
ATP Release from Human Airway Epithelial Cells Exposed to Staphylococcus aureus Alpha-Toxin

 , , and
, , and 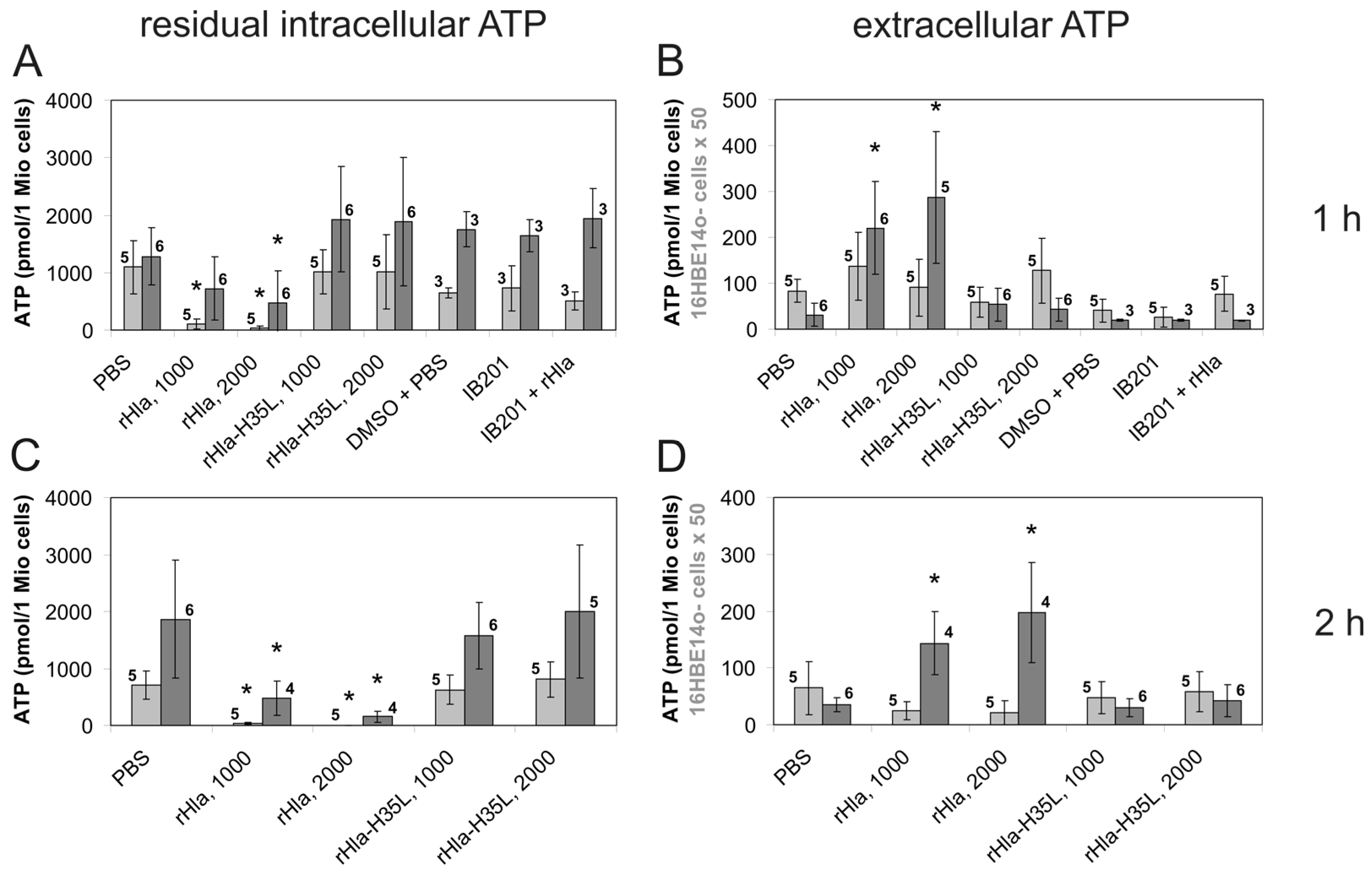{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
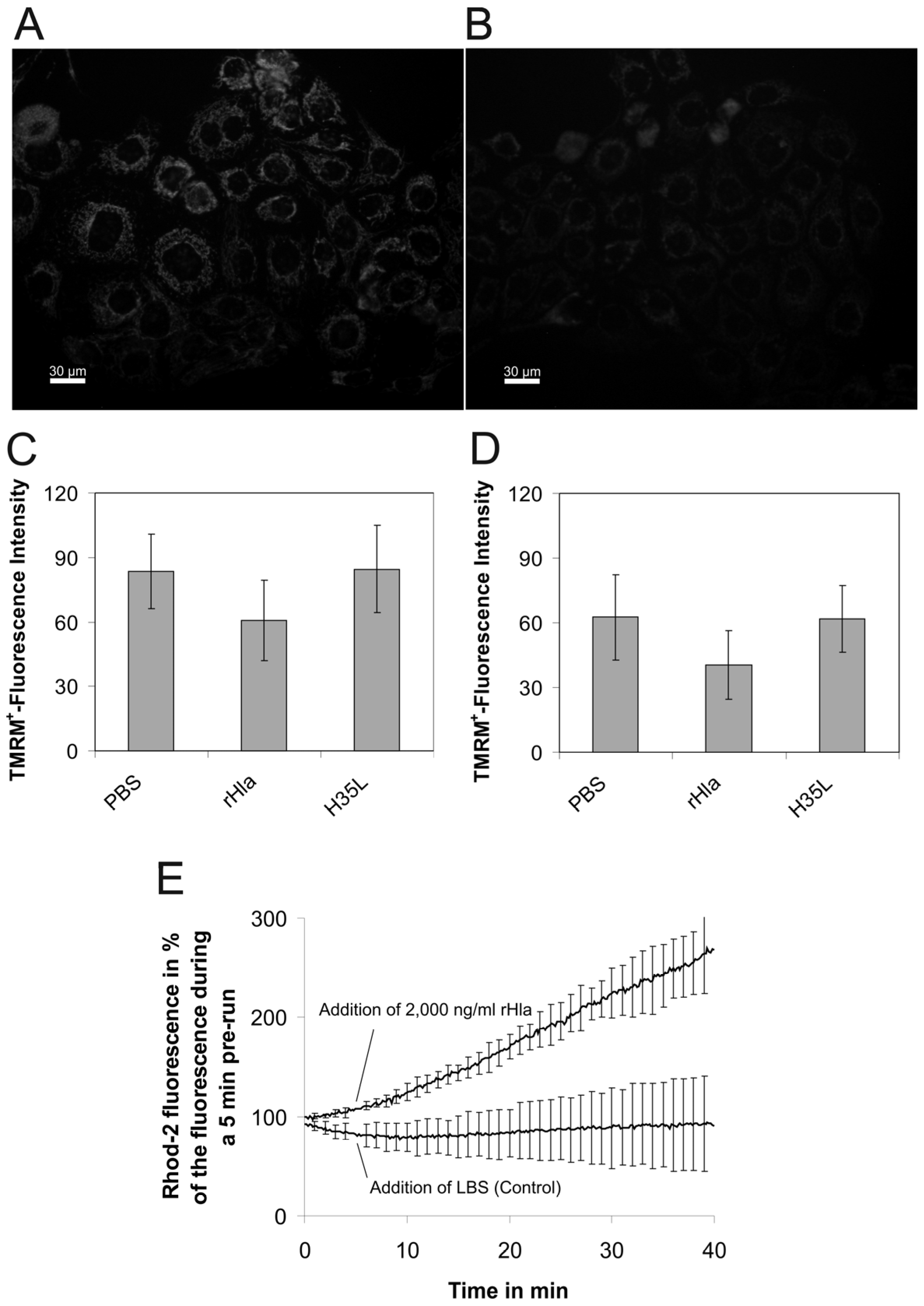
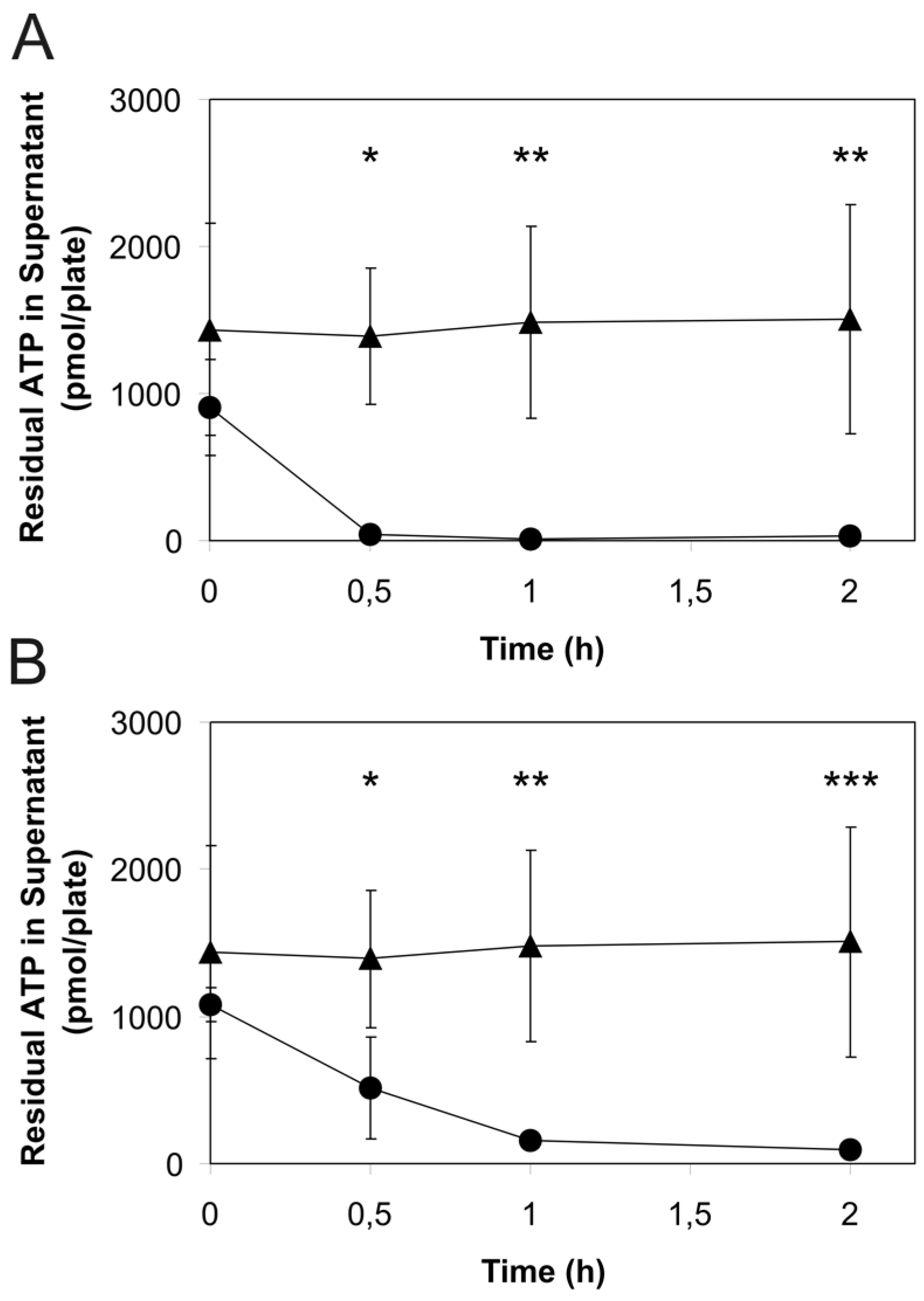
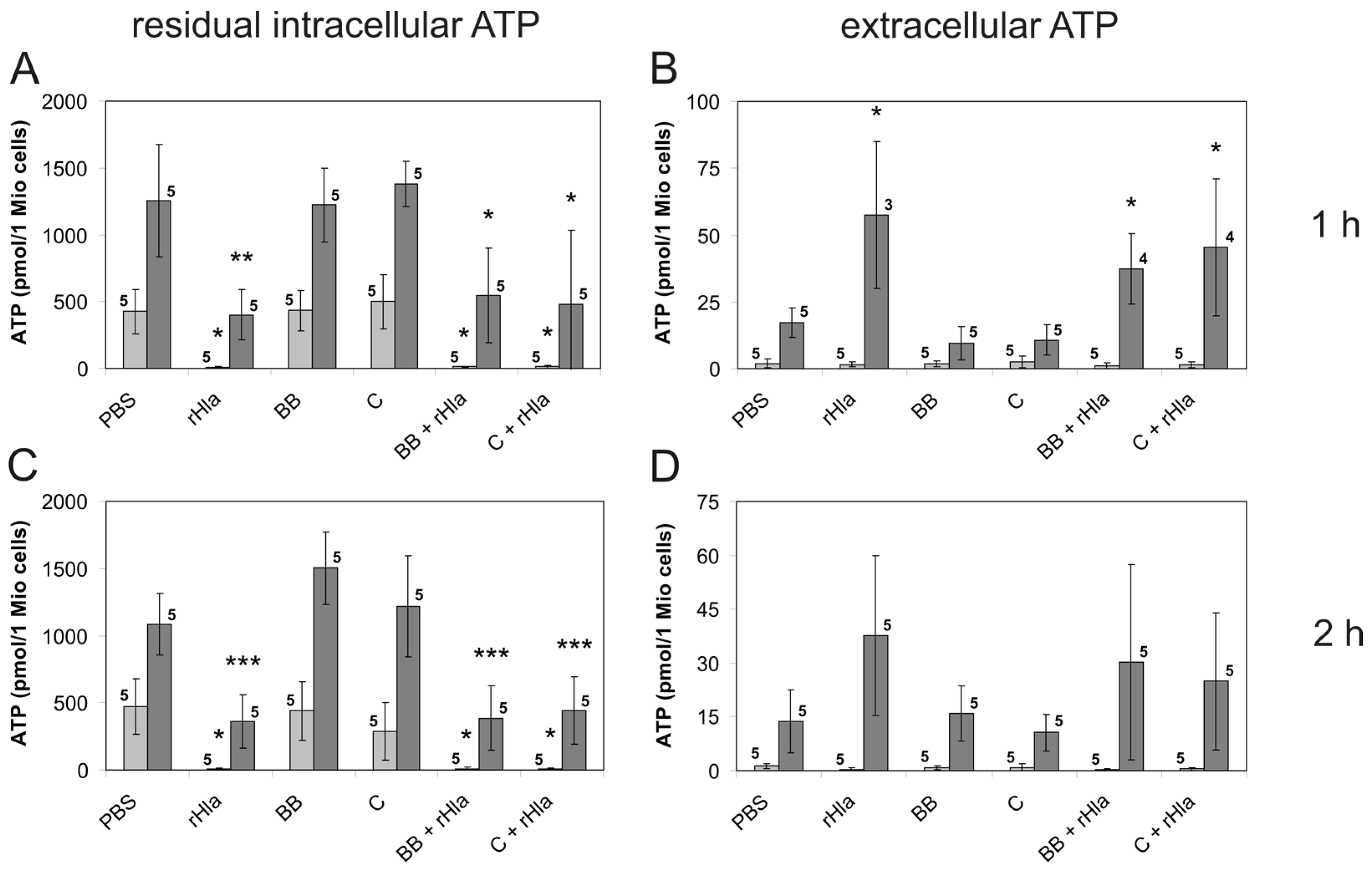
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Mitochondrial Inner Membrane Potential
4.4. Calcium Concentration in the Mitochondrial Matrix
4.5. Sample Preparation for Assaying Intra- and Extracellular ATP Concentrations
4.6. Estimation of the Rates of Extracellular ATP Degradation
4.7. Luminometric ATP Assay
4.8. Data Presentation and Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Menestrina, G.; Dalla Serra, M.; Comai, M.; Coraiola, M.; Viero, G.; Werner, S.; Colin, D.A.; Monteil, H.; Prevost, G. Ion channels and bacterial infection: the case of beta-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett. 2003, 552, 54–60. [Google Scholar] [CrossRef]
- Bubeck Wardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [PubMed]
- Hildebrandt, J.-P. Pore-forming virulence factors of Staphylococcus aureus destabilize epithelial barriers—Effects of alpha-toxin in the early phases of airway infection. AIMS Microbiol. 2015, 1, 11–36. [Google Scholar] [CrossRef]
- Ziebandt, A.-K.; Becher, D.; Ohlsen, K.; Hacker, J.; Hecker, M.; Engelmann, S. The influence of agr and σB in growth phase dependent regulation of virulence factors in Staphylococcus aureus. Proteomics 2004, 4, 3034–3047. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Pohl, M.; Bhakdi, S. Staphylococcus aureus alpha-toxin. Dual mechanism of binding to target cells. J. Biol. Chem. 1991, 266, 17195–17200. [Google Scholar] [PubMed]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Hellmann, N.; Walev, I.; Strand, D.; Plate, M.; Boukhallouk, F.; Brack, A.; Hanada, K.; Decker, H.; Bhakdi, S. Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J. Biol. Chem. 2006, 281, 26014–26021. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, L.; Miles, G.; Bayley, H. Role of the amino latch of staphylococcal alpha-hemolysin in pore formation: A co-operative interaction between the N terminus and position 217. J. Biol. Chem. 2006, 281, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E. α-Hemolysin from Staphylococcus aureus: An archetype of beta-barrel, channel-forming toxins. J. Struct. Biol. 1998, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Walev, I.; Martin, E.; Jonas, D.; Mohamadzadeh, M.; Müller-Klieser, W.; Kunz, L.; Bhakdi, S. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect. Immun. 1993, 61, 4972–4979. [Google Scholar] [PubMed]
- Eichstaedt, S.; Gäbler, K.; Below, S.; Müller, C.; Kohler, C.; Engelmann, S.; Hildebrandt, P.; Völker, U.; Hecker, M.; Hildebrandt, J.-P. Effects of Staphylococcus aureus-hemolysin A on calcium signalling in immortalized human airway epithelial cells. Cell Calcium 2009, 45, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandib, E.; Branton, D.; Deamer, W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, D.; Walev, I.; Berger, T.; Liebetrau, M.; Palmer, M.; Bhakdi, S. Novel path to apoptosis: Small transmembrane pores created by staphylococcal alpha-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect. Immun. 1994, 62, 1304–1312. [Google Scholar] [PubMed]
- Walev, I.; Palmer, M.; Martin, E.; Jonas, D.; Weller, U.; Hohn-Bentz, H.; Husmann, M.; Bhakdi, S. Recovery of human fibroblasts from attack by the pore-forming alpha-toxin of Staphylococcus aureus. Microb. Pathog. 1994, 17, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Lizak, M.; Yarovinsky, T.O. Phospholipid scramblase 1 mediates type I interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe 2012, 11, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Gierok, P.; Harms, M.; Richter, E.; Hildebrandt, J.-P.; Lalk, M.; Mostertz, J.; Hochgräfe, F. Staphylococcus aureus alpha-toxin mediates general and cell type-specific changes in metabolite concentrations of immortalized human airway epithelial cells. PLoS ONE 2014, 9, e94818. [Google Scholar] [CrossRef] [PubMed]
- Schwiebert, E.M.; Zsembery, A. Extracellular ATP as a signaling molecule for epithelial cells. Biochim. Biophys. Acta 2003, 1615, 7–32. [Google Scholar] [CrossRef]
- Walsh, D.E.; Harvey, B.J.; Urbach, V. CFTR regulation of intracellular calcium in normal and cystic fibrosis human airway epithelia. J. Membr. Biol. 2000, 177, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.H.; Lazarowski, E.R.; Picher, M.; Knowles, M.R.; Stutts, M.J.; Boucher, R.C. Basal nucleotide levels, release, and metabolism in normal and cystic fibrosis airways. Mol. Med. 2000, 6, 969–982. [Google Scholar] [PubMed]
- Okada, S.F.; Nicholas, R.A.; Kreda, S.M.; Lazarowski, E.R.; Boucher, R.C. Physiological regulation of ATP release at the apical surface of human airway epithelia. J. Biol. Chem. 2006, 281, 22992–23002. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, C.; Gassmann, O.; Pranskevich, J.N.; Boassa, D.; Smock, A.; Wang, J.; Dahl, G.; Steinem, C.; Sosinsky, G.E. Pannexin1 and pannexin2 channels show quaternary similarities to connexons and different oligomerization numbers from each other. J. Biol. Chem. 2010, 285, 24420–24431. [Google Scholar] [CrossRef] [PubMed]
- Bond, S.R.; Naus, C. The pannexins: Past and present. Front. Physiol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Dahl, G.; Keane, R.W. Pannexin: From discovery to bedside in 11+/−4 years? Brain Res. 2012, 1487, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ransford, G.A.; Fregien, N.; Qiu, F.; Dahl, G.; Conner, G.E.; Salathe, M. Pannexin 1 contributes to ATP release in airway epithelia. Am. J. Respir. Cell Mol. Biol. 2009, 41, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.S.; Min, Y.G.; Rhee, C.S.; Jung, I.H.; Koh, Y.Y.; Jang, T.Y.; Jung, D.H. Effects of alpha-toxin of Staphylococcus aureus on the ciliary activity and ultrastructure of human nasal ciliated epithelial cells. Laryngoscope 1999, 109, 2021–2024. [Google Scholar] [CrossRef] [PubMed]
- Bantel, H.; Sinha, B.; Domschke, W.; Peters, G.; Schulze-Osthoff, K.; Janicke, R.U. α-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J. Cell Biol. 2001, 155, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Button, B.; Boucher, R.C. Role of mechanical stress in regulating airway surface hydration and mucus clearance rates. Respir. Physiol. Neurobiol. 2008, 163, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Douillet, C.D.; Robinson, W.P.; Milano, P.M.; Boucher, R.C.; Rich, P.B. Nucleotides induce IL-6 release from human airway epithelia via P2Y2 and p38 MAPK-dependent pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L734–L746. [Google Scholar] [CrossRef] [PubMed]
- Menzies, B.E.; Kernodle, D.S. Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: Role of histidines in toxin activity in vitro and in a murine model. Infect. Immun. 1994, 62, 1843–1847. [Google Scholar] [PubMed]
- Eiffler, I.; Behnke, J.; Ziesemer, S.; Müller, C.; Hildebrandt, J.-P. Staphylococcus aureus alpha-toxin-mediated cation entry depolarizes membrane potential and activates p38 MAP kinase in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L676–L685. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Schmidtmann, F.; Robinson, T.M.; Yohannes, A.; Fahmi, N.E.; Bezrukov, S.M.; Hecht, S.M. Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg. Med. Chem. 2007, 15, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Ragle, B.E.; Karginov, V.A.; Bubeck Wardenburg, J. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Bayley, H. Interaction of the noncovalent molecular adapter, beta-cyclodextrin, with the staphylococcal alpha-hemolysin pore. Biophys. J. 2000, 79, 1967–1975. [Google Scholar] [CrossRef]
- Wang, J.; Jackson, D.G.; Dahl, G. The food dye FD&C Blue No. 1 is a selective inhibitor of the ATP release channel Panx1. J. Gen. Physiol. 2013, 141, 649–656. [Google Scholar] [PubMed]
- Bruzzone, R.; Barbe, M.T.; Jakob, N.J.; Monyer, H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J. Neurochem. 2005, 92, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, E.R.; Sesma, J.I.; Seminario, L.; Esther, C.R.; Kreda, S.M. Nucleotide release by airway epithelia. Subcell. Biochem. 2011, 55, 1–15. [Google Scholar] [PubMed]
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Seminario-Vidal, L.; Okada, S.F.; Sesma, J.I.; Kreda, S.M.; van Heusden, C.A.; Zhu, Y.; Jones, L.C.; O’Neal, W.K.; Penuela, S.; Laird, D.W.; et al. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J. Biol. Chem. 2011, 286, 26277–26286. [Google Scholar] [CrossRef] [PubMed]
- La Sala, A.; Ferrari, D.; Di Virgilio, F.; Idzko, M.; Norgauer, J.; Girolomoni, G. Alerting and tuning the immune response by extracellular nucleotides. J. Leukoc. Biol. 2003, 73, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Locovei, S.; Scemes, E.; Qiu, F.; Spray, D.C.; Dahl, G. Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 2007, 581, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, A.M.; Ribeiro, C.M.; Boucher, R.C. Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. J. Gen. Physiol. 2001, 117, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Kawakami, M.; Sasaki, S.; Katsumata, T.; Mori, H.; Yoshida, H.; Nakahari, T. ATP regulation of ciliary beat frequency in rat tracheal and distal airway epithelium. Exp. Physiol. 2005, 90, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Ito, Y.; Sato, S.; Ishikawa, T.; Kondo, M.; Nakayama, S.; Shimokata, K.; Kume, H. Apical and basolateral ATP-induced anion secretion in polarized human airway epithelia. Am. J. Respir. Cell Mol. Biol. 2004, 30, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.A.; Sugar, R.A.; Jackson, A.D. Nucleotide-mediated mucin secretion from differentiated human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Danahay, H.; Atherton, H.C.; Jackson, A.D.; Kreindler, J.L.; Poll, C.T.; Bridges, R.J. Membrane capacitance and conductance changes parallel mucin secretion in the human airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L558–L569. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.M.; Pifer, M.A.; Przybylski, R.J.; Dubyak, G.R. Methylene ATP analogs as modulators of extracellular ATP metabolism and accumulation. Br. J. Pharmacol. 2004, 142, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Picher, M.; Boucher, R.C. Human airway ecto-adenylate kinase. A mechanism to propagate ATP signaling on airway surfaces. J. Biol. Chem. 2003, 278, 11256–11264. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.P.; Eltzschig, H.K.; Eckle, T.; Thompson, L.F. Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. 2006, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Parker, W.; Clancy, J.P. Adenosine regulation of cystic fibrosis transmembrane conductance regulator through prostenoids in airway epithelia. Am. J. Respir. Cell Mol. Biol. 2006, 34, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Duchen, M.R. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008, 23, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Schwiering, M.; Brack, A.; Stork, R.; Hellmann, N. Lipid and phase specificity of alpha-toxin from S. aureus. Biochim. Biophys. Acta 2013, 1828, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Below, S.; Konkel, A.; Zeeck, C.; Müller, C.; Kohler, C.; Engelmann, S.; Hildebrandt, J.-P. Virulence factors of Staphylococcus aureus induce Erk-MAP kinase activation and c-Fos expression in S9 and 16HBE14o- human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L470–L479. [Google Scholar] [CrossRef] [PubMed]
- Hermann, I.; Räth, S.; Ziesemer, S.; Volksdorf, T.; Dress, R.J.; Gutjahr, M.; Müller, C.; Beule, A.G.; Hildebrandt, J.-P. Staphylococcus aureus-hemolysin A disrupts cell-matrix adhesions in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Scaduto, R.C.; Grotyohann, L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999, 76, 469–477. [Google Scholar] [CrossRef]
- Shuttleworth, T.J.; Thompson, J.L. Intracellular [Ca2+] and inositol phosphates in avian nasal gland cells. Am. J. Physiol. Cell Physiol. 1989, 257, C1020–C1029. [Google Scholar]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baaske, R.; Richter, M.; Möller, N.; Ziesemer, S.; Eiffler, I.; Müller, C.; Hildebrandt, J.-P. ATP Release from Human Airway Epithelial Cells Exposed to Staphylococcus aureus Alpha-Toxin. Toxins 2016, 8, 365. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8120365
Baaske R, Richter M, Möller N, Ziesemer S, Eiffler I, Müller C, Hildebrandt J-P. ATP Release from Human Airway Epithelial Cells Exposed to Staphylococcus aureus Alpha-Toxin. Toxins. 2016; 8(12):365. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8120365
Chicago/Turabian StyleBaaske, Romina, Mandy Richter, Nils Möller, Sabine Ziesemer, Ina Eiffler, Christian Müller, and Jan-Peter Hildebrandt. 2016. "ATP Release from Human Airway Epithelial Cells Exposed to Staphylococcus aureus Alpha-Toxin" Toxins 8, no. 12: 365. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8120365