Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Clinical Samples
2.3. RNA Extraction, Viral Load Determination, and Full-Genome Amplification
2.4. Illumina Sequencing
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
2.7. Accession Numbers
3. Results
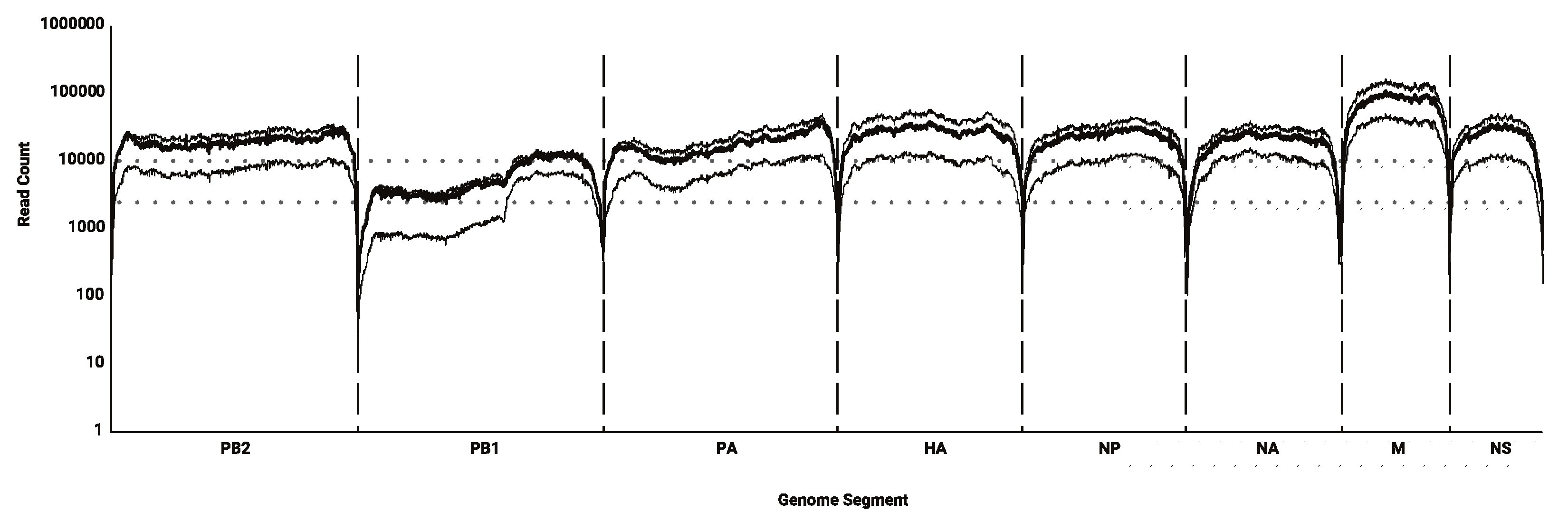
3.1. Sequencing Efficacy
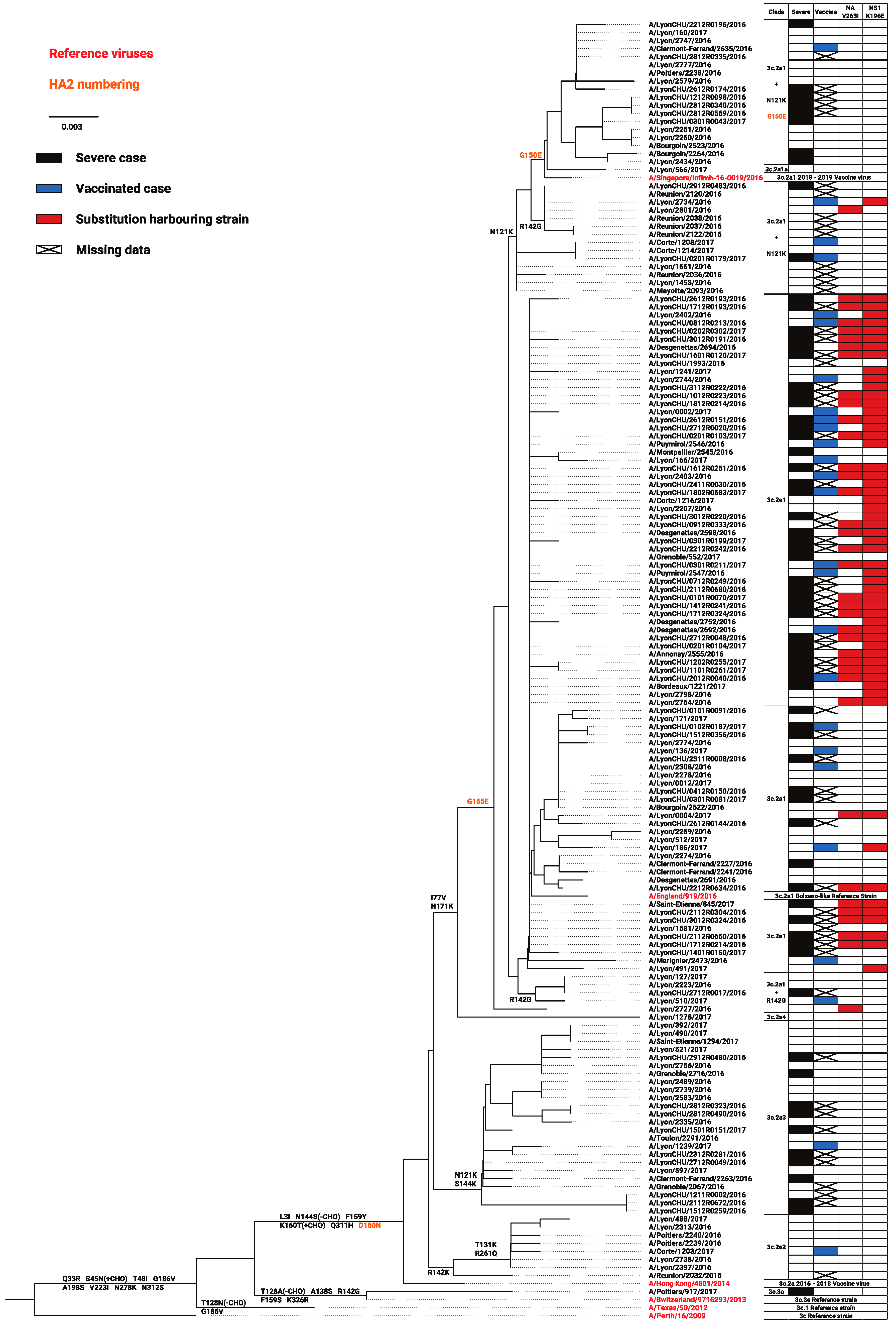
3.2. Distribution of Consensus Variants According to Clades, Severity, or Vaccination
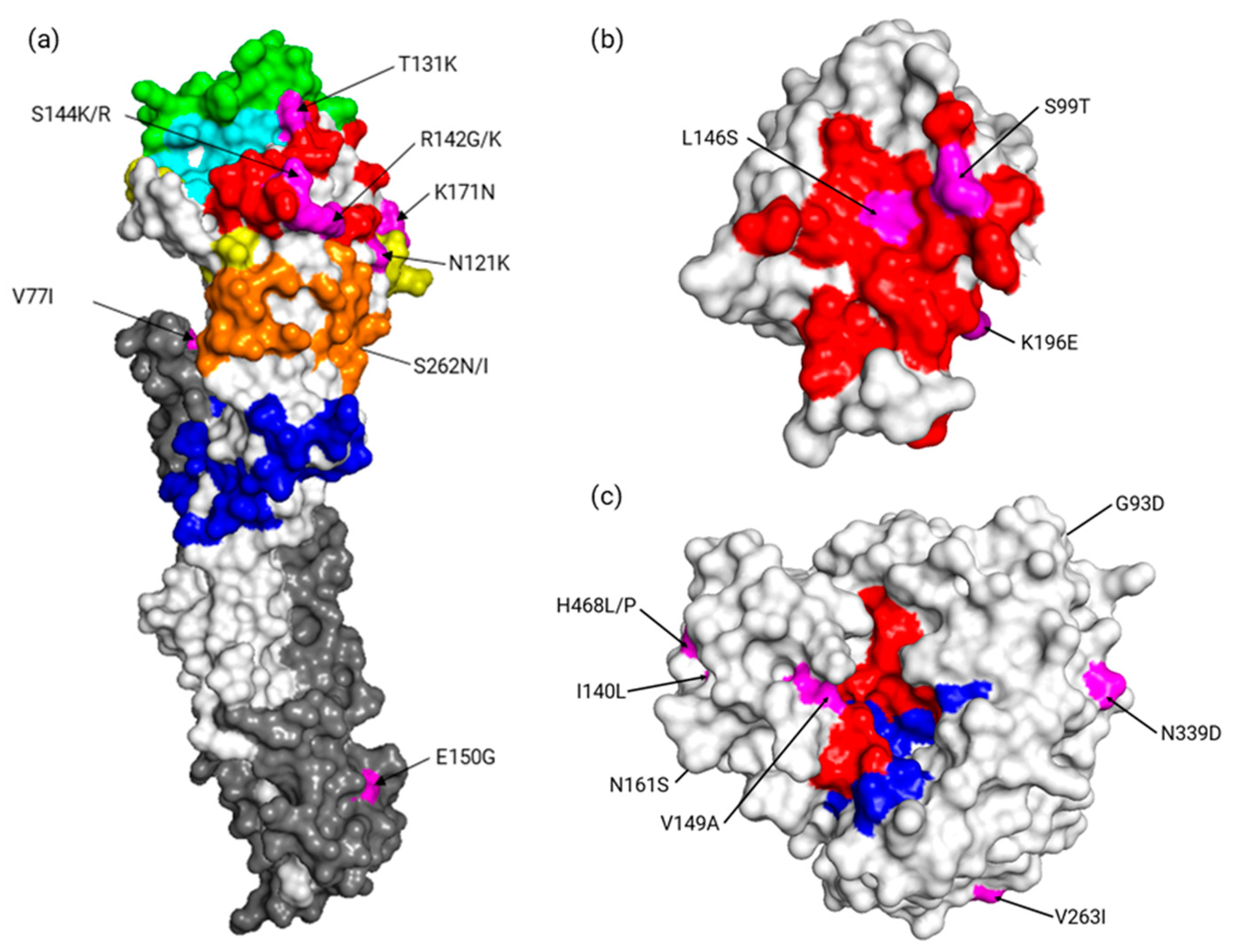
3.3. Diversity and Quasispecies Analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Influenza (Seasonal) Fact Sheet. Prevention and Control of Influenza Pandemics and Annual Epidemics. Available online: http://www.who.int/mediacentre/factsheets/fs211/en/ (accessed on 24 January 2018).
- Uhart, M.; Bricout, H.; Clay, E.; Largeron, N. Public health and economic impact of seasonal influenza vaccination with quadrivalent influenza vaccines compared to trivalent influenza vaccines in Europe. Hum. Vaccin. Immunother. 2016, 12, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Chowell, G.; Bertozzi, S.M.; Colchero, M.A.; Celia, A.A.; Mauricio, H. Severe Respiratory Disease Concurrent with the Circulation of H1N1 Influenza. N. Engl. J. Med. 2009, 361, 674–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, D.J.; Honein, M.A.; Rasmussen, S.A.; Williams, J.L.; Swerdlow, D.L.; Biggerstaff, M.S.; Lindstrom, S.; Louie, J.K.; Christ, C.M.; Bohm, S.R. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet 2009, 374, 451–458. [Google Scholar] [CrossRef]
- Koel, B.F.; Burke, D.F.; Bestebroer, T.M.; van der Vliet, S.; Zondag, G.C.; Vervaet, G.; Skepner, E.; Lewis, N.S.; Spronken, M.I.; Russell, C.A.; et al. Substitutions Near the Receptor Binding Site Determine Major Antigenic Change During Influenza Virus Evolution. Science 2013, 342, 976–979. [Google Scholar] [CrossRef]
- Abed, Y.; Baz, M.; Boivin, G. Impact of neuraminidase mutations conferring influenza resistance to neuraminidase inhibitors in the N1 and N2 genetic backgrounds. Antivir. Ther. 2006, 11, 971–976. [Google Scholar] [PubMed]
- Boivin, G. Detection and management of antiviral resistance for influenza viruses. Influenza Other Respir. Viruses 2013, 7, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedde, M.; Wählisch, S.; Wolff, T.; Schweiger, B. Predominance of HA-222D/G Polymorphism in Influenza A(H1N1) pdm09 Viruses Associated with Fatal and Severe Outcomes Recently Circulating in Germany. PLoS ONE 2013, 8, e57059. [Google Scholar] [CrossRef]
- Hung, I.F.; To, K.K.; Lee, C.K.; Lin, C.K.; Chan, J.F.; Tse, H.; Cheng, V.C.; Chen, H.; Ho, P.L.; Tse, C.W.; et al. Effect of Clinical and Virological Parameters on the Level of Neutralizing Antibody against Pandemic Influenza A Virus H1N1 2009. Clin. Infect. Dis. 2010, 51, 274–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.; Qin, Y.; Cowling, B.J.; Ren, X.; Wardrop, N.A.; Gilbert, M.; Tsang, T.K.; Wu, P.; Feng, L.; Jiang, H.; et al. Global epidemiology of avian influenza A(H5N1) virus infection in humans, 1997–2015: A systematic review. Lancet Infect. Dis. 2016, 16, e108–e118. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Pappas, C.; Yang, H.; Carney, P.J.; Pearce, M.B.; Katz, J.M.; Stevens, J.; Tumpey, T.M. Assessment of transmission, pathogenesis and adaptation of H2 subtype influenza viruses in ferrets. Virology 2015, 477, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Murano, K.; Ohniwa, R.L.; Kawaguchi, A.; Nagata, K. Oseltamivir Expands Quasispecies of Influenza Virus through Cell-to-cell Transmission. Sci. Rep. 2015, 5, 9163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinis, J.M.; Florek, N.W.; Fatola, O.O.; Moncla, L.H.; Mutschler, J.P.; Charlier, O.K.; Meece, J.K.; Belongia, E.A.; Friedrich, T.C. Deep Sequencing Reveals Potential Antigenic Variants at Low Frequencies in Influenza A Virus-Infected Humans. J. Virol. 2016, 90, 3355–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilijevic, J.; Zamarreño, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gómez, G.; Rodriguez, G.; Pérez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathogens. 2017, 13, e1006650. [Google Scholar] [CrossRef]
- Ghedin, E.; Holmes, E.C.; DePasse, J.V.; Pinalla, L.T.; Fitch, A.; Hamelin, M.E.; Papenburg, J.; Boivin, G. Presence of Oseltamivir-Resistant Pandemic A/H1N1 Minor Variants Before Drug Therapy With Subsequent Selection and Transmission. J. Infect. Dis. 2012, 206, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.S.; Hooper, K.A.; Ollodart, A.R.; Dingens, A.S.; Bloom, J.D. Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. eLife 2016, 5, e13974. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.M.; Galvani, A.P.; Bush, R.M. Ecological and immunological determinants of influenza evolution. Nature 2003, 422, 428–433. [Google Scholar] [CrossRef]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef]
- Muscatello, D.J.; Newall, A.T.; Dwyer, D.E.; MacIntyre, C.R. Mortality Attributable to Seasonal and Pandemic Influenza, Australia, 2003 to 2009, Using a Novel Time Series Smoothing Approach. PLoS ONE 2013, 8, e64734. [Google Scholar] [CrossRef]
- Simonsen, L. The global impact of influenza on morbidity and mortality. Vaccine 1999, 17, S3–S10. [Google Scholar] [CrossRef]
- Équipes de surveillance de la grippe. Surveillance de la grippe en France, saison 2016-2017. Bull Epidémiol Hebd. 2017, 466–475. Available online: http://invs.santepubliquefrance.fr/beh/2017/22/2017_22_1.html (accessed on 28 January 2019).
- Zost, S.J.; Parkhouse, K.; Gumina, M.E.; Kim, K.; Diaz Perez, S.; Wilson, P.C.; Treanor, J.J.; Sant, A.J.; Cobey, S.; Hensley, S.E. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 2017, 114, 12578–12583. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichon, M.; Simon, B.; Valette, M.; Bal, A.; Picard, C.; Escuret, V.; Ottmann, M.; Gillet, Y.; Ader, F.; Lina, B.; et al. Evolution of influenza genome diversity during infection in immunocompetent patients. bioRxiv 2018, 435263. [Google Scholar] [CrossRef]
- Rotmistrovsky, K.; Agarwala, R. BMTagger: Best Match Tagger for removing human reads from metagenomics datasets. Available online: ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/ (accessed on 26 April 2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, D.; Von Kuster, G.; Bouvier, E.; Baker, D.; Afgan, E.; Stoler, N.; Taylor, J.; Nekrutenko, A. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014, 15, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.; Kingsford, C. GiRaF: Robust, computational identification of influenza reassortments via graph mining. Nucleic Acids Res. 2011, 39, e34. [Google Scholar] [CrossRef] [PubMed]
- Summary Table of Neuraminidase Amino Acid Substitutions Associated with Reduced Inhibition by Neuraminidase Inhibitors (NAI). Available online: http://www.who.int/influenza/gisrs_laboratory/antiviral_susceptibility/nai_overview/en/ (accessed on 24 January 2018).
- Murcia, P.R.; Hughes, J.; Battista, P.; Lloyd, L.; Baillie, G.J.; Ramirez-Gonzalez, R.H.; Ormond, D.; Olivier, K.; Elton, D.; Mumford, J.A.; et al. Evolution of an Eurasian Avian-like Influenza Virus in Naïve and Vaccinated Pigs. PLoS Pathog. 2012, 8, e1002730. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Tang, J.W.-T.; Kong, D.H.-L.; Loh, T.P.; Chiang, D.K.; Lam, T.T.; Koay, E.S. Comparison of Mutation Patterns in Full-Genome A/H3N2 Influenza Sequences Obtained Directly from Clinical Samples and the Same Samples after a Single MDCK Passage. PLoS ONE 2013, 8, e79252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.M.; Domingues, P.; Zell, R.; Hale, B.G. Structure-Guided Functional Annotation of the Influenza A Virus NS1 Protein Reveals Dynamic Evolution of the p85β-Binding Site During Circulation in Humans. J. Virol. 2017, 91, e01081-17. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Santos, C.; Krug, R.; Subbarao, K. Glutamic Acid at Position 196 of the NS1 Protein is Associated with Both a Block in IRF3 Activation and the Virulence of a HPAI H5N1 Virus in Mice. Fourth ESWI Influenza Conference Poster. Available online: https://core.ac.uk/download/pdf/37965740.pdf (accessed on 24 April 2018).
- Ramírez-Salinas, G.L.; García-Machorro, J.; Quiliano, M.; Zimic, M.; Briz, V.; Rojas-Hernández, S.; Correa-Basurto, J. Molecular modeling studies demonstrate key mutations that could affect the ligand recognition by influenza AH1N1 neuraminidase. J. Mol. Model. 2015, 21, 292. [Google Scholar]
- Wan, H.; Yang, H.; Shore, D.A.; Garten, R.J.; Couzens, L.; Gao, J.; Jiang, L.; Carney, P.J.; Villanueva, J.; Stevens, J.; et al. Structural characterization of a protective epitope spanning A(H1N1) pdm09 influenza virus neuraminidase monomers. Nat. Commun. 2015, 6, 6114. [Google Scholar] [CrossRef] [PubMed]
- Solmone, M.; Vincenti, D.; Prosperi, M.C.F.; Bruselles, A.; Ippolito, G.; Capobianchi, M.R. Use of Massively Parallel Ultradeep Pyrosequencing to Characterize the Genetic Diversity of Hepatitis B Virus in Drug-Resistant and Drug-Naive Patients and To Detect Minor Variants in Reverse Transcriptase and Hepatitis B S Antigen. J. Virol. 2009, 83, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Katano, H.; Nakajima, N.; Tobiume, M.; Ainai, A.; Sekizuka, T.; Hasegawa, H.; Tashiro, M.; Sasaki, Y.; Arakawa, Y.; et al. Characterization of Quasispecies of Pandemic 2009 Influenza A Virus (A/H1N1/2009) by De Novo Sequencing Using a Next-Generation DNA Sequencer. PLoS ONE 2010, 5, e10256. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, B.; Chillemi, G.; Abbate, I.; Bruselles, A.; Rozera, G.; Castrignanò, T.; Paoletti, D.; Picardi, E.; Desideri, A.; Pesole, G.; et al. Assembly and characterization of pandemic influenza A H1N1 genome in nasopharyngeal swabs using high-throughput pyrosequencing. New Microbiol. 2011, 34, 391–397. [Google Scholar] [PubMed]
- Nishijima, N.; Marusawa, H.; Ueda, Y.; Takahashi, K.; Nasu, A.; Osaki, Y.; Kou, T.; Yazumi, S.; Fujiwara, T.; Tsuchiya, S.; et al. Dynamics of Hepatitis B Virus Quasispecies in Association with Nucleos(t)ide Analogue Treatment Determined by Ultra-Deep Sequencing. PLoS ONE 2012, 7, e35052. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto, I.; Maehama, T.; Sekizuka, T.; Ogasawara, Y.; Kondo, K.; Kusumoto-Matsuo, R.; Mori, S.; Ishii, Y.; Takeuchi, T.; Yamaji, T.; et al. Genetic Variation of Human Papillomavirus Type 16 in Individual Clinical Specimens Revealed by Deep Sequencing. PLoS ONE 2013, 8, e80583. [Google Scholar] [CrossRef]
- Ruiz-Carrascoso, G.; Romero-Gómez, M.P.; Plaza, D.; Mingorance, J. Rapid detection and quantitation of ganciclovir resistance in cytomegalovirus quasispecies. J. Med. Virol. 2013, 85, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions within a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; McCrone, J.T.; Petrie, J.G.; Truscon, R.; Johnson, E.; Mantlo, E.K.; Monto, A.S.; Lauring, A.S. Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLoS Pathog. 2017, 13, e1006194. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Mild Outcome | Severe Outcome | ||
|---|---|---|---|---|
| Baseline Demographics | Number of patients (% of total) | 97 (55%) | 79 (45%) | |
| Age in median years (range) | 34 (0–91) | 73 (1–97) | ||
| Sex | Male | 48 | 36 | |
| Female | 48 | 43 | ||
| Origin | Lyon University Hospitals | 6 | 65 | |
| Surveillance network | 91 | 14 | ||
| Sample Characteristics | Sample type | NPA | 2 | 6 |
| NS | 95 | 49 | ||
| TBA | 0 | 13 | ||
| BAL | 0 | 11 | ||
| Median time since onset of symptoms, days (range) | 1 (0–5) | 3 (0–8) | ||
| Viral load—median cycle threshold (range) | 19.7 (14.9–30) | 23.3 (17–35.3) | ||
| Clinical Characteristics | Vaccinated against Influenza for the current season | 21 | 7 | |
| Severe outcome risk factor * | 17 | 65 | ||
| Main severity component | Respiratory | - | 70 | |
| Neurological | 5 | |||
| Multiple organ failure | 4 | |||
| (a) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA1 | HA2 | NP | |||||||||||||
| 255 | 480 | 52 | 614 | 565 | 121 | 131 | 142 | 144 | 171 | 262 | 77 | 150 | 450 | 472 | ||||
| Significant p < | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 0.05 | 0.05 | 0.01 | 0.05 | 0.05 | 0.05 | 0.01 | 0.01 | 0.05 | 0.05 | |||
| Consensus | V | V | R | E | V | N | T | R | S | K | S | V | E | G | A | |||
| Variants | I | I | W/K | D | M | K | K | G/K | K/R | N | N/I | I | G | S/N | T | |||
| Severe cases | - | - | - | - | - | N | T | R | - | - | - | - | - | - | - | |||
| Vaccinated cases | V | V | W/K | D | M | N | - | - | S | K | S | V | E | G | A | |||
| NA | M1 | M2 | NS1 | NS2 | ||||||||||||||
| 93 | 140 | 149 | 161 | 263 | 339 | 468 | 147 | 23 | 99 | 146 | 196 | 224 | 67 | |||||
| Significant p < | 0.05 | 0.01 0.005 | 0.001 0.001 | 0.05 | 1 × 10−4 | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | 1 × 10−4 0.001 | 0.05 | 0.05 | ||||
| Consensus | G | I | V | N | V | N | H | V | S | S | L | K | R | E | ||||
| Variants | D | L | A | S | V | D | L/P | M | N | T | S | E | S/K | A/K | ||||
| Severe cases | - | I | A | - | I | - | - | - | S | - | - | E | R | E | ||||
| Vaccinated cases | G | I | A | N | - | N | H | M | - | S | L | E | - | - | ||||
| (b) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NS1 | NS1 | |||||||||
| Mild | K196 | 196E | Total | Severe | K196 | 196E | Total | |||
| NA | V263 | 64 | 13 | 77 | NA | V263 | 32 | 9 | 41 | |
| 263I | 2 | 8 | 10 | 263I | 0 | 27 | 27 | |||
| Total | 66 | 21 | 87 | Total | 32 | 36 | 68 | |||
| Segment | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||
| Mild/Severe | Threshold | 1 | n | 62/47 | 6/9 | 58/46 | 74/56 | 61/45 | 64/49 | 82/68 | 60/46 |
| Mean | 1.88/2.38 | 2.33/2.55 | 2.03/3.46 | 0.99/1.54 | 2.75/2.73 | 1.64/2.24 | 0.41/0.65 | 0.42/0.59 | |||
| p | 0.17 | 0.81 | <0.001 | <0.005 | 0.88 | 0.03 | 0.53 | 0.17 | |||
| 5 | n | 80/69 | 53/38 | 80/67 | 85/69 | 81/65 | 83/68 | 91/73 | 83/68 | ||
| Mean | 1.68/1.95 | 0.83/1.05 | 1.71/2.84 | 0.93/1.43 | 2.40/2.23 | 1.64/2.07 | 0.41/0.60 | 0.42/0.54 | |||
| p | 0.40 | 0.26 | <0.001 | <0.005 | 0.73 | 0.27 | 0.75 | 0.25 | |||
| Not Vaccinated/Vaccinated | Threshold | 1 | n | 45/23 | 5/3 | 44/22 | 56/26 | 46/22 | 51/23 | 65/26 | 45/19 |
| Mean | 2.22/1.78 | 3.4/1.3 | 2.84/2.13 | 1.20/0.77 | 3.22/2.27 | 1.84/1.47 | 0.51/0.35 | 0.44/0.47 | |||
| p | 0.57 | 0.36 | 0.49 | 0.33 | 0.05 | 0.49 | 0.76 | 0.58 | |||
| 5 | n | 62/26 | 37/17 | 62/26 | 67/27 | 63/26 | 65/27 | 69/27 | 65/25 | ||
| Mean | 1.85/1.69 | 1.14/0.53 | 2.23/2.00 | 1.18/0.74 | 2.68/2.15 | 1.75/1.63 | 0.51/0.33 | 0.45/0.52 | |||
| p | 0.76 | 0.26 | 0.89 | 0.28 | 0.30 | 0.77 | 0.62 | 0.47 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, B.; Pichon, M.; Valette, M.; Burfin, G.; Richard, M.; Lina, B.; Josset, L. Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season. Viruses 2019, 11, 108. https://0-doi-org.brum.beds.ac.uk/10.3390/v11020108
Simon B, Pichon M, Valette M, Burfin G, Richard M, Lina B, Josset L. Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season. Viruses. 2019; 11(2):108. https://0-doi-org.brum.beds.ac.uk/10.3390/v11020108
Chicago/Turabian StyleSimon, Bruno, Maxime Pichon, Martine Valette, Gwendolyne Burfin, Mathilde Richard, Bruno Lina, and Laurence Josset. 2019. "Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season" Viruses 11, no. 2: 108. https://0-doi-org.brum.beds.ac.uk/10.3390/v11020108