Novel Bat Alphacoronaviruses in Southern China Support Chinese Horseshoe Bats as an Important Reservoir for Potential Novel Coronaviruses

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Collection of Bat Samples
2.3. Detection of Bat CoVs by RNA Extraction, RT-PCR and DNA Sequencing
2.4. Viral Culture
2.5. Complete Genome Sequencing of Rs-BatCoV HKU32 and Tr-BatCoV HKU33
2.6. Phylogenetic and Genome Analysis of Rs-BatCoV HKU32 and Tr-BatCoV HKU33
2.7. Expression of ORF10 Accessory Gene and Determination of Leader-Body Junction Sequence
2.8. Accession Number
3. Results
3.1. Bat Coronaviruses Surveillance and Identification of Two Novel Alphacoronaviruses
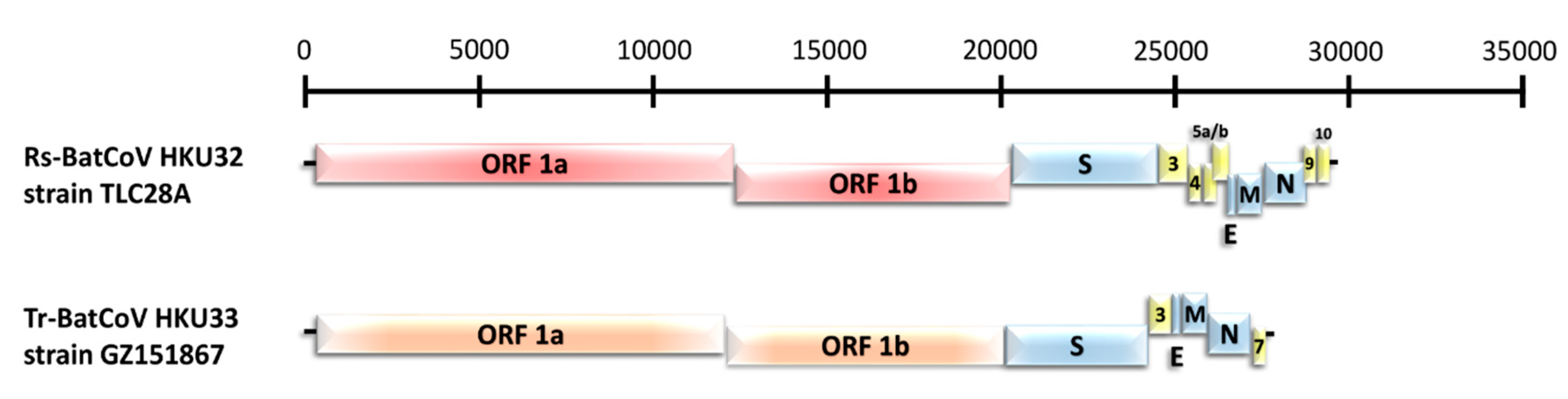
3.2. Genome Features of the Two Novel Alphacoronaviruses, Rs-BatCoV HKU32 and Tr-BatCoV HKU33
3.2.1. Novel alphaCoV Species: Rs-BatCoV HKU32
3.2.2. Novel alphaCoV Species: Tr-BatCoV HKU33
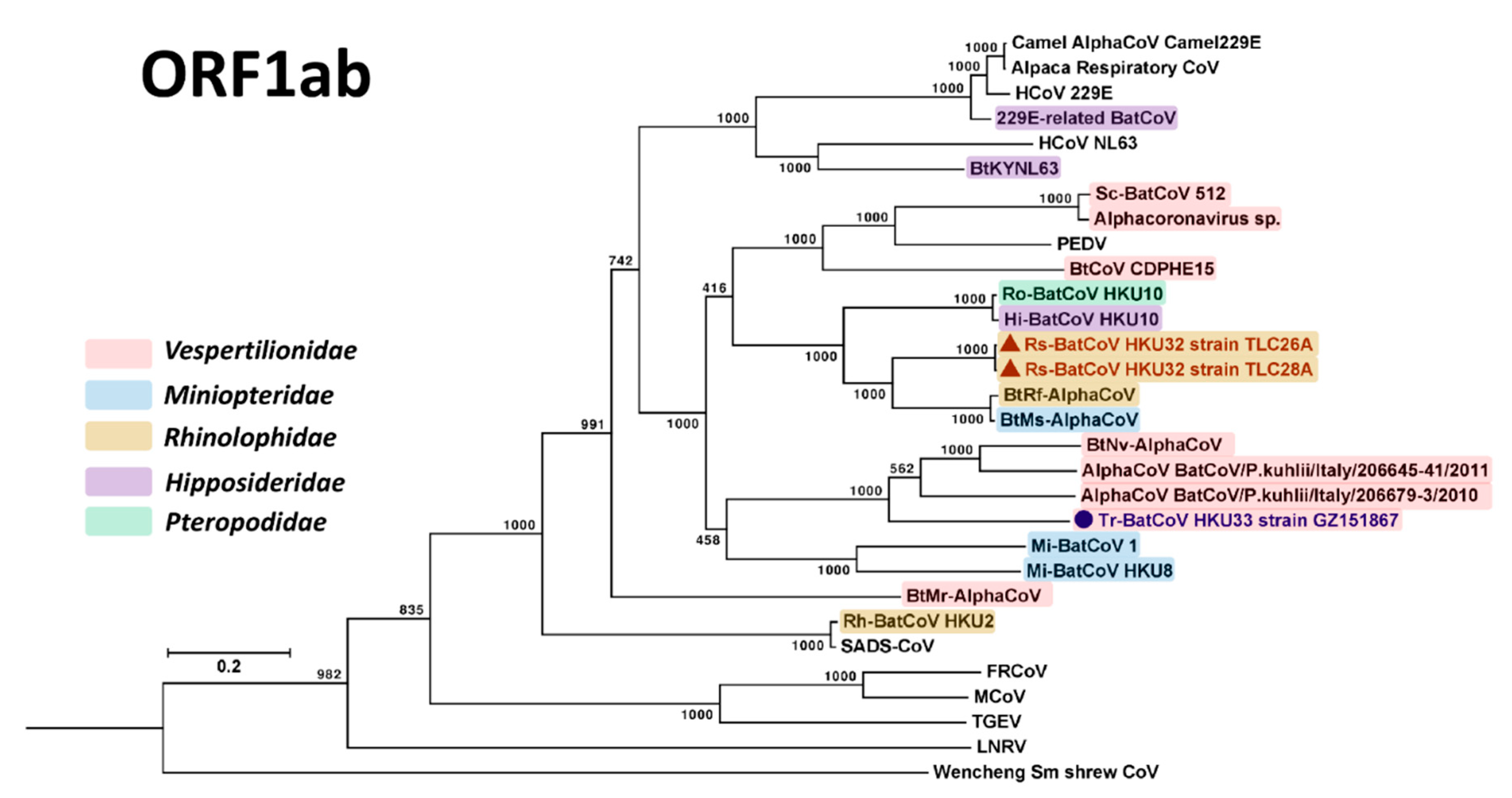
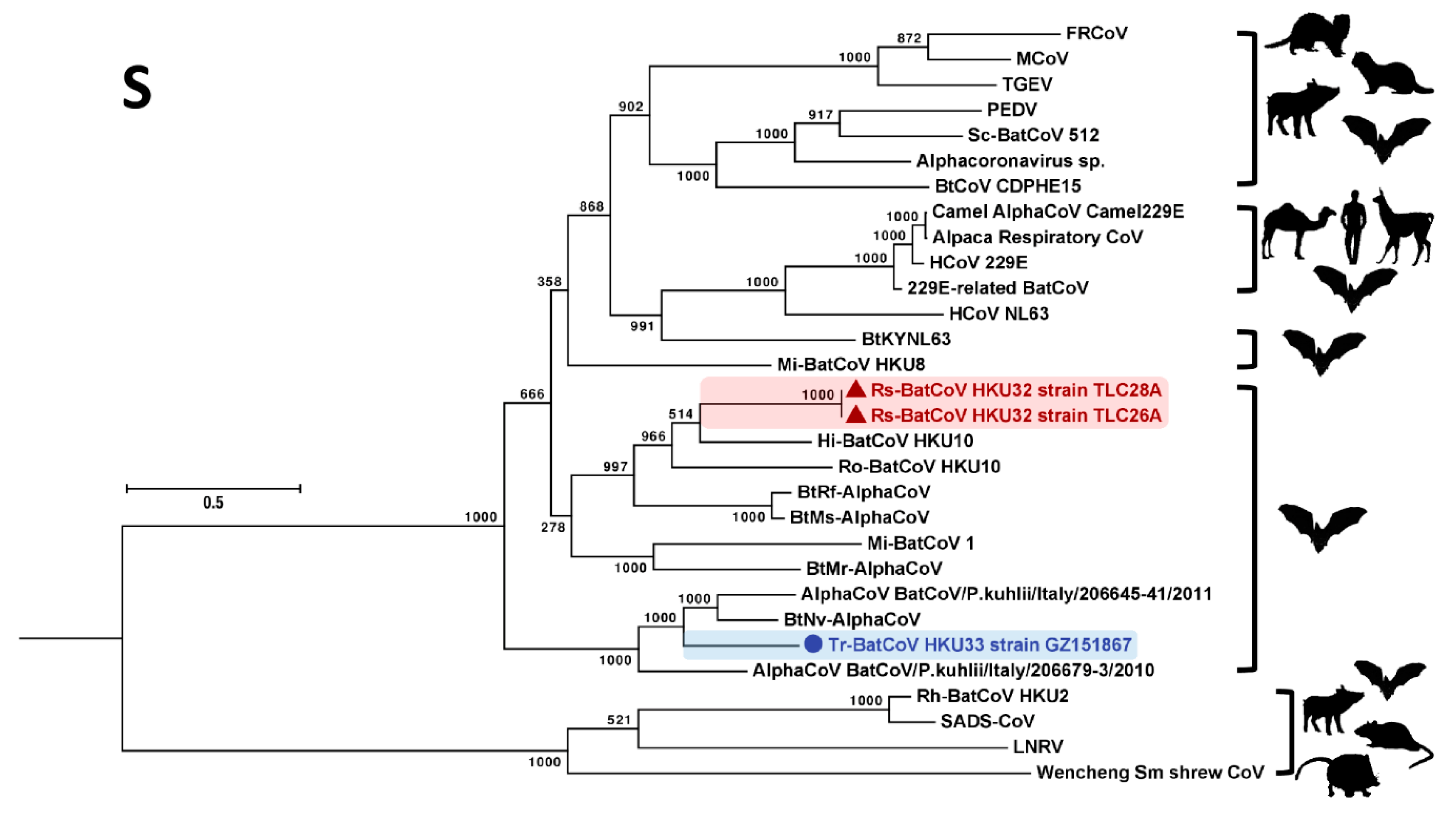
3.3. Phylogenetic Analyses
3.4. Homologous SARSr-CoV ORF7a-Like Accessory Protein in Rs-BatCoV HKU32
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AlphaCoV | Alphacoronavirus |
| BetaCoV | Betacoronavirus |
| bp | Base-pair |
| CoVs | Coronaviruses |
| DeltaCoV | Deltacoronavirus |
| E | Envelope |
| GammaCoV | Gammacoronavirus |
| HCoV | Human coronavirus |
| Hi | Hipposideros |
| ICTV | International Committee on Taxonomy of Viruses |
| M | Membrane |
| MERS-CoV | Middle East Respiratory Syndrome coronavirus |
| N | Nucleocapsid |
| NCBI | National Center for Biotechnology Information |
| ORF | Open reading frame |
| PCR | Polymerase chain reaction |
| Pi | Pipistrellus |
| Pp | Polyprotein |
| RdRp | RNA-dependent RNA polymerase |
| Ro | Rousettus |
| RSK | Rhinolophus sinicus kidney |
| RSL | Rhinolophus sinicus lung |
| RT | Reverse transcription |
| S | Spike |
| SADS-CoV | Swine Acute Diarrhea Syndrome coronavirus |
| SARSr-CoV | Severe Acute Respiratory Syndrome related coronavirus |
| TRS | Transcription regulatory sequence |
| Ty | Tylonycteris |
| µL | Microliter |
References
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef]
- Zhao, G.P. Sars molecular epidemiology: A Chinese fairy tale of controlling an emerging zoonotic disease in the genomics era. Philos. Trans. R Soc. Lond. B Biol. Sci. 2007, 362, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.; Haagmans, B.L.; Muller, M.A.; Gutierrez, C.; Godeke, G.J.; Meyer, B.; Muth, D.; Raj, V.S.; Vries, L.S.; Corman, V.M.; et al. Middle east respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: A comparative serological study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef]
- Haagmans, B.L.; Al Dhahiry, S.H.; Reusken, C.B.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle east respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2013, 14, 140–145. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.-Y. Middle east respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing sars-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Wong, A.C.P.; Lau, T.C.K.; Woo, P.C.Y. Molecular evolution of mers coronavirus: Dromedaries as a recent intermediate host or long-time animal reservoir? Int. J. Mol. Sci. 2017, 18, 2138. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Wang, M.; Lau, S.K.; Xu, H.; Poon, R.W.; Guo, R.; Wong, B.H.; Gao, K.; Tsoi, H.W.; Huang, Y.; et al. Comparative analysis of twelve genomes of three novel group 2C and group 2D coronaviruses reveals unique group and subgroup features. J. Virol. 2007, 81, 1574–1585. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Poon, R.W.S.; Wong, B.H.L.; Tsoi, H.-w.; Yip, B.C.K.; Huang, Y.; Chan, K.-h.; Yuen, K.-y. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Lam, C.S.; Ahmed, S.; Chen, H.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Genetic characterization of betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus hku5 in Japanese pipistrelle: Implications for the origin of the novel middle east respiratory syndrome coronavirus. J. Virol. 2013, 87, 8638–8650. [Google Scholar]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Tsang, A.K.L.; Yuen, K.-Y. Genetic relatedness of the novel human group c betacoronavirus to tylonycteris bat coronavirus hku4 and pipistrellus bat coronavirus hku5. Emerg. Micro. Infect. 2012, 1, e35. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of middle east respiratory syndrome coronavirus by characterization of a conspecific virus from an african bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef]
- Yang, L.; Wu, Z.; Ren, X.; Yang, F.; Zhang, J.; He, G.; Dong, J.; Sun, L.; Zhu, Y.; Zhang, S.; et al. Mers-related betacoronavirus in vespertilio superans bats, China. Emerg. Infect. Dis. 2014, 20, 1260–1262. [Google Scholar] [CrossRef]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. MBio 2017, 8, e00373-17. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Zhang, L.; Luk, H.K.H.; Xiong, L.; Peng, X.; Li, K.S.M.; He, X.; Zhao, P.S.; Fan, R.Y.Y.; Wong, A.C.P.; et al. Receptor usage of a novel bat lineage c betacoronavirus reveals evolution of middle east respiratory syndrome-related coronavirus spike proteins for human dipeptidyl peptidase 4 binding. J. Infect. Dis. 2018, 218, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.M.; Wang, N.; Yang, X.L.; Liu, H.Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.B.; Zhu, G.J.; et al. Discovery of novel bat coronaviruses in south china that use the same receptor as middle east respiratory syndrome coronavirus. J. Virol. 2018, 92, e00116-18. [Google Scholar] [CrossRef]
- Van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.M.; Wolthers, K.C.; Wertheim-van Dillen, P.M.E.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Chu, C.M.; Chan, K.H.; Tsoi, H.W.; Huang, Y.; Wong, B.H.; Poon, R.W.; Cai, J.J.; Luk, W.K.; et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus hku1, from patients with pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Tsang, A.K.L.; Fan, R.Y.Y.; Luk, H.K.H.; Cai, J.-P.; Chan, K.-H.; Zheng, B.-J.; Wang, M.; et al. Discovery of a novel coronavirus, china rattus coronavirus hku24, from norway rats supports the murine origin of betacoronavirus 1 and has implications for the ancestor of betacoronavirus lineage A. J. Virol. 2014, 89, 3076–3092. [Google Scholar] [CrossRef]
- Lai, M.M.C.; Cavanagh, D. The molecular biology of coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 1997; pp. 1–100. [Google Scholar]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. In Current Topics in Microbiology and Immunology; Springer-Verlag: Berlin, Germany, 2005; pp. 1–30. [Google Scholar]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family Coronaviridae. In Virus Taxonomy, Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses, 1st ed.; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2011; pp. 806–828. [Google Scholar]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in saudi arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012, 3, e00473-12. [Google Scholar] [CrossRef]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses nl63 and 229e and their recombination history. J. Virol. 2017, 91, e01953-16. [Google Scholar] [CrossRef]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an ancestral association of human coronavirus 229e with bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Muller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229e in bats, ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Huang, Y.W.; Dickerman, A.W.; Pineyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. MBio 2013, 4, e00737-13. [Google Scholar] [CrossRef]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an hku2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, X.; Qin, P.; Wang, B.; Zhao, P.; Yang, Y.L.; Wang, L.; Wang, D.; Song, Y.; Zhang, X.; et al. Discovery of a novel swine enteric alphacoronavirus (seacov) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef]
- Gong, L.; Li, J.; Zhou, Q.; Xu, Z.; Chen, L.; Zhang, Y.; Xue, C.; Wen, Z.; Cao, Y. A new bat-hku2-like coronavirus in swine, China, 2017. Emerg. Infect. Dis. 2017, 23, 1607. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Wang, M.; Lam, C.S.; Xu, H.; Guo, R.; Chan, K.H.; Zheng, B.J.; et al. Complete genome sequence of bat coronavirus hku2 from chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 2007, 367, 428–439. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Wong, E.Y.M.; Tsang, C.C.; Ahmed, S.S.; Au-Yeung, R.K.H.; Yuen, K.Y.; Wernery, U.; Woo, P.C.Y. Discovery and sequence analysis of four deltacoronaviruses from birds in the middle east reveal interspecies jumping with recombination as a potential mechanism for avian-to-avian and avian-to-mammalian transmission. J. Virol. 2018, 92, e00265-18. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Fan, R.Y.Y.; Huang, Y.; Wang, M.; Guo, R.; Lam, C.S.F.; Tsang, A.K.L.; Lai, K.K.Y.; et al. Isolation and characterization of a novel betacoronavirus subgroup a coronavirus, rabbit coronavirus hku14, from domestic rabbits. J. Virol. 2012, 86, 5481–5496. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. Mafft online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform 2017. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Nelson, C.A.; Pekosz, A.; Lee, C.A.; Diamond, M.S.; Fremont, D.H. Structure and intracellular targeting of the sars-coronavirus ORF7A accessory protein. Structure 2005, 13, 75–85. [Google Scholar] [CrossRef]
- Pekosz, A.; Schaecher, S.R.; Diamond, M.S.; Fremont, D.H.; Sims, A.C.; Baric, R.S. Structure, expression, and intracellular localization of the SARS-COV accessory proteins 7A and 7B. Adv. Exp. Med. Biol. 2006, 581, 115–120. [Google Scholar]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Shek, C.T.; Wang, M.; Choi, G.K.; Guo, R.; Wong, B.H.; Poon, R.W.; Lam, C.S.; et al. Recent transmission of a novel alphacoronavirus, bat coronavirus hku10, from leschenault’s rousettes to pomona leaf-nosed bats: First evidence of interspecies transmission of coronavirus between bats of different suborders. J. Virol. 2012, 86, 11906–11918. [Google Scholar] [CrossRef]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L.; Liu, C.; Wang, L.; Ma, C.; Tang, J.; Baric, R.S.; Jiang, S.; Li, F. Receptor usage and cell entry of bat coronavirus hku4 provide insight into bat-to-human transmission of mers coronavirus. Proc. Natl. Acad. Sci. USA 2014, 111, 12516–12521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qi, J.; Yuan, Y.; Xuan, Y.; Han, P.; Wan, Y.; Ji, W.; Li, Y.; Wu, Y.; Wang, J.; et al. Bat origins of MERS-COV supported by bat coronavirus hku4 usage of human receptor cd26. Cell. Host Microbe 2014, 16, 328–337. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
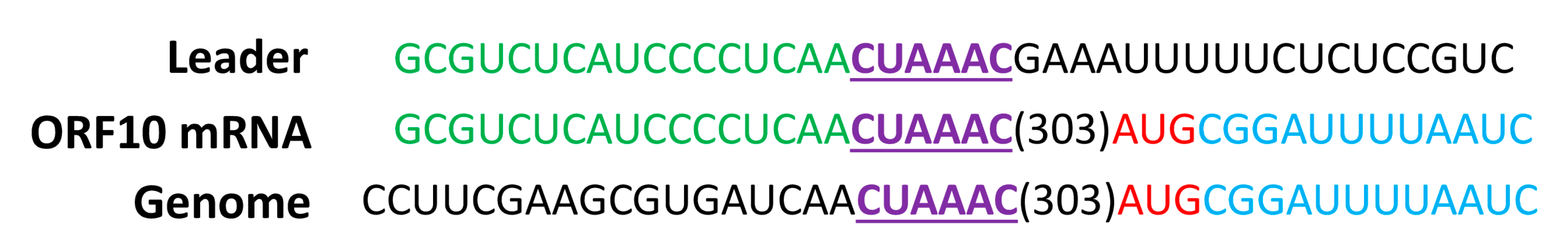{kind=link}
| Scientific Name | Common Name | No. of Bats Captured | No. of Bats Positive for CoV / (%) | CoV Detected | Sampling Location of Bats |
|---|---|---|---|---|---|
| Cynopterus sphinx | Greater short-nosed fruit bat | 3 | 0 | - | SWH |
| Hipposideros armiger | Great roundleaf bat | 3 | 0 | - | GZ |
| Hipposideros larvatus | Intermediate roundleaf bat | 21 | 0 | - | GZ |
| Hipposideros pomona | Pomona leaf-nosed bat | 182 | 2 / (1.1) | Hi-BatCoV HKU10 | TLC13, GD |
| Hypsugo pulveratus | Chinese pipistrelle | 2 | 0 | - | LMHP |
| Miniopterus magnater | Western bent-winged bat | 1 | 0 | - | SK01 |
| Miniopterus pusillus | Small bent-wing bat | 56 | 0 | - | LMH, SWH, SK01 |
| Miniopterus schreibersii | Common bent-wing bat | 23 | 0 | - | SK01 |
| Miniopterus filiginosus | Eastern bent-wing bat | 1 | 0 | - | LMHP |
| Myotis chinensis | Large myotis | 10 | 0 | - | SK01, GZ |
| Myotis ricketti | Rickett’s big-footed bat | 93 | 1 / (1.1) | Coronavirus PREDICT CoV-37 | LMH01, SK01 |
| Nyctalus noctula | Common noctule | 1 | 0 | - | YSO |
| Pipistrellus abramus | Japanese pipistrelle | 6 | 0 | - | MPO, YSO, KKSH |
| Pipistrellus tenuis | Least pipistrelle | 4 | 0 | - | KKSH, YSO, SWH, LMHP |
| Rhinolophus affinis | Intermediate horseshoe bat | 76 | 0 | - | TLC01, TLC13, SK01 |
| Rhinolophus pearsonii | Pearson’s horseshoe bat | 2 | 0 | - | GDP |
| Rhinolophus pusillus | Least horseshoe bat | 17 | 0 | - | TLC13 |
| Rhinolophus sinicus | Chinese horseshoe bat | 272 | 10 / (3.7) | Rs-BatCoV HKU32 (7) SARSr BatCoV (3) | TLC01, GDP |
| Tylonycteris pachypus | Lesser bamboo bat | 240 | 18 / (7.5) | Ty-BatCoV HKU4 | WKT, PFL, SWH, TLC01, GZP |
| Tylonycteris robustula | Greater bamboo bat | 104 | 1 / (0.96) | Tr-BatCoV HKU33 | GZP |
| Putative TRS | ||||||
|---|---|---|---|---|---|---|
| ORF | Nucleotide Positions (Start–End) | No. of Nucleotides | No. of Amino Acids | Frame(s) | Nucleotide Position in Genome | TRS Sequence (Distance (No. of Bases) to AUG) 1 |
| 1ab | 291–20,428 | 20,137 | 6712 | +2, +3 | 69 | AACUAAAC(216)AUG |
| nsp1 | 291–875 | 585 | 195 | +3 | ||
| nsp2 | 876–2963 | 2088 | 696 | +3 | ||
| nsp3 | 2964–7649 | 4686 | 1562 | +3 | ||
| nps4 | 7650–9083 | 1434 | 478 | +3 | ||
| nsp5 | 9084–9989 | 906 | 302 | +3 | ||
| nsp6 | 9990–10817 | 828 | 276 | +3 | ||
| nsp7 | 10,818–11,066 | 249 | 83 | +3 | ||
| nsp8 | 11,067–11,651 | 585 | 195 | +3 | ||
| nsp9 | 11,652–11,975 | 324 | 108 | +3 | ||
| nsp10 | 11,976–12,383 | 408 | 136 | +3 | ||
| nsp11 | 51 | 17 | +3 | |||
| nsp12 | 12,384–15,163 | 2780 | 927 | +2 | ||
| nsp13 | 15,164–16,954 | 1791 | 597 | +2 | ||
| nsp14 | 16,955–18,508 | 1554 | 518 | +2 | ||
| nsp15 | 18,509–19,525 | 1017 | 339 | +2 | ||
| nsp16 | 19,526–20,428 | 903 | 300 | +2 | ||
| S | 20,430–24,485 | 4056 | 1351 | +2 | 20,421 | AACUAAAU(3)AUG |
| ORF3 | 24,485–25,153 | 669 | 222 | +3 | 24,279 | TCCUUAAC(199)AUG |
| ORF4 | 25,184–25,543 | 360 | 119 | +2 | ||
| ORF5a | 25,544–25,888 | 345 | 114 | +2 | 25,540 | GACUAAAUG |
| ORF5b | 25,782–26,225 | 444 | 147 | +3 | ||
| E | 26,209–26,433 | 225 | 74 | +1 | 26,140 | AACUAAAC(64)AUG |
| M | 26,440–27,126 | 687 | 228 | +1 | 26,430 | GTCUAAAC(4)AUG |
| N | 27,137–28,279 | 1143 | 380 | +2 | 27,128 | AACUAAAC(3)AUG |
| ORF9 | 28,251–28,568 | 318 | 105 | +3 | 28,187 | AGCUGAAC(58)AUG |
| ORF10 (SARS-CoV ORF7a-like protein) | 28,593–28,955 | 363 | 120 | +3 | 28,284 | AACUAAAC(303)AUG |
| Amino Acids | |||||
|---|---|---|---|---|---|
| nsp | Putative Function or Domain | Rs-BatCoV HKU32 Strain TLC28A | Ro-BatCoV HKU10 183A | Tr-BatCoV HKU33 Strain GZ151867 | BtNv-AlphaCoV/SC2013 |
| nsp1 | Unknown | M1 – A195 | M1 – A 195 | M1 – A193 | M1 – A193 |
| nsp2 | Unknown | P196 – G891 | K196 – G888 | K194 – G771 | K194 – G771 |
| nsp3 | ADRP, Putative PLpro domains (PL1pro, PL2pro) | G892 – G2453 | S889 – G2518 | G772 – G2339 | G772 – G2338 |
| nsp4 | Hydrophobic domain | S2454 – Q2931 | S2519 – Q 2996 | G2340 – Q2817 | G2339 – Q2815 |
| nsp5 | 3CLpro | S2932 – Q3233 | S2997 – Q3298 | S2818 – Q3119 | A2816 – Q3117 |
| nsp6 | Hydrophobic domain | S3234 – Q3509 | S3299 – Q3574 | G3120 – Q3398 | S3118 – Q3395 |
| nsp7 | Unknown | S3510 – Q3592 | S3575 – Q3657 | S3399 – Q3481 | S3396 – Q3478 |
| nsp8 | Unknown | S3593 – Q3787 | S3658 – Q3852 | S3482 – Q3676 | S3479 – Q3673 |
| nsp9 | Unknown | N3788 – Q3895 | N3853 – Q3960 | N3677 – Q3784 | N3674 – Q3781 |
| nsp10 | Unknown | A3896 – Q4031 | A3961 – Q4097 | A3785 – Q3919 | A3782 –Q3916 |
| nsp11 | Unknown | S4032 – D4048 | A4098 – Q4115 | T3920 – D3936 | A3917 – D3933 |
| nsp12 | RdRp | S4032 – Q4958 | A4098 – Q5024 | T3920 – Q4846 | A3917 – Q4843 |
| nsp13 | Hel | A4959 – Q5555 | S5025 – Q5621 | S4847 – Q5443 | S4844 – Q5440 |
| nsp14 | ExoN, N7-MTase | S5556 – Q6073 | A5622 – Q6139 | A5444 – Q5960 | S5441 – Q5958 |
| nsp15 | NendoU | G6074 – Q6412 | S6140 – Q6478 | S5961 – Q6299 | G5959 – Q6297 |
| nsp16 | O-MT | A6413 – K6712 | S6479 – R6780 | S6300 – Y6591 | S6298 – Y6589 |
| nsp | Cleavage Site | |||
|---|---|---|---|---|
| Rs-BatCoV HKU32 Strain TLC28A | Ro-BatCoV HKU10 183A | Tr-BatCoV HKU33 Strain GZ151867 | BtNv-AlphaCoV/SC2013 | |
| nsp1/nsp2 | A/P | A/K | A/K | A/K |
| nsp2/nsp3 | G/G | G/S | G/G | G/G |
| nsp3/nsp4 | G/S | G/S | G/G | G/G |
| nsp4/nsp5 | Q/S | Q/S | Q/S | Q/A |
| nsp5/nsp6 | Q/S | Q/S | Q/G | Q/S |
| nsp6/nsp7 | Q/S | Q/S | Q/S | Q/S |
| nsp7/nsp8 | Q/S | Q/S | Q/S | Q/S |
| nsp8/nsp9 | Q/N | Q/N | Q/N | Q/N |
| nsp9/nsp10 | Q/A | Q/A | Q/A | Q/A |
| nsp10/nsp12 | Q/S | Q/A | Q/T | Q/A |
| nsp12/nsp13 | Q/A | Q/S | Q/S | Q/S |
| nsp13/nsp14 | Q/S | Q/A | Q/A | Q/S |
| nsp14/nsp15 | Q/G | Q/S | Q/S | Q/G |
| nsp15/nsp16 | Q/A | Q/S | Q/S | Q/S |
| Replicase Polyprotein Domain | Pairwise Sequence Identity with Rs-BatCoV HKU32 Strain TLC28A (%) | ||||
|---|---|---|---|---|---|
| BtRf-AlphaCoV/ HuB2013 | BtMs-AlphaCoV/ GS2013 | Ro-BatCoV HKU10 | PEDV | Tr-BatCoV HKU33 strain GZ151867 | |
| nsp3 | 67.6 | 67.6 | 60.3 | 50.1 | 49.0 |
| nsp5 | 84.8 | 84.8 | 81.5 | 74.2 | 75.2 |
| nsp12 | 92.6 | 92.6 | 90.1 | 83.2 | 83.7 |
| nsp13 | 94.1 | 94.3 | 92.1 | 85.6 | 80.9 |
| nsp14 | 93.4 | 93.4 | 90.0 | 79.9 | 78.8 |
| nsp15 | 89.4 | 89.4 | 83.8 | 76.7 | 78.2 |
| nsp16 | 89.7 | 90.0 | 85.8 | 82.8 | 81.2 |
| 7 Concatenated Domains | 83.2 | 83.3 | 78.6 | 70.3 | 69.5 |
| Overall replicase pp1ab | 80.1 | 80.5 | 75.0 | 65.8 | 63.3 |
| Putative TRS | ||||||
|---|---|---|---|---|---|---|
| ORF | Nucleotide Positions (Start–End) | No. of Nucleotides | No. of Amino Acids | Frame(s) | Nucleotide Position in Genome | TRS Sequence (Distance (No. of Bases) to AUG) 1 |
| 1ab | 278–20,052 | 19,774 | 6591 | +1, +2 | 54 | AACUAAAC(218)AUG |
| nsp1 | 278–856 | 579 | 193 | +2 | ||
| nsp2 | 857–2590 | 1734 | 578 | +2 | ||
| nsp3 | 2591–7294 | 4704 | 1568 | +2 | ||
| nps4 | 7295–8728 | 1434 | 478 | +2 | ||
| nsp5 | 8729–9634 | 906 | 302 | +2 | ||
| nsp6 | 9635–10,471 | 837 | 279 | +2 | ||
| nsp7 | 10,472–10,532 | 249 | 83 | +2 | ||
| nsp8 | 10,533–11,305 | 585 | 195 | +2 | ||
| nsp9 | 11,306–11,629 | 324 | 108 | +2 | ||
| nsp10 | 11,630–12,034 | 405 | 135 | +2 | ||
| nsp11 | 51 | 17 | +2 | |||
| nsp12 | 12,035–14,814 | 2780 | 927 | +1 | ||
| nsp13 | 14,815–16,605 | 1791 | 597 | +1 | ||
| nsp14 | 16,606–18,156 | 1551 | 517 | +1 | ||
| nsp15 | 18,157–19,173 | 1017 | 339 | +1 | ||
| nsp16 | 19,174–20,052 | 879 | 292 | +1 | ||
| S | 20,053–24,150 | 4098 | 1365 | +1 | 20,049 | GACUAAAUG |
| ORF3 | 24,150–24,755 | 606 | 201 | +3 | 23,876 | ATCUCAAC(268)AUG |
| E | 24,777–25,004 | 228 | 75 | +3 | 24,763 | TTCUCAAC(8)AUG |
| M | 25,011–25,697 | 687 | 228 | +3 | 25,001 | GTCUAAAC(4)AUG |
| N | 25,706–26,977 | 1272 | 423 | +2 | 25,699 | AACUAAAC(1)AUG |
| ORF7 | 26,989–27,348 | 360 | 119 | +1 | 26,982 | AACUAAAU(1)AUG |
| Replicase Polyprotein Domain | Pairwise Amino Acid Sequence Identity with Tr-BatCoV HKU33 Strain GZ151867 (%) | ||||
|---|---|---|---|---|---|
| BtNv-AlphaCoV/SC2013 | BtRf-AlphaCoV/HuB2013 | BtMs-AlphaCoV/GS2013 | AlphaCoV BatCoV/P.kuhlii/Italy 206679-3/2010 | Rs-BatCoV HKU32 Strain TLC28A | |
| nsp3 | 58.8 | 48.7 | 48.8 | 57.3 | 49.0 |
| nsp5 | 82.5 | 74.2 | 74.5 | 78.8 | 75.2 |
| nsp12 | 86.3 | 83.5 | 83.6 | 86.4 | 83.7 |
| nsp13 | 87.0 | 79.7 | 80.1 | 82.7 | 80.9 |
| nsp14 | 84.0 | 79.3 | 79.3 | 82.4 | 78.8 |
| nsp15 | 85.8 | 79.6 | 79.0 | 84.7 | 78.2 |
| nsp16 | 87.3 | 80.8 | 80.5 | 84.2 | 81.2 |
| 7 Concatenated Domains | 76.3 | 69.2 | 69.1 | 74.4 | 69.4 |
| Overall replicase pp1ab | 73.4 | 62.9 | 63.0 | 71.6 | 63.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, S.K.P.; Wong, A.C.P.; Zhang, L.; Luk, H.K.H.; Kwok, J.S.L.; Ahmed, S.S.; Cai, J.-P.; Zhao, P.S.H.; Teng, J.L.L.; Tsui, S.K.W.; et al. Novel Bat Alphacoronaviruses in Southern China Support Chinese Horseshoe Bats as an Important Reservoir for Potential Novel Coronaviruses. Viruses 2019, 11, 423. https://0-doi-org.brum.beds.ac.uk/10.3390/v11050423
Lau SKP, Wong ACP, Zhang L, Luk HKH, Kwok JSL, Ahmed SS, Cai J-P, Zhao PSH, Teng JLL, Tsui SKW, et al. Novel Bat Alphacoronaviruses in Southern China Support Chinese Horseshoe Bats as an Important Reservoir for Potential Novel Coronaviruses. Viruses. 2019; 11(5):423. https://0-doi-org.brum.beds.ac.uk/10.3390/v11050423
Chicago/Turabian StyleLau, Susanna K.P., Antonio C.P. Wong, Libao Zhang, Hayes K.H. Luk, Jamie S. L. Kwok, Syed S. Ahmed, Jian-Piao Cai, Pyrear S.H. Zhao, Jade L.L. Teng, Stephen K.W. Tsui, and et al. 2019. "Novel Bat Alphacoronaviruses in Southern China Support Chinese Horseshoe Bats as an Important Reservoir for Potential Novel Coronaviruses" Viruses 11, no. 5: 423. https://0-doi-org.brum.beds.ac.uk/10.3390/v11050423