-S.L., Y.-J.J. and M.-S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Korean government (MIST) (NRF-2018 NRF-2018M3A9H3023077/2021M3A9H3016046) and Ministry of Education (2019R1I1A2A01041221). This work was also carried out with the support of "Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ0150012021)" Rural Development Administration, Republic of Korea.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative program.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the man-uscript, or in the decision to publish the results.
References
- Kaper, J.B.; O’Brien, A.D. Overview and Historical Perspectives. Microbiol Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisot, M.; Parez, N.; Boukhari, R.; Breurec, S.; Jolivet, A. Shigella infection in children under 5 years old in western French Guiana. Epidemiol. Infect. 2018, 146, 980–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotloff, K.L.; Riddle, M.S.; Platts-Mills, J.A.; Pavlinac, P.; Zaidi, A.K.M. Shigellosis. Lancet 2018, 391, 801–812. [Google Scholar] [CrossRef]
- Griffin, P.M.; Tauxe, R.V. The epidemiology of infections caused by Escherichia coli O157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol. Rev. 1991, 13, 60–98. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Scallan, E.; Jones-Bitton, A.; Sargeant, J.M.; Stapleton, J.; Angulo, F.J.; Yeung, D.H.; Kirk, M.D. Global incidence of human Shiga toxin-producing Escherichia coli infections and deaths: A systematic review and knowledge synthesis. Foodborne Pathog. Dis. 2014, 11, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Heredia, N.; García, S. Animals as sources of food-borne pathogens: A review. Anim. Nutr. 2018, 4, 250–255. [Google Scholar] [CrossRef]
- Michino, H.; Araki, K.; Minami, S.; Takaya, S.; Sakai, N.; Miyazaki, M.; Ono, A.; Yanagawa, H. Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. Am. J. Epidemiol. 1999, 150, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 2011, 365, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention (CDC). Available online: https://www.cdc.gov/foodsafety/outbreaks/multistate-outbreaks/outbreaks-list.html (accessed on 1 April 2021).
- Scallan, E.; Mahon, B.E.; Hoekstra, R.M.; Griffin, P.M. Estimates of illnesses, hospitalizations and deaths caused by major bacterial enteric pathogens in young children in the United States. Pediatr. Infect. Dis. J. 2013, 32, 217–221. [Google Scholar] [CrossRef]
- European Food Safety Authority; The European Centre for Disease Prevention and Control. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2017. EFSA J. 2018, 16, e05500. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, H.; Nguyen, L.T.; Lee, E.Y.; Lee, C.Y.; Choi, S.H.; Kim, M.H. Shiga Toxins Trigger the Secretion of Lysyl-tRNA Synthetase to Enhance Proinflammatory Responses. J. Microbiol. Biotechnol. 2016, 26, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.J.; Park, S.K.; Yoon, S.J.; Park, Y.J.; Lee, M.S. Experimental in vivo models of bacterial Shiga toxin-associated hemolytic uremic syndrome. J. Microbiol. Biotechnol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müthing, J.; Meisen, I.; Zhang, W.; Bielaszewska, M.; Mormann, M.; Bauerfeind, R.; Schmidt, M.A.; Friedrich, A.W.; Karch, H. Promiscuous Shiga toxin 2e and its intimate relationship to Forssman. Glycobiology 2012, 22, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Steil, D.; Schepers, C.L.; Pohlentz, G.; Legros, N.; Runde, J.; Humpf, H.U.; Karch, H.; Müthing, J. Shiga toxin glycosphingolipid receptors of Vero-B4 kidney epithelial cells and their membrane microdomain lipid environment. J. Lipid Res. 2015, 56, 2322–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legros, N.; Pohlentz, G.; Steil, D.; Kouzel, I.U.; Liashkovich, I.; Mellmann, A.; Karch, H.; Müthing, J. Membrane assembly of Shiga toxin glycosphingolipid receptors and toxin refractiveness of MDCK II epithelial cells. J. Lipid Res. 2018, 59, 1383–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E.; et al. Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Tesh, V.L. Roles of Shiga Toxins in Immunopathology. Toxins 2019, 11, 212. [Google Scholar] [CrossRef] [Green Version]
- Utskarpen, A.; Massol, R.; van Deurs, B.; Lauvrak, S.U.; Kirchhausen, T.; Sandvig, K. Shiga toxin increases formation of clathrin-coated pits through Syk kinase. PLoS ONE 2010, 5, e10944. [Google Scholar] [CrossRef] [Green Version]
- Renard, H.F.; Garcia-Castillo, M.D.; Chambon, V.; Lamaze, C.; Johannes, L. Shiga toxin stimulates clathrin-independent endocytosis of the VAMP2, VAMP3 and VAMP8 SNARE proteins. J. Cell Sci. 2015, 128, 2891–2902. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, C. Verotoxin Receptor-Based Pathology and Therapies. Front. Cell Infect. Microbiol. 2020, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999, 112 Pt 4, 467–475. [Google Scholar] [CrossRef]
- Leyva-Illades, D.; Cherla, R.P.; Lee, M.S.; Tesh, V.L. Regulation of cytokine and chemokine expression by the ribotoxic stress response elicited by Shiga toxin type 1 in human macrophage-like THP-1 cells. Infect. Immun. 2012, 80, 2109–2120. [Google Scholar] [CrossRef] [Green Version]
- Ohyanagi, H.; Okumura, S.; Yamamoto, M.; Ishida, T.; Kawata, T.; Onoyama, H.; Saitoh, Y. [Epidemiological studies on pancreatic cancer]. Gan To Kagaku Ryoho 1985, 12, 189–199. [Google Scholar]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; De Grandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [CrossRef]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Bekassy, Z.D.; Morgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga toxin activates complement and binds factor H: Evidence for an active role of complement in hemolytic uremic syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef] [Green Version]
- Karpman, D.; Tati, R. Complement contributes to the pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Kidney Int. 2016, 90, 726–729. [Google Scholar] [CrossRef]
- Buelli, S.; Zoja, C.; Remuzzi, G.; Morigi, M. Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Microorganisms 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [Green Version]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Fu, S.; Zhang, J.; Fan, R.; Xu, Y.; Sun, H.; He, X.; Xu, J.; Xiong, Y. Identification and pathogenomic analysis of an Escherichia coli strain producing a novel Shiga toxin 2 subtype. Sci. Rep. 2018, 8, 6756. [Google Scholar] [CrossRef]
- Lacher, D.W.; Gangiredla, J.; Patel, I.; Elkins, C.A.; Feng, P.C. Use of the Escherichia coli Identification Microarray for Characterizing the Health Risks of Shiga Toxin-Producing Escherichia coli Isolated from Foods. J. Food Prot. 2016, 79, 1656–1662. [Google Scholar] [CrossRef]
- FAO; WHO STEC Expert Group. Hazard Identification and Characterization: Criteria for Categorizing Shiga Toxin-Producing Escherichia coli on a Risk Basis(dagger). J. Food Prot. 2019, 82, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Karve, S.S.; Weiss, A.A. Glycolipid binding preferences of Shiga toxin variants. PLoS ONE 2014, 9, e101173. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A. Dysbiosis: A Review Highlighting Obesity and Inflammatory Bowel Disease. J. Clin. Gastroenterol. 2015, 49 (Suppl. S1), S20–S24. [Google Scholar] [CrossRef]
- Yu, L.C.; Shih, Y.A.; Wu, L.L.; Lin, Y.D.; Kuo, W.T.; Peng, W.H.; Lu, K.S.; Wei, S.C.; Turner, J.R.; Ni, Y.H. Enteric dysbiosis promotes antibiotic-resistant bacterial infection: Systemic dissemination of resistant and commensal bacteria through epithelial transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G824–G835. [Google Scholar] [CrossRef]
- Brown, K.; DeCoffe, D.; Molcan, E.; Gibson, D.L. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 2012, 4, 1095–1119. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Zumbrun, S.D.; Melton-Celsa, A.R.; Smith, M.A.; Gilbreath, J.J.; Merrell, D.S.; O’Brien, A.D. Dietary choice affects Shiga toxin-producing Escherichia coli (STEC) O157:H7 colonization and disease. Proc. Natl. Acad. Sci. USA 2013, 110, E2126–E2133. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.; Kurosawa, S.; Stearns-Kurosawa, D.J. Dextran Sulfate Sodium Colitis Facilitates Colonization with Shiga Toxin-Producing Escherichia coli: A Novel Murine Model for the Study of Shiga Toxicosis. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Loconsole, D.; Giordano, M.; Centrone, F.; Accogli, M.; Casulli, D.; De Robertis, A.L.; Morea, A.; Quarto, M.; Parisi, A.; Scavia, G.; et al. Epidemiology of Shiga Toxin-Producing Escherichia coli Infections in Southern Italy after Implementation of Symptom-Based Surveillance of Bloody Diarrhea in the Pediatric Population. Int. J. Environ. Res. Public Health 2020, 17, 5137. [Google Scholar] [CrossRef]
- Tarr, G.A.M.; Shringi, S.; Oltean, H.N.; Mayer, J.; Rabinowitz, P.; Wakefield, J.; Tarr, P.I.; Besser, T.E.; Phipps, A.I. Importance of case age in the purported association between phylogenetics and hemolytic uremic syndrome in Escherichia coli O157:H7 infections. Epidemiol. Infect. 2018, 146, 1550–1555. [Google Scholar] [CrossRef] [Green Version]
- Ringel-Kulka, T.; Cheng, J.; Ringel, Y.; Salojärvi, J.; Carroll, I.; Palva, A.; de Vos, W.M.; Satokari, R. Intestinal microbiota in healthy U.S. young children and adults—A high throughput microarray analysis. PLoS ONE 2013, 8, e64315. [Google Scholar] [CrossRef]
- Chong, Y.; Fitzhenry, R.; Heuschkel, R.; Torrente, F.; Frankel, G.; Phillips, A.D. Human intestinal tissue tropism in Escherichia coli O157: H7--initial colonization of terminal ileum and Peyer’s patches and minimal colonic adhesion ex vivo. Microbiology 2007, 153, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.D.; Navabpour, S.; Hicks, S.; Dougan, G.; Wallis, T.; Frankel, G. Enterohaemorrhagic Escherichia coli O157:H7 target Peyer’s patches in humans and cause attaching/effacing lesions in both human and bovine intestine. Gut 2000, 47, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.P.; Frankel, G.M. The Locus of Enterocyte Effacement and Associated Virulence Factors of Enterohemorrhagic Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Ugalde-Silva, P.; Gonzalez-Lugo, O.; Navarro-Garcia, F. Tight Junction Disruption Induced by Type 3 Secretion System Effectors Injected by Enteropathogenic and Enterohemorrhagic Escherichia coli. Front. Cell Infect. Microbiol. 2016, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Farfan, M.J.; Torres, A.G. Molecular mechanisms that mediate colonization of Shiga toxin-producing Escherichia coli strains. Infect. Immun. 2012, 80, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüller, S. Shiga toxin interaction with human intestinal epithelium. Toxins 2011, 3, 626–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Békássy, Z.D.; Calderon Toledo, C.; Leoj, G.; Kristoffersson, A.; Leopold, S.R.; Perez, M.T.; Karpman, D. Intestinal damage in enterohemorrhagic Escherichia coli infection. Pediatr. Nephrol. 2011, 26, 2059–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle Involvement in Shiga Toxin-Associated Infection. Toxins 2017, 9, 376. [Google Scholar] [CrossRef] [Green Version]
- Karve, S.S.; Pradhan, S.; Ward, D.V.; Weiss, A.A. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS ONE 2017, 12, e0178966. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.G.; Li, Y.; Tutt, C.B.; Xin, L.; Eaves-Pyles, T.; Soong, L. Outer membrane protein A of Escherichia coli O157:H7 stimulates dendritic cell activation. Infect. Immun. 2006, 74, 2676–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, L.M.; Lacher, D.W.; Mammel, M.K.; Leonard, S.R. Comparative Transcriptomics of Shiga Toxin-Producing and Commensal Escherichia coli and Cytokine Responses in Colonic Epithelial Cell Culture Infections. Front. Cell Infect. Microbiol. 2020, 10, 575630. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Lim, J.Y.; Knecht, H.J.; Li, J.; Hovde, C.J. Role of Escherichia coli O157:H7 virulence factors in colonization at the bovine terminal rectal mucosa. Infect. Immun. 2006, 74, 4685–4693. [Google Scholar] [CrossRef] [Green Version]
- Cray, W.C., Jr.; Moon, H.W. Experimental infection of calves and adult cattle with Escherichia coli O157:H7. Appl. Environ. Microbiol. 1995, 61, 1586–1590. [Google Scholar] [CrossRef] [Green Version]
- Wray, C.; McLaren, I.M.; Randall, L.P.; Pearson, G.R. Natural and experimental infection of normal cattle with Escherichia coli O157. Vet. Rec. 2000, 147, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Pruimboom-Brees, I.M.; Morgan, T.W.; Ackermann, M.R.; Nystrom, E.D.; Samuel, J.E.; Cornick, N.A.; Moon, H.W. Cattle lack vascular receptors for Escherichia coli O157:H7 Shiga toxins. Proc. Natl. Acad. Sci. USA 2000, 97, 10325–10329. [Google Scholar] [CrossRef] [Green Version]
- Feehily, C.; Karatzas, K.A. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 2013, 114, 11–24. [Google Scholar] [CrossRef]
- Nguyen, Y.; Sperandio, V. Enterohemorrhagic, E. coli (EHEC) pathogenesis. Front. Cell Infect. Microbiol. 2012, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperandio, V. SdiA sensing of acyl-homoserine lactones by enterohemorrhagic E. coli (EHEC) serotype O157:H7 in the bovine rumen. Gut Microbes 2010, 1, 432–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase-Topping, M.; Gally, D.; Low, C.; Matthews, L.; Woolhouse, M. Super-shedding and the link between human infection and livestock carriage of Escherichia coli O157. Nat. Rev. Microbiol. 2008, 6, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Wang, O.; McAllister, T.A.; Plastow, G.; Stanford, K.; Selinger, B.; Guan, L.L. Interactions of the Hindgut Mucosa-Associated Microbiome with Its Host Regulate Shedding of Escherichia coli O157:H7 by Cattle. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Zaheer, R.; Dugat-Bony, E.; Holman, D.B.; Cousteix, E.; Xu, Y.; Munns, K.; Selinger, L.J.; Barbieri, R.; Alexander, T.; McAllister, T.A.; et al. Changes in bacterial community composition of Escherichia coli O157:H7 super-shedder cattle occur in the lower intestine. PLoS ONE 2017, 12, e0170050. [Google Scholar] [CrossRef] [Green Version]
- Sapountzis, P.; Segura, A.; Desvaux, M.; Forano, E. An Overview of the Elusive Passenger in the Gastrointestinal Tract of Cattle: The Shiga Toxin Producing Escherichia coli. Microorganisms 2020, 8, 877. [Google Scholar] [CrossRef]
- Gigliucci, F.; von Meijenfeldt, F.A.B.; Knijn, A.; Michelacci, V.; Scavia, G.; Minelli, F.; Dutilh, B.E.; Ahmad, H.M.; Raangs, G.C.; Friedrich, A.W.; et al. Metagenomic Characterization of the Human Intestinal Microbiota in Fecal Samples from STEC-Infected Patients. Front. Cell Infect. Microbiol. 2018, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.P.; Parkinson, S.J.; Crupper, S.S.; Robertson, D.C.; Schulz, S.; Waldman, S.A. Cytoplasmic domains mediate the ligand-induced affinity shift of guanylyl cyclase C. Biochemistry 1997, 36, 12921–12929. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.U.; Foata, F.; Philippe, D.; Adolfsson, O.; Eikmanns, B.J.; Blum, S. Anti-inflammatory effects of bifidobacteria by inhibition of LPS-induced NF-kappaB activation. World J. Gastroenterol. 2006, 12, 3729–3735. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Matsui, T.; Itoh, K. Prevention of Escherichia coli O157:H7 infection in gnotobiotic mice associated with Bifidobacterium strains. Antonie Van Leeuwenhoek 2010, 97, 107–117. [Google Scholar] [CrossRef]
- Guo, P.; Zhang, K.; Ma, X.; He, P. Clostridium species as probiotics: Potentials and challenges. J. Anim. Sci. Biotechnol. 2020, 11, 24. [Google Scholar] [CrossRef]
- Xiao, Z.; Liu, L.; Jin, Y.; Pei, X.; Sun, W.; Wang, M. Clostridium tyrobutyricum Protects against LPS-Induced Colonic Inflammation via IL-22 Signaling in Mice. Nutrients 2021, 13, 215. [Google Scholar] [CrossRef]
- Takahashi, M.; Taguchi, H.; Yamaguchi, H.; Osaki, T.; Komatsu, A.; Kamiya, S. The effect of probiotic treatment with Clostridium butyricum on enterohemorrhagic Escherichia coli O157:H7 infection in mice. FEMS Immunol. Med. Microbiol. 2004, 41, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Bjork, S.; Breimer, M.E.; Hansson, G.C.; Karlsson, K.A.; Leffler, H. Structures of blood group glycosphingolipids of human small intestine. A relation between the expression of fucolipids of epithelial cells and the ABO, Le and Se phenotype of the donor. J. Biol. Chem. 1987, 262, 6758–6765. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Iimura, M.; Kaper, J.B.; Torres, A.G.; Kagnoff, M.F. Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell Microbiol. 2006, 8, 869–879. [Google Scholar] [CrossRef]
- Tang, B.; Li, Q.; Zhao, X.H.; Wang, H.G.; Li, N.; Fang, Y.; Wang, K.; Jia, Y.P.; Zhu, P.; Gu, J.; et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy 2015, 11, 344–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.L.; Islur, A.; Haq, R.; Mascarenhas, M.; Karmali, M.A.; Perdue, M.H.; Zanke, B.W.; Sherman, P.M. Escherichia coli Shiga toxins induce apoptosis in epithelial cells that is regulated by the Bcl-2 family. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G811–G819. [Google Scholar] [CrossRef] [Green Version]
- Schüller, S.; Frankel, G.; Phillips, A.D. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell Microbiol. 2004, 6, 289–301. [Google Scholar] [CrossRef]
- Robinson, C.M.; Sinclair, J.F.; Smith, M.J.; O’Brien, A.D. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proc. Natl. Acad. Sci. USA 2006, 103, 9667–9672. [Google Scholar] [CrossRef] [Green Version]
- Mohawk, K.L.; Melton-Celsa, A.R.; Robinson, C.M.; O’Brien, A.D. Neutralizing antibodies to Shiga toxin type 2 (Stx2) reduce colonization of mice by Stx2-expressing Escherichia coli O157:H7. Vaccine 2010, 28, 4777–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laiko, M.; Murtazina, R.; Malyukova, I.; Zhu, C.; Boedeker, E.C.; Gutsal, O.; O’Malley, R.; Cole, R.N.; Tarr, P.I.; Murray, K.F.; et al. Shiga toxin 1 interaction with enterocytes causes apical protein mistargeting through the depletion of intracellular galectin-3. Exp. Cell Res. 2010, 316, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menge, C. Molecular Biology of Escherichia coli Shiga Toxins’ Effects on Mammalian Cells. Toxins 2020, 12, 345. [Google Scholar] [CrossRef] [PubMed]
- Warr, A.R.; Kuehl, C.J.; Waldor, M.K. Shiga toxin remodels the intestinal epithelial transcriptional response to Enterohemorrhagic Escherichia coli. PLoS Pathog. 2021, 17, e1009290. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.S.; Jancic, C.; Garimano, N.; Sacerdoti, F.; Paton, A.W.; Paton, J.C.; Ibarra, C.; Amaral, M.M. Crosstalk between Human Microvascular Endothelial Cells and Tubular Epithelial Cells Modulates Pro-Inflammatory Responses Induced by Shiga Toxin Type 2 and Subtilase Cytotoxin. Toxins 2019, 11, 648. [Google Scholar] [CrossRef] [Green Version]
- Van Setten, P.A.; van Hinsbergh, V.W.; van den Heuvel, L.P.; Preyers, F.; Dijkman, H.B.; Assmann, K.J.; van der Velden, T.J.; Monnens, L.A. Monocyte chemoattractant protein-1 and interleukin-8 levels in urine and serum of patents with hemolytic uremic syndrome. Pediatr. Res. 1998, 43, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, M.M.; Shah, V.; Trompeter, R.S.; Dillon, M.J.; Barratt, T.M. Interleukin-8 and polymorphoneutrophil leucocyte activation in hemolytic uremic syndrome of childhood. Kidney Int. 1992, 42, 951–956. [Google Scholar] [CrossRef] [Green Version]
- Murata, A.; Shimazu, T.; Yamamoto, T.; Taenaka, N.; Nagayama, K.; Honda, T.; Sugimoto, H.; Monden, M.; Matsuura, N.; Okada, S. Profiles of circulating inflammatory- and anti-inflammatory cytokines in patients with hemolytic uremic syndrome due to E. coli O157 infection. Cytokine 1998, 10, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Proulx, F.; Toledano, B.; Phan, V.; Clermont, M.J.; Mariscalco, M.M.; Seidman, E.G. Circulating granulocyte colony-stimulating factor, C-X-C, and C-C chemokines in children with Escherichia coli O157:H7 associated hemolytic uremic syndrome. Pediatr. Res. 2002, 52, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Hodges, K.; Gill, R. Infectious diarrhea: Cellular and molecular mechanisms. Gut Microbes 2010, 1, 4–21. [Google Scholar] [CrossRef] [Green Version]
- Coad, N.A.; Marshall, T.; Rowe, B.; Taylor, C.M. Changes in the postenteropathic form of the hemolytic uremic syndrome in children. Clin. Nephrol. 1991, 35, 10–16. [Google Scholar]
- Herold, S.; Karch, H.; Schmidt, H. Shiga toxin-encoding bacteriophages--genomes in motion. Int. J. Med. Microbiol. 2004, 294, 115–121. [Google Scholar] [CrossRef]
- O’Brien, A.D.; Newland, J.W.; Miller, S.F.; Holmes, R.K.; Smith, H.W.; Formal, S.B. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 1984, 226, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Livny, J.; Neely, M.N.; Acheson, D.W.; Friedman, D.I.; Waldor, M.K. Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol. Microbiol. 2002, 44, 957–970. [Google Scholar] [CrossRef]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Acheson, D.W.; Reidl, J.; Zhang, X.; Keusch, G.T.; Mekalanos, J.J.; Waldor, M.K. In vivo transduction with shiga toxin 1-encoding phage. Infect. Immun. 1998, 66, 4496–4498. [Google Scholar] [CrossRef]
- James, C.E.; Stanley, K.N.; Allison, H.E.; Flint, H.J.; Stewart, C.S.; Sharp, R.J.; Saunders, J.R.; McCarthy, A.J. Lytic and lysogenic infection of diverse Escherichia coli and Shigella strains with a verocytotoxigenic bacteriophage. Appl. Environ. Microbiol. 2001, 67, 4335–4337. [Google Scholar] [CrossRef] [Green Version]
- Kratochvil, S. [Clinical experiences with hypnosis in the Czech republic (II)]. Psychiatr. Neurol. Med. Psychol. Beih 1981, 28, 46–51. [Google Scholar]
- Muniesa, M.; Blanco, J.E.; de Simon, M.; Serra-Moreno, R.; Blanch, A.R.; Jofre, J. Diversity of stx2 converting bacteriophages induced from Shiga-toxin-producing Escherichia coli strains isolated from cattle. Microbiology 2004, 150, 2959–2971. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Bielaszewska, M.; Karch, H. Transduction of enteric Escherichia coli isolates with a derivative of Shiga toxin 2-encoding bacteriophage phi3538 isolated from Escherichia coli O157:H7. Appl. Environ. Microbiol. 1999, 65, 3855–3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, I.; Schmidt, H.; Dow, M.; Malik, A.; Oswald, E.; Nagy, B. Transduction of porcine enteropathogenic Escherichia coli with a derivative of a shiga toxin 2-encoding bacteriophage in a porcine ligated ileal loop system. Appl. Environ. Microbiol. 2003, 69, 7242–7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlen, S.; Dersch, P. Treatment Strategies for Infections With Shiga Toxin-Producing Escherichia coli. Front. Cell Infect. Microbiol. 2020, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Suzuki, R.; Koyanagi, Y.; Isogai, H.; Yoneyama, H.; Isogai, E. Inhibition of enterohemorrhagic Escherichia coli O157:H7 infection in a gnotobiotic mouse model with pre-colonization by Bacteroides strains. Biomed. Rep. 2019, 10, 175–182. [Google Scholar] [CrossRef]
- Eaton, K.A.; Honkala, A.; Auchtung, T.A.; Britton, R.A. Probiotic Lactobacillus reuteri ameliorates disease due to enterohemorrhagic Escherichia coli in germfree mice. Infect. Immun. 2011, 79, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, R.E.; Klopfenstein, T.J.; Erickson, G.E.; Folmer, J.; Hinkley, S.; Moxley, R.A.; Smith, D.R. Effect of Lactobacillus acidophilus strain NP51 on Escherichia coil O157:H7 fecal shedding and finishing performance in beef feedlot cattle. J. Food Prot. 2007, 70, 287–291. [Google Scholar] [CrossRef]
- Le Bihan, G.; Sicard, J.F.; Garneau, P.; Bernalier-Donadille, A.; Gobert, A.P.; Garrivier, A.; Martin, C.; Hay, A.G.; Beaudry, F.; Harel, J.; et al. The NAG Sensor NagC Regulates LEE Gene Expression and Contributes to Gut Colonization by Escherichia coli O157:H7. Front. Cell Infect. Microbiol. 2017, 7, 134. [Google Scholar] [CrossRef]
- Bertin, Y.; Habouzit, C.; Duniere, L.; Laurier, M.; Durand, A.; Duchez, D.; Segura, A.; Thevenot-Sergentet, D.; Baruzzi, F.; Chaucheyras-Durand, F.; et al. Lactobacillus reuteri suppresses E. coli O157:H7 in bovine ruminal fluid: Toward a pre-slaughter strategy to improve food safety? PLoS ONE 2017, 12, e0187229. [Google Scholar] [CrossRef] [Green Version]
- Medellin-Pena, M.J.; Wang, H.; Johnson, R.; Anand, S.; Griffiths, M.W. Probiotics affect virulence-related gene expression in Escherichia coli O157:H7. Appl. Environ. Microbiol. 2007, 73, 4259–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brashears, M.M.; Reilly, S.S.; Gilliland, S.E. Antagonistic action of cells of Lactobacillus lactis toward Escherichia coli O157:H7 on refrigerated raw chicken meat. J. Food Prot. 1998, 61, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Shimizu, K.; Nomoto, K.; Tanaka, R.; Hamabata, T.; Yamasaki, S.; Takeda, T.; Takeda, Y. Inhibition of in vitro growth of Shiga toxin-producing Escherichia coli O157:H7 by probiotic Lactobacillus strains due to production of lactic acid. Int. J. Food Microbiol. 2001, 68, 135–140. [Google Scholar] [CrossRef]
- Massa, S.; Altieri, C.; Quaranta, V.; De Pace, R. Survival of Escherichia coli O157:H7 in yoghurt during preparation and storage at 4 degrees C. Lett. Appl. Microbiol. 1997, 24, 347–350. [Google Scholar] [CrossRef]
- Asahara, T.; Shimizu, K.; Nomoto, K.; Hamabata, T.; Ozawa, A.; Takeda, Y. Probiotic bifidobacteria protect mice from lethal infection with Shiga toxin-producing Escherichia coli O157:H7. Infect. Immun. 2004, 72, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Cooley, M.B.; Chao, D.; Mandrell, R.E. Escherichia coli O157:H7 survival and growth on lettuce is altered by the presence of epiphytic bacteria. J. Food Prot. 2006, 69, 2329–2335. [Google Scholar] [CrossRef]
- Ogawa, M.; Shimizu, K.; Nomoto, K.; Takahashi, M.; Watanuki, M.; Tanaka, R.; Tanaka, T.; Hamabata, T.; Yamasaki, S.; Takeda, Y. Protective effect of Lactobacillus casei strain Shirota on Shiga toxin-producing Escherichia coli O157:H7 infection in infant rabbits. Infect. Immun. 2001, 69, 1101–1108. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, M.; Kheadr, E.E.; Dabour, N.; Richard, D.; Fliss, I. Effect of Bifidobacterium thermacidophilum probiotic feeding on enterohemorrhagic Escherichia coli O157:H7 infection in BALB/c mice. Int. J. Food Microbiol. 2006, 111, 26–33. [Google Scholar] [CrossRef]
- Shu, Q.; Gill, H.S. Immune protection mediated by the probiotic Lactobacillus rhamnosus HN001 (DR20) against Escherichia coli O157:H7 infection in mice. FEMS Immunol. Med. Microbiol. 2002, 34, 59–64. [Google Scholar] [CrossRef]
- Shu, Q.; Gill, H.S. A dietary probiotic (Bifidobacterium lactis HN019) reduces the severity of Escherichia coli O157:H7 infection in mice. Med. Microbiol. Immunol. 2001, 189, 147–152. [Google Scholar] [CrossRef]
- De Sablet, T.; Chassard, C.; Bernalier-Donadille, A.; Vareille, M.; Gobert, A.P.; Martin, C. Human microbiota-secreted factors inhibit shiga toxin synthesis by enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 2009, 77, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordonnier, C.; Le Bihan, G.; Emond-Rheault, J.G.; Garrivier, A.; Harel, J.; Jubelin, G. Vitamin B12 Uptake by the Gut Commensal Bacteria Bacteroides thetaiotaomicron Limits the Production of Shiga Toxin by Enterohemorrhagic Escherichia coli. Toxins 2016, 8, 14. [Google Scholar] [CrossRef]
- Iversen, H.; Lindbäck, T.; L’Abee-Lund, T.M.; Roos, N.; Aspholm, M.; Stenfors Arnesen, L. The gut bacterium Bacteroides thetaiotaomicron influences the virulence potential of the enterohemorrhagic Escherichia coli O103:H25. PLoS ONE 2015, 10, e0118140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, C.M.; Kostrzynska, M.; Ojha, S.; Thompson, S. The effect of probiotics and organic acids on Shiga-toxin 2 gene expression in enterohemorrhagic Escherichia coli O157:H7. J. Microbiol. Methods 2008, 73, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Medellin-Peña, M.J.; Griffiths, M.W. Effect of molecules secreted by Lactobacillus acidophilus strain La-5 on Escherichia coli O157:H7 colonization. Appl. Environ. Microbiol. 2009, 75, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Johnson-Henry, K.C.; Hagen, K.E.; Gordonpour, M.; Tompkins, T.A.; Sherman, P.M. Surface-layer protein extracts from Lactobacillus helveticus inhibit enterohaemorrhagic Escherichia coli O157:H7 adhesion to epithelial cells. Cell Microbiol. 2007, 9, 356–367. [Google Scholar] [CrossRef]
- Sherman, P.M.; Johnson-Henry, K.C.; Yeung, H.P.; Ngo, P.S.; Goulet, J.; Tompkins, T.A. Probiotics reduce enterohemorrhagic Escherichia coli O157:H7- and enteropathogenic E. coli O127:H6-induced changes in polarized T84 epithelial cell monolayers by reducing bacterial adhesion and cytoskeletal rearrangements. Infect. Immun. 2005, 73, 5183–5188. [Google Scholar] [CrossRef] [Green Version]
- Gamage, S.D.; Strasser, J.E.; Chalk, C.L.; Weiss, A.A. Nonpathogenic Escherichia coli can contribute to the production of Shiga toxin. Infect. Immun. 2003, 71, 3107–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamage, S.D.; Patton, A.K.; Strasser, J.E.; Chalk, C.L.; Weiss, A.A. Commensal bacteria influence Escherichia coli O157:H7 persistence and Shiga toxin production in the mouse intestine. Infect. Immun. 2006, 74, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Toshima, H.; Yoshimura, A.; Arikawa, K.; Hidaka, A.; Ogasawara, J.; Hase, A.; Masaki, H.; Nishikawa, Y. Enhancement of Shiga toxin production in enterohemorrhagic Escherichia coli serotype O157:H7 by DNase colicins. Appl. Environ. Microbiol. 2007, 73, 7582–7588. [Google Scholar] [CrossRef] [Green Version]
- Curtis, M.M.; Hu, Z.; Klimko, C.; Narayanan, S.; Deberardinis, R.; Sperandio, V. The gut commensal Bacteroides thetaiotaomicron exacerbates enteric infection through modification of the metabolic landscape. Cell Host Microbe 2014, 16, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, E.A.; Curtis, M.M.; Kumar, A.; Dunny, G.M.; Sperandio, V. Microbiota and Pathogen Proteases Modulate Type III Secretion Activity in Enterohemorrhagic Escherichia coli. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, A.R.; Curtis, M.M.; Ritchie, J.M.; Munera, D.; Waldor, M.K.; Moreira, C.G.; Sperandio, V. Fucose sensing regulates bacterial intestinal colonization. Nature 2012, 492, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Tovaglieri, A.; Sontheimer-Phelps, A.; Geirnaert, A.; Prantil-Baun, R.; Camacho, D.M.; Chou, D.B.; Jalili-Firoozinezhad, S.; de Wouters, T.; Kasendra, M.; Super, M.; et al. Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites. Microbiome 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.A.; Roach, P.J.; Montero, M.; Baroja-Fernández, E.; Muñoz, F.J.; Eydallin, G.; Viale, A.M.; Pozueta-Romero, J. Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 2010, 34, 952–985. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Tyler, P.J.; Starnes, J.; Bratcher, C.L.; Rankins, D.; McCaskey, T.A.; Wang, L. Correlation analysis of Shiga toxin-producing Escherichia coli shedding and faecal bacterial composition in beef cattle. J. Appl. Microbiol. 2013, 115, 591–603. [Google Scholar] [CrossRef]
- Sakamoto, M.; Takagaki, A.; Matsumoto, K.; Kato, Y.; Goto, K.; Benno, Y. Butyricimonas synergistica gen. nov., sp. nov. and Butyricimonas virosa sp. nov., butyric acid-producing bacteria in the family ‘Porphyromonadaceae’ isolated from rat faeces. Int. J. Syst. Evol. Microbiol. 2009, 59, 1748–1753. [Google Scholar] [CrossRef]
- Carlier, J.P.; K’Ouas, G.; Bonne, I.; Lozniewski, A.; Mory, F. Oribacterium sinus gen. nov., sp. nov., within the family ‘Lachnospiraceae’ (phylum Firmicutes). Int. J. Syst. Evol. Microbiol. 2004, 54, 1611–1615. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.D.; Lawson, P.A.; Willems, A.; Cordoba, J.J.; Fernandez-Garayzabal, J.; Garcia, P.; Cai, J.; Hippe, H.; Farrow, J.A. The phylogeny of the genus Clostridium: Proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 1994, 44, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Šimůnek, J.; Hodrová, B.; Bartoňová, H.; Kopečný, J. Chitinolytic bacteria of the mammal digestive tract. Folia Microbiol. 2001, 46, 76–78. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macy, J.M.; Ljungdahl, L.G.; Gottschalk, G. Pathway of succinate and propionate formation in Bacteroides fragilis. J. Bacteriol. 1978, 134, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Crystal Structure of Shiga toxin. (A) Shiga Toxin Type 1 (PDB #1DM0). (B) Shiga Toxin Type 2 (PDB # 1R4P). (C) Shiga Toxin 1 B-subunit with Gb3 receptor (PDB #1BOS). (D) Shiga Toxin 2 B-subunit with Gb3 receptor (PDB #1R4P, deletion of A-subunit). PDB files of all structures were obtained from RCSB PDB (www.rcsb.org) and PDB files were compiled with Chimera 1.10.2 (UCSF Chimera, www.cgl.ucsf.edu/chimera, accessed on 10 June 2021). Reproduced from reference [14]. 2016, MDPI.
Figure 1.
Crystal Structure of Shiga toxin. (A) Shiga Toxin Type 1 (PDB #1DM0). (B) Shiga Toxin Type 2 (PDB # 1R4P). (C) Shiga Toxin 1 B-subunit with Gb3 receptor (PDB #1BOS). (D) Shiga Toxin 2 B-subunit with Gb3 receptor (PDB #1R4P, deletion of A-subunit). PDB files of all structures were obtained from RCSB PDB (www.rcsb.org) and PDB files were compiled with Chimera 1.10.2 (UCSF Chimera, www.cgl.ucsf.edu/chimera, accessed on 10 June 2021). Reproduced from reference [14]. 2016, MDPI.

Figure 2.
Summary of STEC/Stx-induced immunopathology. Dysbiosis, induced for a variety of reasons such as diet, colitis, and inflammation, increases STEC infection and colonization. STEC induces the delivery of Shiga toxins and the production of cytokines and chemokines through colonization in intestinal epithelial cells. In addition to cell death by Stxs, various cells, including neutrophils induced by chemotaxis, induce inflammation in the intestine, which leads to damage. Toxins pass through the intestinal mucosa, enter the bloodstream and travel to target organs such as the kidneys and CNS. After membrane invasion-mediated endocytosis through the toxin receptor Gb3 on the cell surface, Stxs migrate to the Golgi and ER. Shiga toxin acts as a multifunctional bacterial protein, promoting ER stress, ribotoxic stress, pro-inflammatory responses, apoptosis, and autophagy in host cells.
Figure 2.
Summary of STEC/Stx-induced immunopathology. Dysbiosis, induced for a variety of reasons such as diet, colitis, and inflammation, increases STEC infection and colonization. STEC induces the delivery of Shiga toxins and the production of cytokines and chemokines through colonization in intestinal epithelial cells. In addition to cell death by Stxs, various cells, including neutrophils induced by chemotaxis, induce inflammation in the intestine, which leads to damage. Toxins pass through the intestinal mucosa, enter the bloodstream and travel to target organs such as the kidneys and CNS. After membrane invasion-mediated endocytosis through the toxin receptor Gb3 on the cell surface, Stxs migrate to the Golgi and ER. Shiga toxin acts as a multifunctional bacterial protein, promoting ER stress, ribotoxic stress, pro-inflammatory responses, apoptosis, and autophagy in host cells.

Figure 3.
Overview for the Resistance Mechanism of gut microbiota to STEC Infection. The resistance of microbial guns to control STEC works in several ways, and the diagram shows the role of each bacterium in STEC. Bacterioides inhibit Stx production, control direct bacteria and inhibit the colonization of STEC. Bifidobacterium also inhibits STEC colonization and controls STEC proliferation through acetic acid and IgA. Lactobacillus is involved in inhibiting STEC proliferation through the production of hydrogen peroxide, lactic acid, IgA, and leukocyte activity. Pediococcus, Clostridium, and Enterobacter are involved in inhibiting STEC proliferation or controlling Stx production.
Figure 3.
Overview for the Resistance Mechanism of gut microbiota to STEC Infection. The resistance of microbial guns to control STEC works in several ways, and the diagram shows the role of each bacterium in STEC. Bacterioides inhibit Stx production, control direct bacteria and inhibit the colonization of STEC. Bifidobacterium also inhibits STEC colonization and controls STEC proliferation through acetic acid and IgA. Lactobacillus is involved in inhibiting STEC proliferation through the production of hydrogen peroxide, lactic acid, IgA, and leukocyte activity. Pediococcus, Clostridium, and Enterobacter are involved in inhibiting STEC proliferation or controlling Stx production.


{kind=link}
{kind=link}
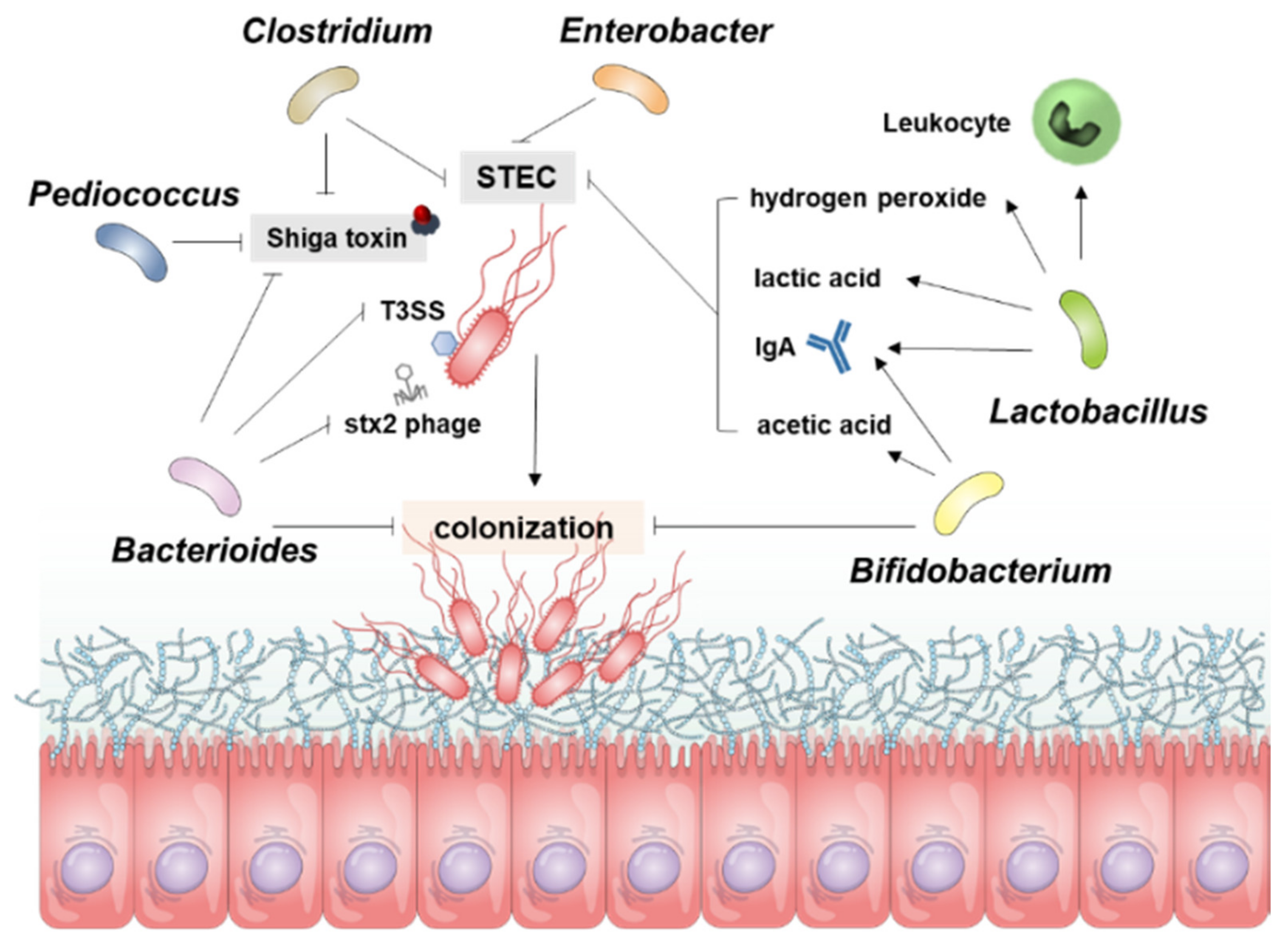{kind=link}
Table 1.
Abilities of probiotics to regulate Stx production and STEC virulence.
| Effect | Activity | Mediator | Model | Intestine In-/Out-Side | Genus | Species | Ref |
|---|---|---|---|---|---|---|---|
| Inhibition | Inhibit growth of E. coli O157:H7 | Butyric, lactic acid | Gnotobiotic mice | Inside | Clostridium | butyricum | [78] |
| N/D | Gnotobiotic mice | Inside | Bacteroides | fragilis | [107] | ||
| N/D | Gnotobiotic mice | Inside | Lactobacillus | reuteri | [108] | ||
| N/D | Cattle | Inside | Lactobacillus | acidophilus | [109] | ||
| N-acetylglucosamine (NAG) and N-acetylneuraminic acid (NANA) | BALB/c mice Bovine rumen | Inside | Bacteroides | thetaiotaomicron | [110] | ||
| Reuterin | fluid | Inside | Lactobacillus | reuteri | [111] | ||
| Reduce autoinducer-2 (AI-2) production | E. coli O157:H7 | N/D | Lactobacillus | acidophilus | [112] | ||
| Hydrogen peroxide | Raw chicken meat | N/D | Lactobacillus | lactis | [113] | ||
| Lactic acid | E. coli O157:H7 | N/D | Lactobacillus | casei | [114] | ||
| Decrease pH | E. coli O157:H7 | N/D | Bifidobacterium | bifidum | [115] | ||
| Acetic acid | BALB/c mice | Inside | Bifidobacterium | breve | [116] | ||
| Nutrition competition (carbon, nitrogen) | Lettuce | N/D | Enterobacter | asburiae | [117] | ||
| Production of anti-Stx1 and -Stx2 IgA in the colon | Infant rabbits | Inside | Lactobacillus | casei | [118] | ||
| IgA | BALB/c mice | Inside | Bifidobacterium | thermacidophilum | [119] | ||
| Regulate host immunity | Upregulate intestinal anti-E. coli IgA responses | BALB/c and C57BL/6 mice | Inside | Lactobacillus | rhamnosus | [120] | |
| Blood leukocyte activity | |||||||
| Inhibit translocation of E. coli O157:H7 | |||||||
| Production of anti-Stx1 and -Stx2 IgA in the colon | Infant rabbits | Inside | Lactobacillus | casei | [118] | ||
| Increase phagocytic activity | BALB/c and C57BL/6 mice | Outside | Bifidobacterium | lactis | [121] | ||
| Increase production of IgA against E. coli O157:H7 | BALB/c and C57BL/6 mice | Inside | Bifidobacterium | lactis | [121] | ||
| Increase production of IgG and IgM against E. coli O157:H7 | BALB/c mice | Outside | Bifidobacterium | thermacidophilum | [119] | ||
| IgA | BALB/c mice | Inside | Bifidobacterium | thermacidophilum | [119] | ||
| Reduce Stx production | N/D | Gnotobiotic mice | Inside | Bifidobacterium | infantis | [75] | |
| N/D | Gnotobiotic mice | Inside | Bifidobacterium | longum | [75] | ||
| Butyric, lactic acid | Gnotobiotic mice | Inside | Clostridium | butyricum | [78] | ||
| N/D | Gnotobiotic mice | Inside | Bacteroides | fragilis | [107] | ||
| Acetic acid | BALB/c mice | Inside | Bifidobacterium | breve | [116] | ||
| Production of anti-Stx1 and -Stx2 IgA in the colon | Infant rabbits | Inside | Lactobacillus | casei | [118] | ||
| N/D | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [122] | ||
| Uptake vitamin B12 | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [123] | ||
| Suppress kidney necrosis induced by E. coli O157:H7 | N/D | Gnotobiotic mice | Inside | Lactobacillus | reuteri | [108] | |
| Repress T3SS of E. coli O157:H7 | NANA and NAG | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [110] | |
| Reduce intestinal injuries after E. coli O157:H7 infection | Production of anti-Stx1 and -Stx2 IgA in the colon | Infant rabbits | Inside | Lactobacillus | casei | [118] | |
| Increased production of IgG and IgM against E. coli O157:H7 | BALB/c mice | Inside | Bifidobacterium | thermacidophilum | [119] | ||
| Inhibit stx2 phage particle release | N/D | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [122] | |
| Reduce Stx2 gene expression | Inhibition of phage production | E. coli O153:H25 | N/D | Bacteroides | thetaiotaomicron | [124] | |
| Organic acid produced by probiotics | E. coli O157:H7 | N/D | Pediococcus | pentosaceus | [125] | ||
| Lactobacillus | rhamnosus GG | ||||||
| Bifidobacterium | thermophilum | ||||||
| Reduce attaching and effacing lesions of E. coli O157:H7 | Spent medium | ICR mice | Inside | Lactobacillus | acidophilus | [126] | |
| HeLa cells | N/D | ||||||
| Hep-2 cells | |||||||
| S-layer protein | Hep-2 cells | N/D | Lactobacillus | helveticus | [127] | ||
| T84 cells | |||||||
| N/D | Hep-2 cells | N/D | Lactobacillus | rhamnosus | [128] | ||
| T84 cells | |||||||
| Enhancement | Increase toxin receptor expression on host cells | Butyrate | E. coli O157:H7 | N/D | N/D | N/D | [45] |
| Enhance toxin production | Bacteriophage transfer | CD-1 mice | Inside | Escherichia | coli | [129] | |
| [130] | |||||||
| Damage of E. coli O157:H7 DNA using DNase colicins | E. coli O157:H7 | N/D | Escherichia | coli | [131] | ||
| Increase the expression of the virulence genes of E. coli O157:H7 | Regulate Cra, a transcription factor for virulence genes of E. coli O157:H7 | C3H/HeJ mice | Inside | Bacteroides | thetaiotaomicron | [132] | |
| Exacerbate weight loss after infection | E. coli O157:H7 | Inside | Bacteroides | thetaiotaomicron | [132] | ||
| Enhance colonization | Butyrate | E. coli O157:H7 | N/D | N/D | N/D | [45] | |
| Secrete proteases that cleave the translocon of the T3SS | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [133] | ||
| Fucose | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [134] | ||
| Enhance T3SS of E. coli O157:H7 | Secrete proteases that cleave the translocon of the T3SS | E. coli O157:H7 | N/D | Bacteroides | thetaiotaomicron | [133] | |
| Increase E. coli O157:H7 motility | 4-Methyl benzoic acid | E. coli O157:H7 | N/D | N/D | N/D | [135] | |
| 3,4-Dimethylbenzoic acid | |||||||
| Hexanoic acid | |||||||
| Heptanoic acid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, K.-S.; Jeong, Y.-J.; Lee, M.-S. Escherichia coli Shiga Toxins and Gut Microbiota Interactions. Toxins 2021, 13, 416. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060416
AMA Style
Lee K-S, Jeong Y-J, Lee M-S. Escherichia coli Shiga Toxins and Gut Microbiota Interactions. Toxins. 2021; 13(6):416. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060416
Chicago/Turabian StyleLee, Kyung-Soo, Yu-Jin Jeong, and Moo-Seung Lee. 2021. "Escherichia coli Shiga Toxins and Gut Microbiota Interactions" Toxins 13, no. 6: 416. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13060416
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.