Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity


Laboratory of Viral Diseases, NIAID, NIH, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Viruses 2019, 11(4), 346; https://0-doi-org.brum.beds.ac.uk/10.3390/v11040346
Submission received: 19 March 2019
/
Revised: 11 April 2019
/
Accepted: 13 April 2019
/
Published: 16 April 2019
(This article belongs to the Special Issue What’s New with Flu?)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Influenza A virions possess two surface glycoproteins—the hemagglutinin (HA) and neuraminidase (NA)—which exert opposite functions. HA attaches virions to cells by binding to terminal sialic acid residues on glycoproteins/glycolipids to initiate the infectious cycle, while NA cleaves terminal sialic acids, releasing virions to complete the infectious cycle. Antibodies specific for HA or NA can protect experimental animals from IAV pathogenesis and drive antigenic variation in their target epitopes that impairs vaccine effectiveness in humans. Here, we review progress in understanding HA/NA co-evolution as each acquires epistatic mutations to restore viral fitness to mutants selected in the other protein by host innate or adaptive immune pressure. We also discuss recent exciting findings that antibodies to HA can function in vivo by blocking NA enzyme activity to prevent nascent virion release and enhance Fc receptor-based activation of innate immune cells.
1. Introduction
Year in and year out, influenza A virus (IAV) imposes an enormous economic and health burden, with the potential to cause periodic catastrophic pandemics. IAV is an enveloped negative stranded RNA virus with a segmented genome belonging to the Orthomyxoviridae family. Eight gene segments code for 10 structural and at least 9 nonstructural/regulatory proteins [1,2,3]. PB1, PB2, PA, NP, M1, NS1, and NEP are present inside the lipid envelope, while M2, hemagglutinin (HA), and neuraminidase (NA) are embedded in the envelope and available for antibody (Ab) binding.
Inactivated IAV vaccines induce antibody (Ab) responses to the HA, although it is now appreciated that NA might be an important target as well [4]. The high mutational tolerance [5] of these surface glycoproteins, both structurally and functionally compared to other IAV gene products [6], facilitates their “antigenic drift”—immune escape from Ab responses based on mutant selection [7]. Glycoprotein change is enhanced by the segmented nature of IAV genome, which facilitates intergenic epistasis through rapid recombination of mutant genes. Such recombination occurs rapidly in vivo [8,9,10,11,12] and enables antigenic shift, a process that introduces novel HA and NA genes from the enormous animal reservoir into the human IAV virome [9].
Eighteen hemagglutinin and 11 neuraminidase subtypes are known to exist in nature. All but H17N10 and H18N11 subtypes, found to date in Peruvian bats [13,14], circulate in wild aquatic birds, which is by far the largest of the known natural IAV reservoirs, which also include humans, swine, horses, dogs, and seals. Based on sequencing data available in GenBank, out of the 144 possible HA-NA combinations in non-bat IAVs, over 120 combinations have been documented in nature [15,16]. While many combinations are possible, far fewer are prevalent in nature, consistent with the co-evolution of HA and NA subtypes. Here we review the functional, genetic, and immunological interactions of the HA and NA.
2. HA Attaches, NA Releases
HA is a homotrimer whose globular domain contains a receptor binding site (RBS) specific for sialic acid (SA), which terminates many host glycans. The RBS is surrounded by antigenic sites recognized by the most potent virus neutralizing Abs. HA initiates infection by attaching virus to SA and possibly other receptors on the target cell surface [17,18]. Attachment is a complex process influenced by multiple parameters. The avidity of a single HA trimer for SA is low, with mM to high μM Kd values. However, multivalent binding of multiple HA trimers on the virion results in 104- to 106-fold increase in avidity [19,20,21,22], making attachment essentially irreversible in the absence of mitigating factors (e.g., NA or attachment blocking Abs). The effect of virion morphology on binding is potentially important, as freshly isolated viruses are typically filamentous, becoming more spherical (~100 nm diameter) during adaptation to cultured cells or eggs [23,24]. While intuition suggests that filaments should bind cells better than spheres, the available data suggest otherwise [25,26].
The specificity of HA for various types of SA linkage is a major contributor to their host and organ tropism. HA from human isolates generally prefer α2,6-linked SAs, while avian lineage HAs favor α2,3 linkages [25,27]. α2,6-linked SA glycan preference appears to dictate a requirement for fibronectin to initiate infection post attachment [28], pointing to unappreciated subtleties in how HA-mediated attachment leads to productive infection. The α2,6-α2,3-linked human–avian dichotomy is a gross oversimplification of HA specificity, and there is evidence that HA specificity can vary successively among human isolates. While an α2,3-linked SAs preference is associated with enhanced pathogenicity, it can also impair replication and aerosolization [29,30,31]. On the other hand, there are reports that SA-binding specificity has no apparent effect on IAV transmissibility or pathogenicity [32,33,34], suggesting that receptor binding preference is not a sole determinant of these functions. It is clear that HA acquisition of glycans during its evolution in humans can influence HA binding avidity, typically [35,36,37], but not always [22], decreasing binding. As the H3 HA has accumulated glycans, HA specificity has modulated towards branched glycans with extended poly-N-acetyl-lactosamine chains capable of bridging two RBSs of single HA trimer to enhance avidity [32,38].
Internalization of cell-associated virions occurs via multiple mechanisms including clathrin/caveolin-dependent and -independent endocytosis or, in the case of filamentous strains, macropinocytosis [39,40,41,42,43]. When the lowering endosomal pH reaches a trigger point that varies with HA strain, HA conformation changes to expose a domain that mediates fusion of viral and cellular membranes, releasing the viral core into the cytosol [44,45,46,47,48,49,50]. Proper acid triggered exposure of the fusion peptide requires cleavage of the HA into two subunits (HA1 and HA2), normally performed by furin or furin-like proteases during or after HA egress through the late secretory pathway [51,52].
Unlike HA, NA is not needed to initiate infection [17], and indeed, inhibiting NA can enhance infectivity [53]. NA plays an essential role in completing the infectious cycle by enabling virion release and preventing HA-mediated aggregation of virions by desialylating HA (and other NA molecules) [54,55,56,57] and possibly virion glycolipids as well. For some strains, HA desialylation is required for HA cleavage by proteases to activate fusion activity [58,59].
3. HA/NA Coevolution: NA Perspective
As a functional HA antagonist, NA walks a fine line. It must have sufficient activity to release and disaggregate virions, but not so much as to reduce HA-mediated attachment to initiate infection. The earliest and most basic evidence for HA/NA co-evolution is the non-random association of HA and NA subtypes that occur in natural IAV isolates, since it is clear that reassortment is a highly robust process in nature [8,10,11,12] and that co-infections commonly occur in many species with viruses of various subtypes. In one of the first studies to understand this preference, Baum and Paulson reported that N2 NA specificity evolved to match HA specificity during evolution of human N2 viruses from 1957 to 1987 [60]. In the most extreme example of HA–NA cooperation, NA is capable of evolving a sialic acid binding site that functionally replaces the HA binding site (Figure 1A) [32,61,62,63]. This renders IAV into a paramyxovirus-like configuration with a fusion protein (HA = paramyxovirus F protein) and a combined SA receptor/neuraminidase protein (NA = paramyxovirus HN). As with the HN, the NA receptor site is physically separated from the catalytic site. NA is also capable of modifying its active site to simultaneously serve as a sialic receptor for viral attachment, as occurred during the recent human H3N2 viruses [32,64]. Infection of cultured cell by such viruses can be blocked by the NA active site inhibitor oseltamivir, demonstrating functionally that NA replaces HA receptor function, at least in vitro.
NA also evolves subtler alterations that modulate NA specificity or catalytic properties. Before delving into this topic, it is important to recognize that specificity and catalysis are so intimately related that changes to one property can inevitably change the other (this also applies to HA specificity and avidity), given sufficiently discriminatory assays. As the actual in vivo ligands for HA and NA are uncertain and are likely to display enormous heterogeneity and vary considerably based on the precise locale of a given virion, it is wise not to make firm conclusions regarding the functional consequences of measured alterations in NA/HA-glycan interactions.
The most direct mechanism for NA to adjust its active site properties is to substitute residues in and about the active site. The high conservation of these residues (Figure 2) [55,65] in the absence of NA inhibitor-driven selection indicates that fitness costs limit variation in this region. A less obvious location for functional variation is the fibrous stalk that attaches the globular domain to the membrane. Stalk length governs the height of the globular domain and hence its access to substrates and also its interactions with HA (Figure 1A). Furthermore, it might also allosterically modulate NA activity [66]. Early NA RNA sequencing studies revealed that N1 from viruses isolated between 1933 and 1935 differed in stalk lengths by 11 to 16 residues [67]. Later, it was reported that NA stalk length shortening is frequently observed during adaptation of IAV between avian species [68,69], where it can be associated with increased lethality and transmission [70,71]. Deletions in the NA coding region occurred in H3N2 strains with narrowed receptor specificity, also generating NA inhibitor resistance [72,73]. Molecularly, it is easy to understand how stalk deletions can occur. Indeed, the IAV polymerase may even have evolved a tendency towards bulk nucleotide deletions, as they are a prominent feature of defective interfering (DI) particles, which are present in all virus preparations, and dominate the virion population when the virus is propagated at a high multiplicity of infection (MOI) [74]. The polymerase is also, however, capable of nucleotide insertions by recombination of IAV genes [75] and even by introduction of ribosomal RNA (and likely other host) sequences [76]. Providing one of the first demonstrations of the co-evolution of HA-NA at the molecular level, Mitnaul et al. reported that shortened NA stalk length reduced NA activity to cellular receptors and fitness, with fitness restored by mutations in HA that altered its receptor binding properties [75].
NA function can also be controlled at the level of NA expression to compensate for modified receptor binding and other alterations in HA function. Monoclonal Ab (mAb) selection of HA escape mutations in some instances co-selected NA mutants with single amino acid substitutions that reduce NA virion incorporation, likely due to interference with normal NA assembly or trafficking (Figure 1B) [77,78]. Importantly, some of these NA mutations modified NA sensitivity to clinically used inhibitors and/or modified NA antigenicity. This makes the critical point that NA-HA epistasis muddies interpretation of in vivo evolutionary selection pressures. Thus, it is possible that some of the observed antigenic drift in NA in human IAV evolution is due to selection by HA-specific Abs (and vice versa). A useful guiding philosophy is that extreme caution should be exercised in interpreting the selection pressures responsible for the evolution of genetic alterations.
Adaptation of the mouse A/Puerto Rico/8/1934 (H1N1) strain to guinea pigs revealed another mechanism of controlling NA incorporation into virions, based on a single amino acid substitution in NP that reduced NA mRNA and vRNA expression, reducing NA synthesis and impairing NA gene segment packaging into progeny virions (Figure 1C) [8]. Such semi-infectious (SI) virions lacking one or more gene segments outnumber intact virions in most virus preparations, with the NA segment being most frequently absent [79,80]. SI viruses are relevant in infection since they produce progeny that are easily rescued by recombination, which occurs at high frequency in animal models [8,10,11,12].
4. HA/NA Coevolution: HA Perspective
As a functional NA antagonist, HA also walks a fine line. It must have sufficient avidity to attach virus to cells, but not so much as to reduce NA mediated release from cells or to aggregate viruses that retain terminal SA due to incomplete NA action. In the presence of exogenously supplemented NA, IAV is able to replicate while rapidly acquiring HA substitutions at the tip of RBS (S193R and V205M) that decrease HA–SA binding [81]. Gradually decreasing exogenous NA during virus passage selected an unusual NA-independent strain with HA mutations associated with much weaker SA binding (V135A, S145N located in or near the RBS, and R220K located at the trimer interface). Similarly, the HA substitutions K173E and I260M are selected in oseltamivir resistant H3N2 clinical isolates that, quite remarkably, lack the NA gene segment (Figure 3A) [82].
The widespread clinical use of NA inhibitors provides an opportunity to study how mutations in NA epistatically select for mutations in HA (and potentially other genes) during IAV evolution in humans. Phylogenetic analysis reveals that mutations in HA can favor the emergence of NA inhibitors-resistant variants [83]. Initial in vitro studies found that that NA inhibitor resistant mutations occur in otherwise conserved structural and catalytic NA residues [84,85,86,87]. Surprisingly, however, predominant substitutions, even in some cases with unaltered NA, occurred in multiple HA residues near or in the RBS (N145S, N150S, V90A, L240Q, E116G, T155A, V229I, R229S/I, S165N, K222T, S186F) [84,88,89,90], demonstrating the extensive epitasis between HA and NA.
5. Antibody Response to HA and NA
Influenza virions comprise four major structural proteins—M1, HA, NP, and NA—present at a molar ratio, respectively, of ~ 100, 26, 22, and 3 [2]. Under experimental settings, immunization with virions in diverse species (chickens, mice, guinea pigs) results in predominant Ab responses to HA, NA, and NP, as determined by ELISA titers (respectively 55%, 35%, and 10%) [91]. Remarkably, this immunodominance hierarchy is maintained even in lampreys, despite generating Abs using a completely different family of receptors from the Ig receptors used in jawed vertebrates [91].
The vast majority of anti-HA Abs induced by virus infections, virions, or human vaccines recognize the globular domain, which consists of five canonical antigen sites (Sa, Sb, Ca1, Ca2, and Cb for H1 and A, B, C, D, and E for H3) [92,93,94,95]. A point often missed is that in Caton et al.’s original description of mAb-defined HA antigenicity [96], the full binding of 39 of 89 mAbs is diminished by substitutions in amino acids present in two or more sites. For all but the Sb Ag site, HA trimerization is required to attain full antigenicity [97]. Once formed, neither acid-induced HA head dissociation [98], nor release of the head domain by proteolytic treatment fully disrupts antibody recognition of any of the five major antigenic sites (Figure 4) [97].
While the HA globular domain is clearly immunodominant, the much more phylogenetically conserved stem also elicits Abs in humans and experimental animals following infection or vaccination. In initial studies by Russ and colleagues, stem specific polyclonal Abs were found to be specific for denatured HA and exhibited no clear biological activity [99,100,101,102,103,104]. In 1993, however, Okuno et al. [105] identified a stem-specific mAb that neutralized H1 and H2 viruses and later showed that this mAb could provide broad protection in vivo for viruses with group I HA [106]. Although far less abundant than head-specific Abs in human or animal sera [107], Abs specific for the stem domain are induced in all species examined and can attain reasonable, and even high, titers when elicited by native stem vaccines that lack the globular domain [108,109,110] or when the globular domain antigenic sites on intact HA vaccine are blocked by N-linked glycosylation [111]. Repeated exposure to dissimilar HA strains greatly enhances anti-stem responses (Figure 5) [112,113,114], based on the preferential activation of memory vs. naïve B cells. Although exposure of the HA to low pH destroys many stem epitopes recognized by protective Abs [115,116], the biogenesis of several epitopes was shown to be trimerization independent (Figure 4) [117]. While this is potentially good news for producing effective monomeric HA stem immunogens [118], it should be noted that many or even most stem epitopes require HA trimerization to confer full Abs binding [110,117,119,120].
Surprisingly little is known about the immunodominance hierarchy of antigenic sites in the HA globular domain, i.e., the prevalence of Abs and B cells specific for each site. A recent study provided a foundation for mechanistically dissecting B cell and Ab immunodominance in the mouse model. Angeletti et al. [121] created a panel of mAb-selected viruses lacking four of five of the head antigenic sites, enabling measurement of B cells and Abs to individual antigenic sites. This revealed that Abs for the Cb site, as initially reported [122,123], dominate the response early after infection, with anti-Sb Abs attaining dominance by 21 d post infection and continued diversification over time to greatly increase responses to the other antigenic sites. Immunodominance after vaccination with inactivated virus differs, being focused on HA tip region, particularly at Sa and/or Sb sites [121].
The Ab immunodominance hierarchy is important since it is likely to play a central role in determining the selection pressure that drives antigenic drift. Ab selection pressure is the product of the magnitude of Ab response and the functional activity of the Abs against the different sites. Abs that bind the Sa, Sb, Ca1, and Ca2 sites most efficiently block attachment [124,125], with Cb-specific antibodies exhibiting only low potency, consistent with the distal location of the Cb-site from the RBS. Parsing how anti-HA Abs actually work in vivo is a difficult task. Even tip-specific Abs may principally block infection at the level of virus fusion in the endosome activities of at endosomal fusion inhibition [125,126,127], possibly due to a requirement for HA–SA binding to trigger synchronized multi-HA fusion [47] in the late endosome.
Despite the clear anti-virus activity of NA-specific Abs known for 50 years [128] and their well-documented protective effects in humans [129,130,131], NA antigenicity is far less characterized than HA antigenicity. Early studies delineated a number of antigenic sites on N2 HA [132,133,134,135], and recent studies have similarly identified a few epitopes in contemporary N1 NAs [136,137]. The extent to which these epitopes contribute to immunodominant antigenic sites remains to be established.
On a molar basis, NA is the most immunogenic IAV protein in naïve animals [91]. Immunizing with equal molar amounts on HA and NA in humans reveals comparable immunogenicity [138,139]. In IAV-exposed individuals, however, HA immunodominance is enhanced due to immunogen competition, likely based on HA-specific B cells capturing more virions and recruiting more memory Tfh cells, which limit B cell responses [140,141,142].
NA Abs classically function by blocking NA activity against biologically relevant SA-containing substrates that interact with HA. These include cell surface receptors, which otherwise would sequester nascent virions on the cell surface and virion HA itself, which serves as a virion aggregant. NA Abs presumably also function in Fc-based humoral and cellular anti-viral immunity, including complement-based mechanisms, virion phagocytosis, and NK cell mediated effector functions.
With the substantial evidence supporting a protective role for NA antibodies in human immunity to IAV [129,130,131,141,143,144,145], the reader might be surprised to learn that the amount of NA present in human vaccines is not regulated by the FDA, nor publicly reported by the vaccine manufacturers, though it clearly varies enormously between various vaccine formulations. This is difficult to understand, since measuring native NA is far easier than measuring native HA. As an enzyme that requires native structure to maintain enzymatic activity, NA is measured inexpensively and rapidly, simply by its ability to cleave a fluorogenic substrate. In a single afternoon, even a marginal scientist (e.g., an aging PI with waning experimental skills), could measure the NA content of all vaccines used in the world in a given year. In addition, the enzyme-linked lectin assay (ELLA) enables a high throughput determination of functional anti-NA antibodies in serum [146]. This information could then be used to correlate NA vaccine content with the induction of anti-NA Abs and vaccine efficacy.
6. Ab-Based NA–HA Cross Talk
The proximity between HA and NA on the limited surface area of the virion raises the possibility that Abs to HA could affect NA function. Note that the opposite event, NA-specific Abs modulating HA function, is much less likely based on the ~10:1 molar dominance of HA. Even at Ab saturation, only a minor fraction of HA will be in proximity to NA-bound Abs, an effect amplified by NA clustering on the virion surface [147,148,149]. After Paniker [150] first reported that anti-HA Abs could inhibit NA activity 50 years ago, Russ et al. [151] showed that this required the physical association of HA with NA on virions, as would be expected. Re-examining this phenomenon using mAbs, mAbs specific for sites at the HA tip (Sa and Sb) were far less efficient at blocking NA activity to fetuin, a 48 kDa soluble glycoprotein substrate, than mAbs that bind to sites lower on HA spike (Ca, Cb) [124].
Extending these findings, mAbs specific for the HA stem were reported to efficiently block NA activity against either fetuin- or SA-containing surfaces, including the cell surface [152,153]. This provided an explanation for the ability of stem-specific Abs to prevent virus release and to thereby reduce viral infectivity [57]. Exploiting the flexible length of the NA stalk, Kosik et al. found that the anti-NA activity of HA-stem specific Abs is inversely proportional to the height of the NA globular domain, which they used to demonstrate the importance of this effect in reducing IAV pathology in mouse lungs and mortality [153]. In as much as the protective capacity of anti-HA stem Abs in mice appears to result from Fc receptor-dependent, innate immune cells [154,155], these findings imply that anti-stem Abs function in vivo by negating the inhibitory effects of virion NA activity on innate immune cell activation. This phenomenon was first reported by Bar-On et al. [156] and is consistent with a requirement for HA engagement of innate immune cell SA residues, as shown by the blocking effects of anti-HA Abs [157,158]. Indeed, the ability of anti-stem mAbs to activate NK reporter cells was proportional to their steric inhibition of NA activity [153]. As stem-specific Abs are possibly the basis for universal vaccination, it is critical to understand how they mediate protection.
These findings underscore the complexity of humoral immunity to IAV and the difficulties in assessing the contribution of various Ab-based effector mechanisms to protection, as well as to the evolution of HA and NA. Pragmatically, there is a potential danger in vaccination that leads to a stem response whose protection is abrogated by a head response sufficient to block innate immune cell activation but insufficient to protect via standard virus neutralization.
7. Future Directions
It is clear that HA and NA intimately function as a unit to enhance virus transmission and consequently co-evolve to optimize their overall fitness. Without doubt the segmentation of IAV facilitates this co-evolution as it enhances the ability of a virus population to sample permutations of HA and NA genes through co-infection, which appears to be a common event in vivo. Indeed, the abundance of semi-infectious virions lacking the NA gene [80] ensures that HA genes will widely sample NA genes to generate fully infectious viruses capable of inter-host transmission, which is severely bottlenecked in humans [3]. The intertwined relationship between HA and NA is reflected in the function of anti-HA Abs, which sterically block NA access to biological substrates in a manner dependent on the location of their epitopes on the HA spike and the length of the NA stalk.
These findings raise numerous questions for future studies, including:
- (1)
- How does the geometric distribution of HA and NA on the virion effect their functions? Why is NA clustered on virions? Is the extent of clustering variable between IAV strains? How does the relationship between NA and HA alter in filamentous vs. spherical virions? How does the stoichiometry of virion HA and NA influence viral function? What mechanisms does the virus use to control HA–NA virion content?
- (2)
- How do Ab-heavy chains influence the functional activities of anti-HA Abs on NA activity, particularly the larger oligomeric structure of IgM and IgG? Further, what is the functional impact of the effect of Ab binding molecules such as complement?
- (3)
- What is it exactly about HA and NA that enables their rapid antigenic evolution, while analogous proteins on other viruses evolve much more slowly? Is it the freedom for independent mutations afforded by a segmented genome? Is it some inherent resistance to in vivo Ab-mediated neutralization in humans that is not apparent in animal models? Is it something about the immunodominance hierarchy in individuals that enables sequential escape across a population?
Humanity has been studying IAV for nearly 90 years. While great strides have been made in understanding many aspects of viral replication, immunity, and pathogenesis, essential information needed to improve vaccination remains elusive. Despite its diminutive genome, IAV is a formidable foe. Deciphering its secrets will take a concerted effort and requires a reappraisal of what we think we know. The field should welcome with open arms young minds and those from different backgrounds who will be less burdened by preconceived opinions and more likely to take creative approaches to answering known questions and posing new ones.
Acknowledgments
The authors are supported by the Division of Intramural Research, NIAID, Bethesda, MD.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muramoto, Y.; Noda, T.; Kawakami, E.; Akkina, R.; Kawaoka, Y. Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 2013, 87, 2455–2462. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martinez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; Malosh, R.E.; Monto, A.S.; Lauring, A.S. Stochastic processes constrain the within and between host evolution of influenza virus. eLife 2018, 7. [Google Scholar] [CrossRef]
- Eichelberger, M.C.; Wan, H. Influenza neuraminidase as a vaccine antigen. Curr. Top. Microbiol. Immunol. 2015, 386, 275–299. [Google Scholar]
- Thyagarajan, B.; Bloom, J.D. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. eLife 2014, 3. [Google Scholar] [CrossRef]
- Visher, E.; Whitefield, S.E.; McCrone, J.T.; Fitzsimmons, W.; Lauring, A.S. The Mutational Robustness of Influenza A Virus. PLoS Pathog. 2016, 12, e1005856. [Google Scholar] [CrossRef]
- Altman, M.O.; Angeletti, D.; Yewdell, J.W. Antibody Immunodominance: The Key to Understanding Influenza Virus Antigenic Drift. Viral Immunol. 2018, 31, 142–149. [Google Scholar] [CrossRef]
- Brooke, C.B.; Ince, W.L.; Wei, J.; Bennink, J.R.; Yewdell, J.W. Influenza A virus nucleoprotein selectively decreases neuraminidase gene-segment packaging while enhancing viral fitness and transmissibility. Proc. Natl. Acad. Sci. USA 2014, 111, 16854–16859. [Google Scholar] [CrossRef] [Green Version]
- Ince, W.L.; Gueye-Mbaye, A.; Bennink, J.R.; Yewdell, J.W. Reassortment complements spontaneous mutation in influenza A virus NP and M1 genes to accelerate adaptation to a new host. J. Virol. 2013, 87, 4330–4338. [Google Scholar] [CrossRef]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Ann. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef]
- Phipps, K.L.; Marshall, N.; Tao, H.; Danzy, S.; Onuoha, N.; Steel, J.; Lowen, A.C. Seasonal H3N2 and 2009 Pandemic H1N1 Influenza A Viruses Reassort Efficiently but Produce Attenuated Progeny. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Richard, M.; Herfst, S.; Tao, H.; Jacobs, N.T.; Lowen, A.C. Influenza A virus reassortment is limited by anatomical compartmentalization following co-infection via distinct routes. J. Virol. 2018. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, Y.; Tefsen, B.; Shi, Y.; Gao, G.F. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 2014, 22, 183–191. [Google Scholar] [CrossRef]
- Tsai, K.N.; Chen, G.W. Influenza genome diversity and evolution. Microbes Infect. 2011, 13, 479–488. [Google Scholar] [CrossRef]
- Rejmanek, D.; Hosseini, P.R.; Mazet, J.A.; Daszak, P.; Goldstein, T. Evolutionary Dynamics and Global Diversity of Influenza A Virus. J. Virol. 2015, 89, 10993–11001. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Eichelberger, M.C.; Compans, R.W.; Air, G.M. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J. Virol. 1995, 69, 1099–1106. [Google Scholar]
- Stray, S.J.; Cummings, R.D.; Air, G.M. Influenza virus infection of desialylated cells. Glycobiology 2000, 10, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem. Int. Ed. Engl. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Song, H.; Qi, J.; Xiao, H.; Bi, Y.; Zhang, W.; Xu, Y.; Wang, F.; Shi, Y.; Gao, G.F. Avian-to-Human Receptor-Binding Adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep. 2017, 20, 1201–1214. [Google Scholar] [CrossRef]
- Wang, F.; Qi, J.; Bi, Y.; Zhang, W.; Wang, M.; Zhang, B.; Wang, M.; Liu, J.; Yan, J.; Shi, Y.; et al. Adaptation of avian influenza A (H6N1) virus from avian to human receptor-binding preference. EMBO J. 2015, 34, 1661–1673. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Ince, W.L.; Gentles, L.E.; Oler, A.J.; Kosikova, M.; Angel, M.; Magadan, J.G.; Xie, H.; Brooke, C.B.; Yewdell, J.W. Correction: Influenza A virus hemagglutinin glycosylation compensates for antibody escape fitness costs. PLoS Pathog. 2018, 14, e1007141. [Google Scholar] [CrossRef]
- Badham, M.D.; Rossman, J.S. Filamentous Influenza Viruses. Curr. Clin. Microbiol. Rep. 2016, 3, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Calder, L.J.; Wasilewski, S.; Berriman, J.A.; Rosenthal, P.B. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 10685–10690. [Google Scholar] [CrossRef] [Green Version]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 2000, 74, 8502–8512. [Google Scholar] [CrossRef]
- Dadonaite, B.; Vijayakrishnan, S.; Fodor, E.; Bhella, D.; Hutchinson, E.C. Filamentous influenza viruses. J. Gen. Virol. 2016, 97, 1755–1764. [Google Scholar] [CrossRef]
- Chen, L.M.; Blixt, O.; Stevens, J.; Lipatov, A.S.; Davis, C.T.; Collins, B.E.; Cox, N.J.; Paulson, J.C.; Donis, R.O. In vitro evolution of H5N1 avian influenza virus toward human-type receptor specificity. Virology 2012, 422, 105–113. [Google Scholar] [CrossRef]
- Leung, H.S.; Li, O.T.; Chan, R.W.; Chan, M.C.; Nicholls, J.M.; Poon, L.L. Entry of influenza A Virus with a alpha2,6-linked sialic acid binding preference requires host fibronectin. J. Virol. 2012, 86, 10704–10713. [Google Scholar] [CrossRef]
- Chutinimitkul, S.; Herfst, S.; Steel, J.; Lowen, A.C.; Ye, J.; van Riel, D.; Schrauwen, E.J.; Bestebroer, T.M.; Koel, B.; Burke, D.F.; et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 2010, 84, 11802–11813. [Google Scholar] [CrossRef]
- Watanabe, T.; Kiso, M.; Fukuyama, S.; Nakajima, N.; Imai, M.; Yamada, S.; Murakami, S.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Sakoda, Y.; et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 2013, 501, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Belser, J.A.; Gustin, K.M.; Pearce, M.B.; Maines, T.R.; Zeng, H.; Pappas, C.; Sun, X.; Carney, P.J.; Villanueva, J.M.; Stevens, J.; et al. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature 2013, 501, 556–559. [Google Scholar] [CrossRef]
- Gulati, S.; Smith, D.F.; Cummings, R.D.; Couch, R.B.; Griesemer, S.B.; St George, K.; Webster, R.G.; Air, G.M. Human H3N2 Influenza Viruses Isolated from 1968 To 2012 Show Varying Preference for Receptor Substructures with No Apparent Consequences for Disease or Spread. PLoS ONE 2013, 8, e66325. [Google Scholar]
- Yang, H.; Carney, P.J.; Chang, J.C.; Guo, Z.; Villanueva, J.M.; Stevens, J. Structure and receptor binding preferences of recombinant human A(H3N2) virus hemagglutinins. Virology 2015, 477, 18–31. [Google Scholar] [CrossRef] [Green Version]
- De Graaf, M.; Fouchier, R.A. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 2014, 33, 823–841. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.P.; Xiong, X.L.; Wharton, S.A.; Martin, S.R.; Coombs, P.J.; Vachieri, S.G.; Christodoulou, E.; Walker, P.A.; Liu, J.F.; Skehel, J.J.; et al. Evolution of the receptor binding properties of the influenza A(H3N2) hemagglutinin. Proc. Natl. Acad. Sci. USA 2012, 109, 21474–21479. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Ulett, K.B.; Boyd, K.L.; Madsen, J.; Hawgood, S.; McCullers, J.A. N-linked glycosylation attenuates H3N2 influenza viruses. J. Virol. 2007, 81, 8593–8600. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; David, A.; Schmidt, L.; Gibbs, J.S.; Puigbo, P.; Ince, W.L.; Bennink, J.R.; Yewdell, J.W. Fitness costs limit influenza A virus hemagglutinin glycosylation as an immune evasion strategy. Proc. Natl. Acad. Sci. USA 2011, 108, E1417–E1422. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.J.; de Vries, R.P.; Grant, O.C.; Thompson, A.J.; McBride, R.; Tsogtbaatar, B.; Lee, P.S.; Razi, N.; Wilson, I.A.; Woods, R.J.; et al. Recent H3N2 Viruses Have Evolved Specificity for Extended, Branched Human-type Receptors, Conferring Potential for Increased Avidity. Cell Host Microbe 2017, 21, 23–34. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, C. Influenza A virus H5N1 entry into host cells is through clathrin-dependent endocytosis. Sci. China Life Sci. 2009, 52, 464–469. [Google Scholar] [CrossRef]
- Sun, E.Z.; Liu, A.A.; Zhang, Z.L.; Liu, S.L.; Tian, Z.Q.; Pang, D.W. Real-Time Dissection of Distinct Dynamin-Dependent Endocytic Routes of Influenza A Virus by Quantum Dot-Based Single-Virus Tracking. ACS Nano 2017, 11, 4395–4406. [Google Scholar] [CrossRef]
- Rossman, J.S.; Leser, G.P.; Lamb, R.A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 2012, 86, 10950–10960. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Bizebard, T.; Gigant, B.; Rigolet, P.; Rasmussen, B.; Diat, O.; Bosecke, P.; Wharton, S.A.; Skehel, J.J.; Knossow, M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995, 376, 92–94. [Google Scholar] [CrossRef]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Das, D.K.; Govindan, R.; Nikic-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018. [Google Scholar] [CrossRef]
- Leikina, E.; Ramos, C.; Markovic, I.; Zimmerberg, J.; Chernomordik, L.V. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 2002, 21, 5701–5710. [Google Scholar] [CrossRef] [Green Version]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar] [CrossRef]
- Ivanovic, T.; Choi, J.L.; Whelan, S.P.; van Oijen, A.M.; Harrison, S.C. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2013, 2, e00333. [Google Scholar] [CrossRef]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef]
- Steinhauer, D.A. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 1999, 258, 1–20. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Bennink, J.R.; Hosaka, Y. Cells process exogenous proteins for recognition by cytotoxic T lymphocytes. Science 1988, 239, 637–640. [Google Scholar] [CrossRef]
- Basak, S.; Tomana, M.; Compans, R.W. Sialic-Acid Is Incorporated into Influenza Hemagglutinin Glycoproteins in the Absence of Viral Neuraminidase. Virus Res. 1985, 2, 61–68. [Google Scholar] [CrossRef]
- Romero-Beltran, L.; Baker, S.F.; Puerto-Solis, M.; Gonzalez-Losa, R.; Conde-Ferraez, L.; Alvarez-Sanchez, L.C.; Martinez-Sobrido, L.; Ayora-Talavera, G. Mutations at highly conserved residues in influenza A(H1N1)pdm09 virus affect neuraminidase activity. Virus Res. 2016, 225, 1–9. [Google Scholar] [CrossRef]
- Chockalingam, A.K.; Hickman, D.; Pena, L.; Ye, J.Q.; Ferrero, A.; Echenique, J.R.; Chen, H.J.; Sutton, T.; Perez, D.R. Deletions in the Neuraminidase Stalk Region of H2N2 and H9N2 Avian Influenza Virus Subtypes Do Not Affect Postinfluenza Secondary Bacterial Pneumonia. J. Virol. 2012, 86, 3564–3573. [Google Scholar] [CrossRef] [Green Version]
- Yamayoshi, S.; Uraki, R.; Ito, M.; Kiso, M.; Nakatsu, S.; Yasuhara, A.; Oishi, K.; Sasaki, T.; Ikuta, K.; Kawaoka, Y. A Broadly Reactive Human Anti-hemagglutinin Stem Monoclonal Antibody That Inhibits Influenza A Virus Particle Release. Ebiomedicine 2017, 17, 182–191. [Google Scholar] [CrossRef]
- Yamamoto-Goshima, F.; Maeno, K. Approach to the involvement of influenza B neuraminidase in the cleavage of HA by host cell protease using low pH-induced cell fusion reaction. Microbiol. Immunol. 1994, 38, 819–822. [Google Scholar] [CrossRef]
- Li, S.; Schulman, J.; Itamura, S.; Palese, P. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J. Virol. 1993, 67, 6667–6673. [Google Scholar]
- Baum, L.G.; Paulson, J.C. The N2 neuraminidase of human influenza virus has acquired a substrate specificity complementary to the hemagglutinin receptor specificity. Virology 1991, 180, 10–15. [Google Scholar] [CrossRef]
- Laver, W.G.; Colman, P.M.; Webster, R.G.; Hinshaw, V.S.; Air, G.M. Influenza virus neuraminidase with hemagglutinin activity. Virology 1984, 137, 314–323. [Google Scholar] [CrossRef]
- Lin, Y.P.; Gregory, V.; Collins, P.; Kloess, J.; Wharton, S.; Cattle, N.; Lackenby, A.; Daniels, R.; Hay, A. Neuraminidase Receptor Binding Variants of Human Influenza A(H3N2) Viruses Resulting from Substitution of Aspartic Acid 151 in the Catalytic Site: A Role in Virus Attachment? J. Virol. 2010, 84, 6769–6781. [Google Scholar] [CrossRef]
- Uhlendorff, J.; Matrosovich, T.; Klenk, H.D.; Matrosovich, M. Functional significance of the hemadsorption activity of influenza virus neuraminidase and its alteration in pandemic viruses. Arch. Virol. 2009, 154, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Mögling, R.; Richard, M.J.; Vliet, S.v.d.; Beek, R.v.; Schrauwen, E.J.A.; Spronken, M.I.; Rimmelzwaan, G.F.; Fouchier, R.A.M. Neuraminidase-mediated haemagglutination of recent human influenza A(H3N2) viruses is determined by arginine 150 flanking the neuraminidase catalytic site. J. Gen. Virol. 2017, 98, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Air, G.M. Influenza neuraminidase. Influenza Other Respir. Viruses 2012, 6, 245–256. [Google Scholar] [CrossRef]
- Durrant, J.D.; Bush, R.M.; Amaro, R.E. Microsecond Molecular Dynamics Simulations of Influenza Neuraminidase Suggest a Mechanism for the Increased Virulence of Stalk-Deletion Mutants. J. Phys. Chem. B 2016, 120, 8590–8599. [Google Scholar] [CrossRef]
- Blok, J.; Air, G. Block deletions in the neuraminidase genes from some influenza A viruses of the N1 subtype. Virology 1982, 118, 229–234. [Google Scholar] [CrossRef]
- Li, J.; Zu Dohna, H.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and genetic variation of neuraminidase stalk deletions in avian influenza viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef]
- Sorrell, E.M.; Song, H.; Pena, L.; Perez, D.R. A 27-amino-acid deletion in the neuraminidase stalk supports replication of an avian H2N2 influenza A virus in the respiratory tract of chickens. J. Virol. 2010, 84, 11831–11840. [Google Scholar] [CrossRef]
- Stech, O.; Veits, J.; Abdelwhab el, S.M.; Wessels, U.; Mettenleiter, T.C.; Stech, J. The Neuraminidase Stalk Deletion Serves as Major Virulence Determinant of H5N1 Highly Pathogenic Avian Influenza Viruses in Chicken. Sci. Rep. 2015, 5, 13493. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, S.; Zhang, X.; Fu, Q.; Zhang, Z.; Shi, S.; Zhu, Y.; Gu, M.; Peng, D.; Liu, X. A 20-amino-acid deletion in the neuraminidase stalk and a five-amino-acid deletion in the NS1 protein both contribute to the pathogenicity of H5N1 avian influenza viruses in mallard ducks. PLoS ONE 2014, 9, e95539. [Google Scholar] [CrossRef]
- Gulati, S.; Smith, D.F.; Air, G.M. Deletions of neuraminidase and resistance to oseltamivir may be a consequence of restricted receptor specificity in recent H3N2 influenza viruses. Virol. J. 2009, 6, 22. [Google Scholar] [CrossRef]
- Eshaghi, A.; Shalhoub, S.; Rosenfeld, P.; Li, A.; Higgins, R.R.; Stogios, P.J.; Savchenko, A.; Bastien, N.; Li, Y.; Rotstein, C.; et al. Multiple influenza A (H3N2) mutations conferring resistance to neuraminidase inhibitors in a bone marrow transplant recipient. Antimicrob. Agents Chemother. 2014, 58, 7188–7197. [Google Scholar] [CrossRef]
- Brooke, C.B. Population Diversity and Collective Interactions during Influenza Virus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Mitnaul, L.J.; Matrosovich, M.N.; Castrucci, M.R.; Tuzikov, A.B.; Bovin, N.V.; Kobasa, D.; Kawaoka, Y. Balanced hemagglutinin and neuraminidase activities are critical for efficient replication of influenza A virus. J. Virol. 2000, 74, 6015–6020. [Google Scholar] [CrossRef]
- Khatchikian, D.; Orlich, M.; Rott, R. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature 1989, 340, 156–157. [Google Scholar] [CrossRef]
- Hensley, S.E.; Das, S.R.; Gibbs, J.S.; Bailey, A.L.; Schmidt, L.M.; Bennink, J.R.; Yewdell, J.W. Influenza A virus hemagglutinin antibody escape promotes neuraminidase antigenic variation and drug resistance. PLoS ONE 2011, 6, e15190. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; Ince, W.L.; Brooke, C.B.; Subba, A.; Delboy, M.G.; Russ, G.; Gibbs, J.S.; Bennink, J.R.; Yewdell, J.W. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe 2013, 13, 314–323. [Google Scholar]
- Brooke, C.B. Biological activities of ‘noninfectious’ influenza A virus particles. Future Virol. 2014, 9, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Brooke, C.B.; Ince, W.L.; Wrammert, J.; Ahmed, R.; Wilson, P.C.; Bennink, J.R.; Yewdell, J.W. Most influenza a virions fail to express at least one essential viral protein. J. Virol. 2013, 87, 3155–3162. [Google Scholar] [CrossRef]
- Hughes, M.T.; Matrosovich, M.; Rodgers, M.E.; McGregor, M.; Kawaoka, Y. Influenza A viruses lacking sialidase activity can undergo multiple cycles of replication in cell culture, eggs, or mice. J. Virol. 2000, 74, 5206–5212. [Google Scholar] [CrossRef]
- Moules, V.; Ferraris, O.; Terrier, O.; Giudice, E.; Yver, M.; Rolland, J.P.; Bouscambert-Duchamp, M.; Bergeron, C.; Ottmann, M.; Fournier, E.; et al. In vitro characterization of naturally occurring influenza H3NA- viruses lacking the NA gene segment: Toward a new mechanism of viral resistance? Virology 2010, 404, 215–224. [Google Scholar] [CrossRef]
- Neverov, A.D.; Kryazhimskiy, S.; Plotkin, J.B.; Bazykin, G.A. Coordinated Evolution of Influenza A Surface Proteins. PLoS Genetics 2015, 11, e1005404. [Google Scholar] [CrossRef]
- Staschke, K.A.; Colacino, J.M.; Baxter, A.J.; Air, G.M.; Bansal, A.; Hornback, W.J.; Munroe, J.E.; Laver, W.G. Molecular basis for the resistance of influenza viruses to 4-guanidino-Neu5Ac2en. Virology 1995, (2), 642–646. [Google Scholar] [CrossRef]
- Carr, J.; Ives, J.; Kelly, L.; Lambkin, R.; Oxford, J.; Mendel, D.; Tai, L.; Roberts, N. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antiviral Res. 2002, 54, 79–88. [Google Scholar] [CrossRef]
- Ives, J.A.; Carr, J.A.; Mendel, D.B.; Tai, C.Y.; Lambkin, R.; Kelly, L.; Oxford, J.S.; Hayden, F.G.; Roberts, N.A. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Res. 2002, 55, 307–317. [Google Scholar] [CrossRef]
- Pizzorno, A.; Abed, Y.; Plante, P.L.; Carbonneau, J.; Baz, M.; Hamelin, M.E.; Corbeil, J.; Boivin, G. Evolution of oseltamivir resistance mutations in Influenza A(H1N1) and A(H3N2) viruses during selection in experimentally infected mice. Antimicrob. Agents Chemother. 2014, 58, 6398–6405. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L.; Blick, T.J.; Sahasrabudhe, A.; Tiong, T.; Marshall, D.; Hart, G.J.; Bethell, R.C.; Penn, C.R. Generation and characterization of variants of NWS/G70C influenza virus after in vitro passage in 4-amino-Neu5Ac2en and 4-guanidino-Neu5Ac2en. Antimicrob. Agents Chemother. 1996, 40, 40–46. [Google Scholar] [CrossRef]
- Blick, T.J.; Sahasrabudhe, A.; McDonald, M.; Owens, I.J.; Morley, P.J.; Fenton, R.J.; McKimm-Breschkin, J.L. The interaction of neuraminidase and hemagglutinin mutations in influenza virus in resistance to 4-guanidino-Neu5Ac2en. Virology 1998, 246, 95–103. [Google Scholar] [CrossRef]
- Barnett, J.M.; Cadman, A.; Burrell, F.M.; Madar, S.H.; Lewis, A.P.; Tisdale, M.; Bethell, R. In vitro selection and characterisation of influenza B/Beijing/1/87 isolates with altered susceptibility to zanamivir. Virology 1999, 265, 286–295. [Google Scholar] [CrossRef]
- Altman, M.O.; Bennink, J.R.; Yewdell, J.W.; Herrin, B.R. Lamprey VLRB response to influenza virus supports universal rules of immunogenicity and antigenicity. eLife 2015, 4. [Google Scholar] [CrossRef]
- Gerhard, W.; Yewdell, J.; Frankel, M.E.; Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 1981, 290, 713–717. [Google Scholar] [CrossRef]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E., Jr.; Wilson, I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010, 328, 357–360. [Google Scholar] [CrossRef]
- Wiley, D.C.; Wilson, I.A.; Skehel, J.J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981, 289, 373–378. [Google Scholar] [CrossRef]
- Suntronwong, N.; Klinfueng, S.; Vichiwattana, P.; Korkong, S.; Thongmee, T.; Vongpunsawad, S.; Poovorawan, Y. Genetic and antigenic divergence in the influenza A(H3N2) virus circulating between 2016 and 2017 in Thailand. PLoS ONE 2017, 12, e0189511. [Google Scholar] [CrossRef]
- Caton, A.J.; Brownlee, G.G.; Yewdell, J.W.; Gerhard, W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 1982, 31, 417–427. [Google Scholar] [CrossRef]
- Magadan, J.G.; Khurana, S.; Das, S.R.; Frank, G.M.; Stevens, J.; Golding, H.; Bennink, J.R.; Yewdell, J.W. Influenza A Virus Hemagglutinin Trimerization Completes Monomer Folding and Antigenicity. J. Virol. 2013. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Gerhard, W.; Bachi, T. Monoclonal anti-hemagglutinin antibodies detect irreversible antigenic alterations that coincide with the acid activation of influenza virus A/PR/834-mediated hemolysis. J. Virol. 1983, 48, 239–248. [Google Scholar]
- Vareckova, E.; Mucha, V.; Ciampor, F.; Betakova, T.; Russ, G. Monoclonal antibodies demonstrate accessible HA2 epitopes in minor subpopulation of native influenza virus haemagglutinin molecules. Arch. Virol. 1993, 130, 45–56. [Google Scholar] [CrossRef]
- Kostolansky, F.; Styk, B.; Russ, G. Inhibition of influenza virus haemolytic and haemagglutination activities by monoclonal antibodies to haemagglutinin glycopolypeptides HA1 and HA2. Acta Virol. 1989, 33, 504–512. [Google Scholar]
- Russ, G.; Polakova, K.; Kostolansky, F.; Styk, B.; Vancikova, M. Monoclonal antibodies to glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. Acta Virol. 1987, 31, 374–386. [Google Scholar]
- Styk, B.; Russ, G.; Polakova, K. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. IV. Immunogenic properties of separated haemagglutinin glycopolypeptides. Acta Virol. 1979, 23, 9–20. [Google Scholar]
- Russ, G.; Styk, B.; Polakova, K. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. II. Reactivity with rabbit sera against intact virus and purified undissociated haemagglutinin. Acta Virol. 1978, 22, 371–382. [Google Scholar]
- Polakova, K.; Russ, G.; Styk, B. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. I. Gel filtration in 6 M guanidine hydrochloride. Acta Virol. 1978, 22, 362–370. [Google Scholar]
- Okuno, Y.; Isegawa, Y.; Sasao, F.; Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 1993, 67, 2552–2558. [Google Scholar]
- Okuno, Y.; Matsumoto, K.; Isegawa, Y.; Ueda, S. Protection against the mouse-adapted A/FM/1/47 strain of influenza A virus in mice by a monoclonal antibody with cross-neutralizing activity among H1 and H2 strains. J. Virol. 1994, 68, 517–520. [Google Scholar]
- Sui, J.; Sheehan, J.; Hwang, W.C.; Bankston, L.A.; Burchett, S.K.; Huang, C.Y.; Liddington, R.C.; Beigel, J.H.; Marasco, W.A. Wide prevalence of heterosubtypic broadly neutralizing human anti-influenza A antibodies. Clin. Infect. Dis. 2011, 52, 1003–1009. [Google Scholar] [CrossRef]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.J.; Kanekiyo, M.; Kong, W.P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G.; et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015, 21, 1065–1070. [Google Scholar] [CrossRef]
- Mallajosyula, V.V.; Citron, M.; Ferrara, F.; Lu, X.; Callahan, C.; Heidecker, G.J.; Sarma, S.P.; Flynn, J.A.; Temperton, N.J.; Liang, X.; et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc. Natl. Acad. Sci. USA 2014, 111, E2514–E2523. [Google Scholar] [CrossRef]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.; van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef]
- Eggink, D.; Goff, P.H.; Palese, P. Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J. Virol. 2014, 88, 699–704. [Google Scholar] [CrossRef]
- Wrammert, J.; Koutsonanos, D.; Li, G.M.; Edupuganti, S.; Sui, J.; Morrissey, M.; McCausland, M.; Skountzou, I.; Hornig, M.; Lipkin, W.I.; et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011, 208, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Pica, N.; Hai, R.; Krammer, F.; Wang, T.T.; Maamary, J.; Eggink, D.; Tan, G.S.; Krause, J.C.; Moran, T.; Stein, C.R.; et al. Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 2573–2578. [Google Scholar] [CrossRef] [Green Version]
- Tete, S.M.; Krammer, F.; Lartey, S.; Bredholt, G.; Wood, J.; Skrede, S.; Cox, R.J. Dissecting the hemagglutinin head and stalk-specific IgG antibody response in healthcare workers following pandemic H1N1 vaccination. Npj Vaccines 2016, 1. [Google Scholar] [CrossRef]
- Tan, G.S.; Lee, P.S.; Hoffman, R.M.; Mazel-Sanchez, B.; Krammer, F.; Leon, P.E.; Ward, A.B.; Wilson, I.A.; Palese, P. Characterization of a broadly neutralizing monoclonal antibody that targets the fusion domain of group 2 influenza A virus hemagglutinin. J. Virol. 2014, 88, 13580–13592. [Google Scholar] [CrossRef]
- Chai, N.; Swem, L.R.; Reichelt, M.; Chen-Harris, H.; Luis, E.; Park, S.; Fouts, A.; Lupardus, P.; Wu, T.D.; Li, O.; et al. Two Escape Mechanisms of Influenza A Virus to a Broadly Neutralizing Stalk-Binding Antibody. PLoS Pathog. 2016, 12, e1005702. [Google Scholar] [CrossRef]
- Magadan, J.G.; Altman, M.O.; Ince, W.L.; Hickman, H.D.; Stevens, J.; Chevalier, A.; Baker, D.; Wilson, P.C.; Ahmed, R.; Bennink, J.R.; et al. Biogenesis of influenza a virus hemagglutinin cross-protective stem epitopes. PLoS Pathog. 2014, 10, e1004204. [Google Scholar] [CrossRef]
- Seok, J.H.; Kim, J.; Lee, D.B.; Cho, K.J.; Lee, J.H.; Bae, G.; Chung, M.S.; Kim, K.H. Conformational modulation of influenza virus hemagglutinin: Characterization and in vivo efficacy of monomeric form. Sci. Rep. 2017, 7, 7540. [Google Scholar] [CrossRef]
- Harris, A.K.; Myerson, J.R.; Matsuoka, Y.; Kuybeda, O.; Moran, A.; Bliss, D.; Das, S.R.; Yewdell, J.W.; Sapiro, G.; Subbarao, K.; et al. Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc. Natl. Acad. Sci USA 2013, in press. [Google Scholar] [CrossRef]
- Dreyfus, C.; Ekiert, D.C.; Wilson, I.A. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 influenza virus hemagglutinin. J. Virol. 2013, 87, 7149–7154. [Google Scholar] [CrossRef]
- Angeletti, D.; Gibbs, J.S.; Angel, M.; Kosik, I.; Hickman, H.D.; Frank, G.M.; Das, S.R.; Wheatley, A.K.; Prabhakaran, M.; Leggat, D.J.; et al. Defining B cell immunodominance to viruses. Nat. Immunol. 2017, 18, 456–463. [Google Scholar] [CrossRef]
- Kavaler, J.; Caton, A.J.; Staudt, L.M.; Schwartz, D.; Gerhard, W. A set of closely related antibodies dominates the primary antibody response to the antigenic site CB of the A/PR/8/34 influenza virus hemagglutinin. J. Immunol. 1990, 145, 2312–2321. [Google Scholar] [PubMed]
- Staudt, L.M.; Gerhard, W. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. I. Significant variation in repertoire expression between individual mice. J. Exp. Med. 1983, 157, 687–704. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Yewdell, J.W. Influenza A virus hemagglutinin specific antibodies interfere with virion neuraminidase activity via two distinct mechanisms. Virology 2017, 500, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.J.; Dimmock, N.J. Two influenza A virus-specific Fabs neutralize by inhibiting virus attachment to target cells, while neutralization by their IgGs is complex and occurs simultaneously through fusion inhibition and attachment inhibition. Virology 2000, 278, 423–435. [Google Scholar] [CrossRef]
- Edwards, M.J.; Dimmock, N.J. A haemagglutinin (HA1)-specific FAb neutralizes influenza A virus by inhibiting fusion activity. J. Gen. Virol. 2001, 82, 1387–1395. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.J.; Dimmock, N.J. Hemagglutinin 1-specific immunoglobulin G and Fab molecules mediate postattachment neutralization of influenza A virus by inhibition of an early fusion event. J. Virol. 2001, 75, 10208–10218. [Google Scholar] [CrossRef]
- Kilbourne, E.D.; Laver, W.G.; Schulman, J.L.; Webster, R.G. Antiviral activity of antiserum specific for an influenza virus neuraminidase. J. Virol. 1968, 2, 281–288. [Google Scholar]
- Ogra, P.L.; Chow, T.; Beutner, K.R.; Rubi, E.; Strussenberg, J.; DeMello, S.; Rizzone, C. Clinical and immunologic evaluation of neuraminidase-specific influenza A virus vaccine in humans. J. Infect. Dis. 1977, 135, 499–506. [Google Scholar] [CrossRef]
- Beutner, K.R.; Chow, T.; Rubi, E.; Strussenberg, J.; Clement, J.; Ogra, P.L. Evaluation of a neuraminidase-specific influenza A virus vaccine in children: Antibody responses and effects on two successive outbreaks of natural infection. J. Infect. Dis. 1979, 140, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.R.; Kasel, J.A.; Chanock, R.M. Association of serum anti-neuraminidase antibody with resistance to influenza in man. N. Engl. J. Med. 1972, 286, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Air, G.M.; Els, M.C.; Brown, L.E.; Laver, W.G.; Webster, R.G. Location of antigenic sites on the three-dimensional structure of the influenza N2 virus neuraminidase. Virology 1985, 145, 237–248. [Google Scholar] [CrossRef]
- Webster, R.G.; Brown, L.E.; Laver, W.G. Antigenic and biological characterization of influenza virus neuraminidase (N2) with monoclonal antibodies. Virology 1984, 135, 30–42. [Google Scholar] [CrossRef]
- Webster, R.G.; Hinshaw, V.S.; Laver, W.G. Selection and analysis of antigenic variants of the neuraminidase of N2 influenza viruses with monoclonal antibodies. Virology 1982, 117, 93–104. [Google Scholar] [CrossRef]
- Laver, W.G.; Air, G.M.; Webster, R.G.; Markoff, L.J. Amino acid sequence changes in antigenic variants of type A influenza virus N2 neuraminidase. Virology 1982, 122, 450–460. [Google Scholar] [CrossRef]
- Wan, H.; Gao, J.; Xu, K.; Chen, H.; Couzens, L.K.; Rivers, K.H.; Easterbrook, J.D.; Yang, K.; Zhong, L.; Rajabi, M.; et al. Molecular basis for broad neuraminidase immunity: Conserved epitopes in seasonal and pandemic H1N1 as well as H5N1 influenza viruses. J. Virol. 2013, 87, 9290–9300. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fantoni, G.; Couzens, L.; Gao, J.; Plant, E.; Ye, Z.; Eichelberger, M.C.; Wan, H. Comparative Efficacy of Monoclonal Antibodies That Bind to Different Epitopes of the 2009 Pandemic H1N1 Influenza Virus Neuraminidase. J. Virol. 2016, 90, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.E.; Kilbourne, E.D. Dissociation of influenza virus hemagglutinin and neuraminidase eliminates their intravirionic antigenic competition. J. Virol. 1993, 67, 5721–5723. [Google Scholar] [PubMed]
- Johansson, B.E.; Bucher, D.J.; Kilbourne, E.D. Purified influenza virus hemagglutinin and neuraminidase are equivalent in stimulation of antibody response but induce contrasting types of immunity to infection. J. Virol. 1989, 63, 1239–1246. [Google Scholar]
- Kendal, A.P.; Noble, G.R.; Dowdle, W.R. Neuraminidase content of influenza vaccines and neuraminidase antibody responses after vaccination of immunologically primed and unprimed populations. J. Infect. Dis. 1977, 136 (Suppl. 3), S415–S424. [Google Scholar] [CrossRef]
- Kilbourne, E.D.; Cerini, C.P.; Khan, M.W.; Mitchell, J.W., Jr.; Ogra, P.L. Immunologic response to the influenza virus neuraminidase is influenced by prior experience with the associated viral hemagglutinin. I. Studies in human vaccinees. J. Immunol. 1987, 138, 3010–3013. [Google Scholar]
- Johansson, B.E.; Moran, T.M.; Bona, C.A.; Popple, S.W.; Kilbourne, E.D. Immunologic response to influenza virus neuraminidase is influenced by prior experience with the associated viral hemagglutinin. II. Sequential infection of mice simulates human experience. J. Immunol. 1987, 139, 2010–2014. [Google Scholar]
- Rajendran, M.; Nachbagauer, R.; Ermler, M.E.; Bunduc, P.; Amanat, F.; Izikson, R.; Cox, M.; Palese, P.; Eichelberger, M.; Krammer, F. Analysis of Anti-Influenza Virus Neuraminidase Antibodies in Children, Adults, and the Elderly by ELISA and Enzyme Inhibition: Evidence for Original Antigenic Sin. mBio 2017, 8. [Google Scholar] [CrossRef]
- Monto, A.S.; Petrie, J.G.; Cross, R.T.; Johnson, E.; Liu, M.; Zhong, W.; Levine, M.; Katz, J.M.; Ohmit, S.E. Antibody to Influenza Virus Neuraminidase: An Independent Correlate of Protection. J. Infect. Dis. 2015, 212, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Couch, R.B.; Kasel, J.A.; Gerin, J.L.; Schulman, J.L.; Kilbourne, E.D. Induction of partial immunity to influenza by a neuraminidase-specific vaccine. J. Infect Dis. 1974, 129, 411–420. [Google Scholar] [CrossRef]
- Couzens, L.; Gao, J.; Westgeest, K.; Sandbulte, M.; Lugovtsev, V.; Fouchier, R.; Eichelberger, M. An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J. Virol. Methods 2014, 210, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef] [Green Version]
- Murti, K.G.; Webster, R.G. Distribution of hemagglutinin and neuraminidase on influenza virions as revealed by immunoelectron microscopy. Virology 1986, 149, 36–43. [Google Scholar] [CrossRef]
- Fontana, J.; Steven, A.C. Influenza virus-mediated membrane fusion: Structural insights from electron microscopy. Arch. Biochem. Biophys. 2015, 581, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Paniker, C.K. Serological relationships between the neuraminidases in influenza viruses. J. Gen. Virol. 1968, 2, 385–394. [Google Scholar] [CrossRef]
- Russ, G.; Varekova, E.; Styk, B. Steric effects in the reaction of influenza virus neuraminidases with antibodies. Acta Virologica. 1974, 18, 299–306. [Google Scholar]
- Chen, Y.-Q.; Lan, L.Y.-L.; Huang, M.; Henry, C.; Wilson, P.C. Hemagglutinin stalk-reactive antibodies interfere with influenza virus neuraminidase activity by steric hindrance. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Angeletti, D.; Gibbs, J.S.; Angel, M.; Takeda, K.; Kosikova, M.; Nair, V.; Hickman, H.D.; Xie, H.; Brooke, C.B.; et al. Neuraminidase inhibition contributes to influenza A virus neutralization by anti-hemagglutinin stem antibodies. J. Exp. Med. 2019, 216, 304–316. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest. 2016, 126, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiLillo, D.J.; Tan, G.S.; Palese, P.; Ravetch, J.V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat. Med. 2014, 20, 143–151. [Google Scholar] [CrossRef]
- Bar-On, Y.; Seidel, E.; Tsukerman, P.; Mandelboim, M.; Mandelboim, O. Influenza Virus Uses Its Neuraminidase Protein to Evade the Recognition of Two Activating NK Cell Receptors. J. Infect. Dis. 2014, 210, 410–418. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Tan, G.S.; Mullarkey, C.E.; Lee, A.J.; Lam, M.M.; Krammer, F.; Henry, C.; Wilson, P.C.; Ashkar, A.A.; Palese, P.; et al. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc. Natl. Acad. Sci. USA 2016, 113, 11931–11936. [Google Scholar] [CrossRef] [Green Version]
- Leon, P.E.; He, W.; Mullarkey, C.E.; Bailey, M.J.; Miller, M.S.; Krammer, F.; Palese, P.; Tan, G.S. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact. Proc. Natl. Acad. Sci. USA 2016, 113, E5944–E5951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Naturally observed mechanisms to optimize HA–NA stoichiometry. (A) The HA H1 (pdb entry 3lzg) is colored pink with wt-RBS in black and mutated RBS with decreased binding in red. The NA N2 is colored turquoise (the model was created by superimposition of pdb entries 2hty and 6crd-containing tetrabrachion stalk) with the receptor destroying/catalytic site in blue, receptor binding site in magenta, and combined receptor binding/destroying site in yellow. (B) After the HA (pink) acquires an avidity decreasing mutation (blue), the NA (green) follows with a mutation (bright green) that impairs NA intracellular trafficking and incorporation into virions. (C) During host adaptation an NP mutation (red-orange) can decrease NA gene segment incorporation leading to diminished amounts of virion NA to rebalance HA–NA levels. UCSF Chimera 1.13 was used to render the 3-D structures.
Figure 1.
Naturally observed mechanisms to optimize HA–NA stoichiometry. (A) The HA H1 (pdb entry 3lzg) is colored pink with wt-RBS in black and mutated RBS with decreased binding in red. The NA N2 is colored turquoise (the model was created by superimposition of pdb entries 2hty and 6crd-containing tetrabrachion stalk) with the receptor destroying/catalytic site in blue, receptor binding site in magenta, and combined receptor binding/destroying site in yellow. (B) After the HA (pink) acquires an avidity decreasing mutation (blue), the NA (green) follows with a mutation (bright green) that impairs NA intracellular trafficking and incorporation into virions. (C) During host adaptation an NP mutation (red-orange) can decrease NA gene segment incorporation leading to diminished amounts of virion NA to rebalance HA–NA levels. UCSF Chimera 1.13 was used to render the 3-D structures.

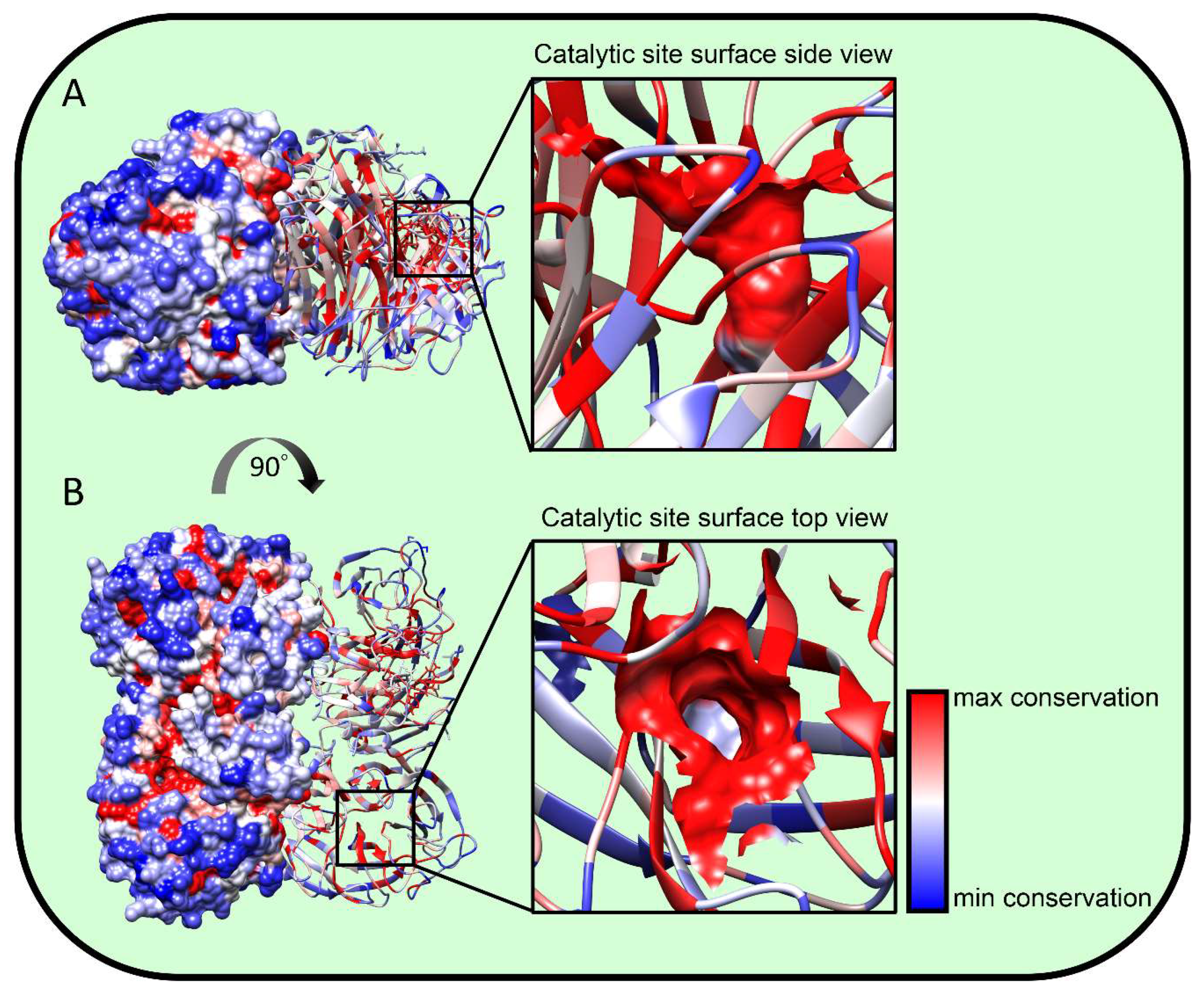
Figure 2.
NA structural conservation. We aligned roughly 400 NA amino acid sequences from IAV strains spanning the last 100 years (200 hundred H1N1, 50 H5N1, and 150 H3N2) using MUSCLE software at the fludb website (www.fludb.org). We projected residue conservation onto the N2 NA A/Perth/16/2009 H3N2 (pdb entry 6br5) structure using UCSF Chimera 1.13 software. Blue represents residues with maximal variability while red represents minimal variability. (A) Side view showing surface rendering of half the structure with the other half showing ribbon rendering, demonstrating the variability of NA surface residues vs. the conservation of internal residues. (B) Top view. The black squares show the catalytic site, which is magnified on the right. The residues forming the catalytic site are surface-rendered; note the high conservation except at the base, which is variable and consistent with its lack of substrate interaction.
Figure 2.
NA structural conservation. We aligned roughly 400 NA amino acid sequences from IAV strains spanning the last 100 years (200 hundred H1N1, 50 H5N1, and 150 H3N2) using MUSCLE software at the fludb website (www.fludb.org). We projected residue conservation onto the N2 NA A/Perth/16/2009 H3N2 (pdb entry 6br5) structure using UCSF Chimera 1.13 software. Blue represents residues with maximal variability while red represents minimal variability. (A) Side view showing surface rendering of half the structure with the other half showing ribbon rendering, demonstrating the variability of NA surface residues vs. the conservation of internal residues. (B) Top view. The black squares show the catalytic site, which is magnified on the right. The residues forming the catalytic site are surface-rendered; note the high conservation except at the base, which is variable and consistent with its lack of substrate interaction.

Figure 3.
Naturally observed mechanisms of NA–HA epistasis. (A) Some oseltamivir-resistant IAV clinical isolates completely lack the NA gene segment and adapt by acquiring mutations that reduce HA receptor avidity. HA is colored pink, NA green, NA inhibitor yellow, HA mutation blue. (B) NA inhibitors can select escape mutants with changes exclusively in HA that alter HA receptor binding properties. The H1 HA (pdb entry 3lzg) colored pink with wt-RBS black and mutated RBS red, rendered as side view. The N1 NA colored turquoise (pdb entry 3ti6) with or without oseltamivir (yellow-green) bound to receptor destroying site (blue) rendered as top view. UCSF Chimera 1.13 software was used to visualize the molecules.
Figure 3.
Naturally observed mechanisms of NA–HA epistasis. (A) Some oseltamivir-resistant IAV clinical isolates completely lack the NA gene segment and adapt by acquiring mutations that reduce HA receptor avidity. HA is colored pink, NA green, NA inhibitor yellow, HA mutation blue. (B) NA inhibitors can select escape mutants with changes exclusively in HA that alter HA receptor binding properties. The H1 HA (pdb entry 3lzg) colored pink with wt-RBS black and mutated RBS red, rendered as side view. The N1 NA colored turquoise (pdb entry 3ti6) with or without oseltamivir (yellow-green) bound to receptor destroying site (blue) rendered as top view. UCSF Chimera 1.13 software was used to visualize the molecules.

Figure 4.
Biogenesis of HA antigenic sites. The HA head antigenic site Sb and HA stem protective epitopes are formed on HA monomers during biogenesis, while for other sites on the HA head, full maturation requires HA trimerization. Contrary to HA stem epitopes, which are destroyed by conformational changes triggered by low pH, HA head antigenic sites are remarkably resistant and remain intact even after proteolytic liberation of the monomeric head domain. The pdb entries 1htm, 1ibn, and 2vir (A/Aichi/2/1968 H3N2) were used to visualize various conformational stages during the fusion process. The HA1 is colored bright blue, HA2 orange, and fusion peptide red. Approximate localization of the canonical antigenic sites and the HA stem epitopes are highlighted by translucent purple with dashed edges. UCSF Chimera 1.13 software was used to visualize the molecules.
Figure 4.
Biogenesis of HA antigenic sites. The HA head antigenic site Sb and HA stem protective epitopes are formed on HA monomers during biogenesis, while for other sites on the HA head, full maturation requires HA trimerization. Contrary to HA stem epitopes, which are destroyed by conformational changes triggered by low pH, HA head antigenic sites are remarkably resistant and remain intact even after proteolytic liberation of the monomeric head domain. The pdb entries 1htm, 1ibn, and 2vir (A/Aichi/2/1968 H3N2) were used to visualize various conformational stages during the fusion process. The HA1 is colored bright blue, HA2 orange, and fusion peptide red. Approximate localization of the canonical antigenic sites and the HA stem epitopes are highlighted by translucent purple with dashed edges. UCSF Chimera 1.13 software was used to visualize the molecules.

Figure 5.
The mechanisms of humoral response modulation towards HA stem domain. If regular HA (top left) is used as an immunogen, the dominant antibody response is directed towards the head domain (dark red). Immunization with hyperglycosylated HA head (complex glycans colored bright green) or sequential immunization with dissimilar head (olive green, dark red, bright blue), identical stem (dim gray) constructs (top middle), or physically separated stem (top right) all result in anti-stem dominated Ab responses.
Figure 5.
The mechanisms of humoral response modulation towards HA stem domain. If regular HA (top left) is used as an immunogen, the dominant antibody response is directed towards the head domain (dark red). Immunization with hyperglycosylated HA head (complex glycans colored bright green) or sequential immunization with dissimilar head (olive green, dark red, bright blue), identical stem (dim gray) constructs (top middle), or physically separated stem (top right) all result in anti-stem dominated Ab responses.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, 11, 346. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040346
AMA Style
Kosik I, Yewdell JW. Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses. 2019; 11(4):346. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040346
Chicago/Turabian StyleKosik, Ivan, and Jonathan W. Yewdell. 2019. "Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity" Viruses 11, no. 4: 346. https://0-doi-org.brum.beds.ac.uk/10.3390/v11040346
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.