Interaction of Bacteriophage l with Its E. coli Receptor, LamB

Abstract
:1. Introduction
2. Structure of the LamB Receptor and Interaction with Bacteriophage λ



{kind=link}
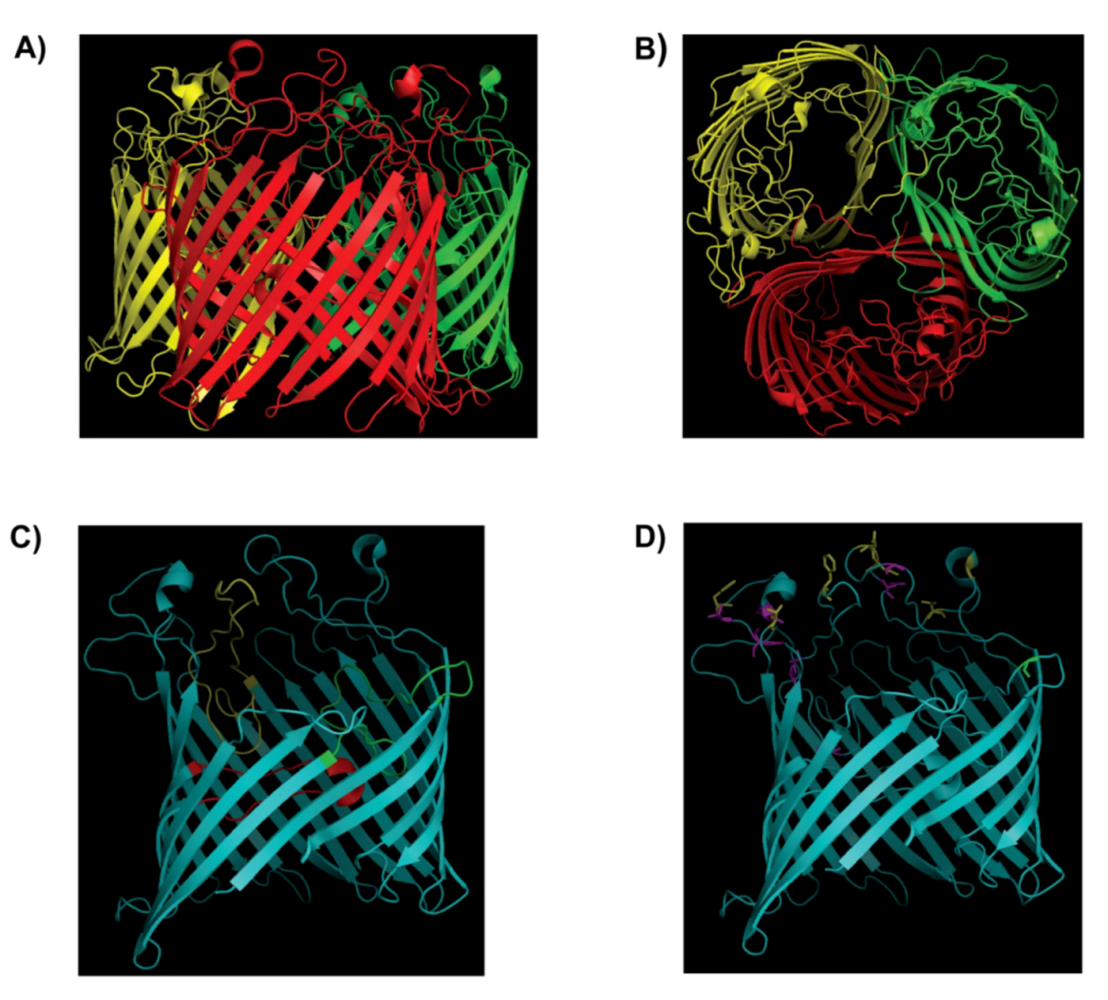{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue(s) | Substitution |
|---|---|
| 18 | Gly → Val |
| 148 | Glu → Lys |
| 151 | Gly → Asp |
| 152 | Ser → Phe |
| 154 | Ser → Phe |
| 155 | Phe → Ser |
| 163 | Tyr → Asp |
| 164 | Thr → Pro |
| 245 | Gly → Arg |
| 245 | Gly → Val |
| 247 | Ser → Leu |
| 249 | Gly → Asp |
| 250 | Ser → Phe |
| 259 | Phe → Val |
| 259 | Phe → Val |
| 382 | Gly → Asp |
| 382 | Gly → Val |
| 401 | Gly → Asp |
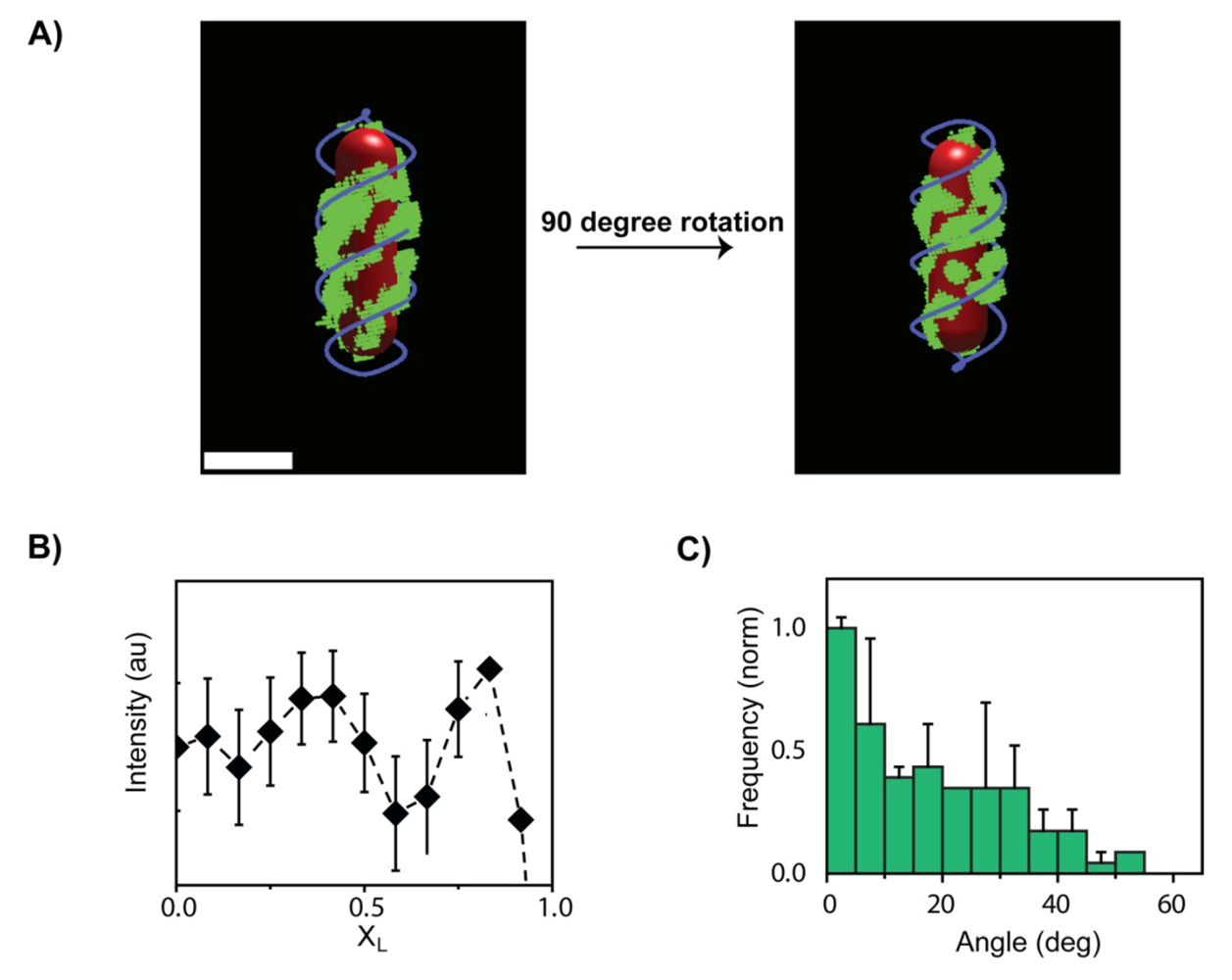
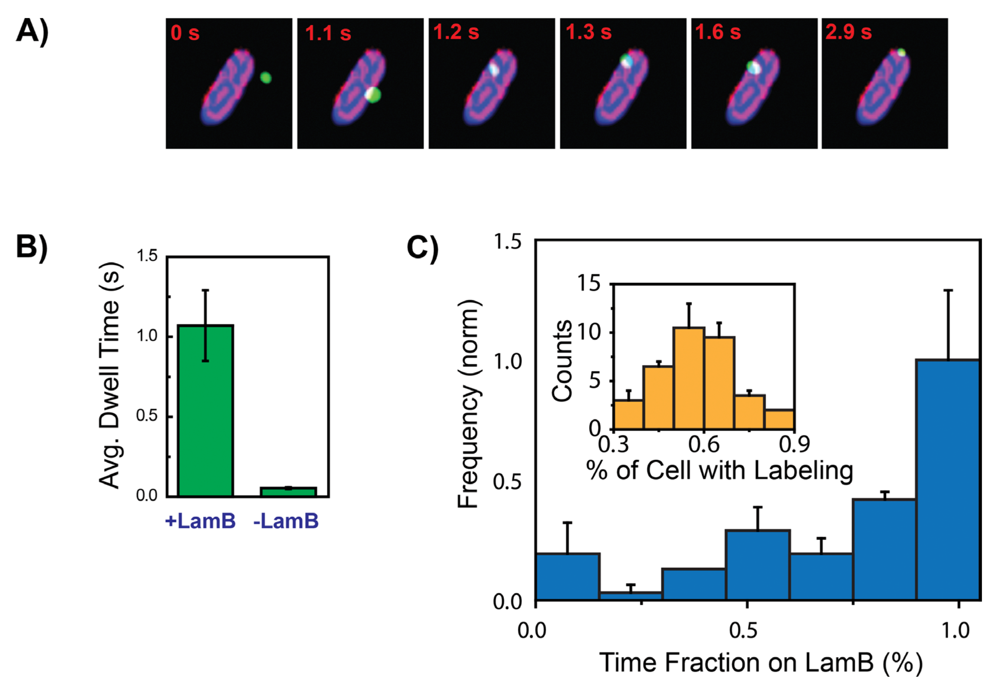
3. The λ Phage LamB Target Finding Process
| Phage: Host | Foci at the pole and mid-cell (%) | Foci in other location (%) |
|---|---|---|
| λ: E. coli | 69 | 31 |
| T7: E. coli | 71 | 29 |
| P1: E. coli | 78 | 22 |
| T4: E. coli | 95 | 5 |
| λØ80: E. coli | 68 | 32 |
| KVP40: V. cholera | 73 | 27 |
| ØA1122: Y. pseudotuberculosis | 68 | 32 |



4. Theoretical Model for Viral Target Finding

5. Summary
Conflict of Interest
References and Notes
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef]
- Mudhakir, D.; Harashima, H. Learning from the viral journey: How to enter cells and how to overcome intracellular barriers to reach the nucleus. AAPS J. 2009, 11, 65–77. [Google Scholar] [CrossRef]
- Smith, A.E.; Helenius, A. How viruses enter animal cells. Science 2004, 304, 237–242. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122. [Google Scholar] [CrossRef]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Friedman, D.I.; Court, D.L. Bacteriophage lambda: Alive and well and still doing its thing. Curr. Opin. Microbiol. 2001, 4, 201–207. [Google Scholar]
- Hendrix, R.W. Bacteriophage genomics. Curr. Opin. Microbiol. 2003, 6, 506–511. [Google Scholar] [CrossRef]
- Hendrix, R.; Roberts, J.; Stahl, F.; Wesberg, R. Lambda II; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1983. [Google Scholar]
- Ptasne, M. A Genetic Switch, Gene Control and Phage Lambda; Blackwell Scientific and Cell Press: Palo Alto, CA, USA, 1986. [Google Scholar]
- Ptasne, M. A Gentic Switch: Phage Lambda Revisited; Cold Spring Harbor: New York, NY, USA, 2004. [Google Scholar]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in bacteriophage lambda development. Annu. Rev. Genet. 2005, 39, 409–429. [Google Scholar] [CrossRef]
- Randall-Hazelbauer, L.; Schwartz, M. Isolation of the bacteriophage lambda receptor from Escherichia coli. J. Bacteriol. 1973, 116, 1436–1446. [Google Scholar]
- Schwartz, M. Reversible interaction between coliphage lambda and its receptor protein. J. Mol. Biol. 1975, 99, 185–201. [Google Scholar]
- Schwartz, M. The adsorption of coliphage lambda to its host: Effect of variations in the surface density of receptor and in phage-receptor affinity. J. Mol. Biol. 1976, 103, 521–536. [Google Scholar] [CrossRef]
- Schwartz, M. Interaction of Phages with Their Receptor Proteins; Chapman and Hall: London, UK, 1980. [Google Scholar]
- Berkane, E.; Orlik, F.; Stegmeier, J.F.; Charbit, A.; Winterhalter, M.; Benz, R. Interaction of bacteriophage lambda with its cell surface receptor: An in vitro study of binding of the viral tail protein gpJ to LamB (Maltoporin). Biochemistry 2006, 45, 2708–2720. [Google Scholar]
- Charbit, A.; Clement, J.M.; Hofnung, M. Further sequence analysis of the phage lambda receptor site. Possible implications for the organization of the lamB protein in Escherichia coli K12. J. Mol. Biol. 1984, 175, 395–401. [Google Scholar] [CrossRef]
- Charbit, A.; Werts, C.; Michel, V.; Klebba, P.E.; Quillardet, P.; Hofnung, M. A role for residue 151 of LamB in bacteriophage lambda adsorption: Possible steric effect of amino acid substitutions. J. Bacteriol. 1994, 176, 3204–3209. [Google Scholar]
- Clement, J.M.; Lepouce, E.; Marchal, C.; Hofnung, M. Genetic study of a membrane protein: DNA sequence alterations due to 17 lamB point mutations affecting adsorption of phage lambda. EMBO J. 1983, 2, 77–80. [Google Scholar]
- Hofnung, M.; Hatfield, D.; Schwartz, M. malB region in Escherichia coli K-12: Characterization of new mutations. J. Bacteriol. 1974, 117, 40–47. [Google Scholar]
- Hofnung, M.; Jezierska, A.; Braun-Breton, C. lamB mutations in E. coli K12: Growth of lambda host range mutants and effect of nonsense suppressors. Mol. Gen. Genet. 1976, 145, 207–213. [Google Scholar]
- Scandella, D.; Arber, W. An Escherichia coli mutant which inhibits the injection of phage lambda DNA. Virology 1974, 58, 504–513. [Google Scholar] [CrossRef]
- Wang, J.; Hofnung, M.; Charbit, A. The C-terminal portion of the tail fiber protein of bacteriophage lambda is responsible for binding to LamB, its receptor at the surface of Escherichia coli K-12. J. Bacteriol. 2000, 182, 508–512. [Google Scholar] [CrossRef]
- Wang, J.; Michel, V.; Hofnung, M.; Charbit, A. Cloning of the J gene of bacteriophage lambda, expression and solubilization of the J protein: First in vitro studies on the interactions between J and LamB, its cell surface receptor. Res. Microbiol. 1998, 149, 611–624. [Google Scholar] [CrossRef]
- Rothenberg, E.; Sepulveda, L.A.; Skinner, S.O.; Zeng, L.; Selvin, P.R.; Golding, I. Single-virus tracking reveals a spatial receptor-dependent search mechanism. Biophys. J. 2011, 100, 2875–2882. [Google Scholar]
- Golding, I. Decision making in living cells: Lessons from a simple system. Annu. Rev. Biophys. 2011, 40, 63–80. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R.; Hong, G.F.; Hill, D.F.; Petersen, G.B. Nucleotide sequence of bacteriophage lambda DNA. J. Mol. Biol. 1982, 162, 729–773. [Google Scholar]
- Hendrix, R. Lambda II; Cold Spring Harbor Laboratory: New York, NY, USA, 1985. [Google Scholar]
- Weigle, J. Assembly of phage lambda in vitro. Proc. Natl. Acad. Sci. U. S. A. 1966, 55, 1462–1466. [Google Scholar] [CrossRef]
- Meyer, J.R.; Dobias, D.T.; Weitz, J.S.; Barrick, J.E.; Quick, R.T.; Lenski, R.E. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 2012, 335, 428–432. [Google Scholar]
- Hendrix, R.W.; Duda, R.L. Bacteriophage lambda PaPa: Not the mother of all lambda phages. Science 1992, 258, 1145–1148. [Google Scholar]
- Roa, M.; Scandella, D. Multiple steps during the interaction between coliphage lambda and its receptor protein in vitro. Virology 1976, 72, 182–194. [Google Scholar] [CrossRef]
- Weigle, J. Studies on head-tail union in bacteriophage lambda. J. Mol. Biol. 1968, 33, 483–489. [Google Scholar]
- Smit, J.; Kamio, Y.; Nikaido, H. Outer membrane of Salmonella typhimurium: Chemical analysis and freeze-fracture studies with lipopolysaccharide mutants. J. Bacteriol. 1975, 124, 942–958. [Google Scholar]
- Kamio, Y.; Nikaido, H. Outer membrane of Salmonella typhimurium: Accessibility of phospholipid head groups to phospholipase c and cyanogen bromide activated dextran in the external medium. Biochemistry 1976, 15, 2561–2570. [Google Scholar] [CrossRef]
- Nikaido, H. Porins and specific channels of bacterial outer membranes. Mol. Microbiol. 1992, 6, 435–442. [Google Scholar]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Nikaido, H.; Vaara, M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [Google Scholar]
- Zhai, Y.; Saier, M.H., Jr. The beta-barrel finder (BBF) program, allowing identification of outer membrane beta-barrel proteins encoded within prokaryotic genomes. Protein Sci. 2002, 11, 2196–2207. [Google Scholar]
- Engelhardt, H.; Heinz, C.; Niederweis, M. A tetramericporin limits the cell wall permeability of Mycobacterium smegmatis. J. Biol. Chem. 2002, 277, 37567–37572. [Google Scholar]
- Heinz, C.; Engelhardt, H.; Niederweis, M. The core of the tetramericmycobacterialporinMspA is an extremely stable beta-sheet domain. J. Biol. Chem. 2003, 278, 8678–8685. [Google Scholar]
- Model, K.; Prinz, T.; Ruiz, T.; Radermacher, M.; Krimmer, T.; Kuhlbrandt, W.; Pfanner, N.; Meisinger, C. Protein translocase of the outer mitochondrial membrane: Role of import receptors in the structural organization of the TOM complex. J. Mol. Biol. 2002, 316, 657–666. [Google Scholar] [CrossRef]
- Schleiff, E.; Eichacker, L.A.; Eckart, K.; Becker, T.; Mirus, O.; Stahl, T.; Soll, J. Prediction of the plant beta-barrel proteome: A case study of the chloroplast outer envelope. Protein Sci. 2003, 12, 748–759. [Google Scholar]
- Parker, M.W.; Buckley, J.T.; Postma, J.P.; Tucker, A.D.; Leonard, K.; Pattus, F.; Tsernoglou, D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 1994, 367, 292–295. [Google Scholar] [CrossRef]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptamerictransmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef]
- Hancock, R.E.; Reeves, P. Lipopolysaccharide-deficient, bacteriophage-resistant mutants of Escherichia coli K-12. J. Bacteriol. 1976, 127, 98–108. [Google Scholar]
- Roa, M. Interaction of bacteriophage K10 with its receptor, the lamB protein of Escherichia coli. J. Bacteriol. 1979, 140, 680–686. [Google Scholar]
- Wandersman, C.; Schwartz, M. Protein Ia and the lamB protein can replace each other in the constitution of an active receptor for the same coliphage. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 5636–5639. [Google Scholar]
- Ishii, J.N.; Okajima, Y.; Nakae, T. Characterization of lamB protein from the outer membrane of Escherichia coli that forms diffusion pores selective for maltose-maltodextrins. FEBS Lett. 1981, 134, 217–220. [Google Scholar] [CrossRef]
- Nakae, T.; Ishii, J.N. Molecular weights and subunit structure of LamB proteins. Annalesde Microbiologie 1982, 133A, 21–25. [Google Scholar]
- Neuhaus, J.M. The receptor protein of phage lambda: Purification, characterization and preliminary electrical studies in planar lipid bilayers. Annalesde Microbiologie 1982, 133A, 27–32. [Google Scholar]
- Schirmer, T.; Keller, T.A.; Wang, Y.F.; Rosenbusch, J.P. Structural basis for sugar translocation through maltoporin channels at 3.1 A resolution. Science 1995, 267, 512–514. [Google Scholar]
- Dutzler, R.; Wang, Y.F.; Rizkallah, P.; Rosenbusch, J.P.; Schirmer, T. Crystal structures of various maltooligosaccharides bound to maltoporin reveal a specific sugar translocation pathway. Structure 1996, 4, 127–134. [Google Scholar] [CrossRef]
- Charbit, A.; Gehring, K.; Nikaido, H.; Ferenci, T.; Hofnung, M. Maltose transport and starch binding in phage-resistant point mutants of maltoporin. Functional and topological implications. J. Mol. Biol. 1988, 201, 487–496. [Google Scholar] [CrossRef]
- Fuerst, C.R.; Bingham, H. Genetic and physiological characterization of the J gene of bacteriophage lambda. Virology 1978, 87, 437–458. [Google Scholar]
- Adam, G.; Delbruck, M. Reduction of dimensionality in biological diffusion processes. In Structural Chemistry and Molecular Biology; W.H. Freeman & Company: San Francisco, NC, USA, 1968. [Google Scholar]
- Berg, H.C.; Purcell, E.M. Physics of chemoreception. Biophys. J. 1977, 20, 193–219. [Google Scholar] [CrossRef]
- Moldovan, R.; Chapman-McQuiston, E.; Wu, X.L. On kinetics of phage adsorption. Biophys. J. 2007, 93, 303–315. [Google Scholar] [CrossRef]
- Gibbs, K.A.; Isaac, D.D.; Xu, J.; Hendrix, R.W.; Silhavy, T.J.; Theriot, J.A. Complex spatial distribution and dynamics of an abundant Escherichia coli outer membrane protein, LamB. Mol. Microbiol. 2004, 53, 1771–1783. [Google Scholar] [CrossRef]
- Edgar, R.; Rokney, A.; Feeney, M.; Semsey, S.; Kessel, M.; Goldberg, M.B.; Adhya, S.; Oppenheim, A.B. Bacteriophage infection is targeted to cellular poles. Mol. Microbiol. 2008, 68, 1107–1116. [Google Scholar] [CrossRef]
- Kaiser, D.; Dworkin, M. Gene transfer to myxobacterium by Escherichia coli phage P1. Science 1975, 187, 653–654. [Google Scholar]
- Hashemolhosseini, S.; Holmes, Z.; Mutschler, B.; Henning, U. Alterations of receptor specificities of coliphages of the T2 family. J. Mol. Biol. 1994, 240, 105–110. [Google Scholar] [CrossRef]
- Steven, A.C.; Trus, B.L.; Maizel, J.V.; Unser, M.; Parry, D.A.; Wall, J.S.; Hainfeld, J.F.; Studier, F.W. Molecular substructure of a viral receptor-recognition protein. The gp17 tail-fiber of bacteriophage T7. J. Mol. Biol. 1988, 200, 351–365. [Google Scholar] [CrossRef]
- Garcia, E.; Elliott, J.M.; Ramanculov, E.; Chain, P.S.; Chu, M.C.; Molineux, I.J. The genome sequence of Yersiniapestisbacteriophage phiA1122 reveals an intimate history with the coliphage T3 and T7 genomes. J. Bacteriol. 2003, 185, 5248–5262. [Google Scholar] [CrossRef]
- Miller, E.S.; Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Durkin, A.S.; Ciecko, A.; Feldblyum, T.V.; White, O.; Paulsen, I.T.; Nierman, W.C.; et al. Complete genome sequence of the broad-host-range vibriophage KVP40: Comparative genomics of a T4-related bacteriophage. J. Bacteriol. 2003, 185, 5220–5233. [Google Scholar]
- Osborn, M.J.; Rothfield, L. Cell shape determination in Escherichia coli. Curr. Opin. Microbiol. 2007, 10, 606–610. [Google Scholar] [CrossRef]
- Vats, P.; Shih, Y.L.; Rothfield, L. Assembly of the MreB-associated cytoskeletal ring of Escherichia coli. Mol. Microbiol. 2009, 72, 170–182. [Google Scholar] [CrossRef]
- Oddershede, L.; Dreyer, J.K.; Grego, S.; Brown, S.; Berg-Sorensen, K. The motion of a single molecule, the lambda-receptor, in the bacterial outer membrane. Biophys. J. 2002, 83, 3152–3161. [Google Scholar] [CrossRef]
- Brown, S. Engineered iron oxide-adhesion mutants of the Escherichia coli phage lambda receptor. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 8651–8655. [Google Scholar] [CrossRef]
- Golding, I.; Cox, E.C. Physical nature of bacterial cytoplasm. Phys. Rev. Lett. 2006, 96, 098102. [Google Scholar] [CrossRef]
- Holyst, R.; Plewczynski, D.; Aksimentiev, A.; Burdzy, K. Diffusion on curved, periodic surfaces. Phys. Rev. E 1999, 60, 302–307. [Google Scholar] [CrossRef]
- Axelrod, D.; Wang, M.D. Reduction-of-dimensionality kinetics at reaction-limited cell surface receptors. Biophys. J. 1994, 66, 588–600. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chatterjee, S.; Rothenberg, E. Interaction of Bacteriophage l with Its E. coli Receptor, LamB. Viruses 2012, 4, 3162-3178. https://0-doi-org.brum.beds.ac.uk/10.3390/v4113162
Chatterjee S, Rothenberg E. Interaction of Bacteriophage l with Its E. coli Receptor, LamB. Viruses. 2012; 4(11):3162-3178. https://0-doi-org.brum.beds.ac.uk/10.3390/v4113162
Chicago/Turabian StyleChatterjee, Sujoy, and Eli Rothenberg. 2012. "Interaction of Bacteriophage l with Its E. coli Receptor, LamB" Viruses 4, no. 11: 3162-3178. https://0-doi-org.brum.beds.ac.uk/10.3390/v4113162