NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana †



1
Małopolska Centre of Biotechnology, Jagiellonian University, 30-387 Kraków, Poland
2
National Research Institute of Animal Production, 32-083 Balice, Poland
3
Department of Fundamental and Preclinical Sciences, Faculty of Biology and Veterinary Sciences, Nicolaus Copernicus University in Toruń, 87-100 Toruń, Poland
*
Author to whom correspondence should be addressed.
†
Presented at the First International Electronic Conference on Genes: Theoretical and Applied Genomics, 2–30 November 2020; Available online: https://iecge.sciforum.net/.
Proceedings 2021, 76(1), 7; https://0-doi-org.brum.beds.ac.uk/10.3390/IECGE-07155
Published: 2 November 2020
(This article belongs to the Proceedings of The 1st International Electronic Conference on Genes: Theoretical and Applied Genomics)
{kind=link}
{kind=link}
Abstract
:Peroxisomes are the type of organelles in eukaryotic cells that are involved in different biochemical pathways depending on the type of cell. We have isolated a number of peroxisome unusual positioning (peup) mutants, which display the accumulation of abnormal peroxisomes, and demonstrated that autophagy is involved in removing damaged organelles. These peup mutants also show defects of other autophagy-related processes, such as the recovery from dark-senescence, and also failed to induce vacuole-related vesicle formations during microautophagy under nutrient deprivations. The aim of this study was to identify the causative gene of the peup33 mutant using next-generation sequencing (NGS) as a tool. Identification of mutations with NGS will allow us to save time compared to the conventional mapping method. Here, we present the workflow of the experiment, the procedure of bioinformatic analysis and the software applied to the sequence data produced by NGS.
Published: 2 November 2020
1. Introduction
Peroxisomes are a type of enzymatic organelles in eukaryotic cells that are involved in different biochemical pathways depending on the type of cell and its function. Based on these criteria we can distinguish, e.g., glyoxysomes, leaf peroxisomes, root peroxisomes or unspecialized peroxisomes. Peroxisomes play two main roles in plants. In germinating seeds, glyoxysomes are involved in the β-oxidation of fatty acids to carbohydrates, which provide energy to germinate. In leaves, peroxisomes accumulate enzymes for glycolate pathway, which metabolizes by-products of photosynthesis. Large amounts of hydrogen peroxide (H2O2) produced during enzymatic reactions gradually oxidize and damage peroxisomes [1]. Damaged peroxisomes are constantly removed on the way of selective autophagy (pexophagy), which is a part of the cell quality control system.
While screening for novel peroxisome mutants of Arabidopsis from the pool of EMS (ethylmethane sulfonate) mutagenized GFP-PTS1 (expressing green fluorescence protein as a peroxisome marker) seeds, it has been observed that peroxisomes are usually dispersed in a cell and co-localize with chloroplasts. Based on this finding, different peroxisome unusual positioning (peup) mutants were isolated. A part of the peup mutants demonstrated increased number of peroxisomes, which formed aggregates containing damaged peroxisomes. Additionally, ATG8 autophagosome marker co-localized with the aggregates, indicating that autophagy is involved in removing of damaged peroxisomes [1]. Indeed, we identified the peup1, peup2 and peup4 mutants as the mutants defective in ATG2, ATG18a and ATG7, respectively, revealing that pexophagy requires the core set of autophagy genes [1]. Recently, we reported new causative mutations of peup17 and peup22 in ATG5 and ATG7 genes [2].
For many decades, genetic mapping of mutations using chromosome walking has functioned as a fundamental procedure of identification of causative mutations in forward genetic approach, in plants as well as in other organisms. However, this process may be sometimes challenging, because mutagenesis procedures, such as chemical treatment, can induce a large number of mutations. In fine mapping of recessive mutation and after phenotype screening, the region where the mutation localizes in must be determined, e.g., by using PCR-based methods. This step is crucial for further investigation and, at the same time, the most time-consuming procedure [3].
Recent advances in next-generation sequencing, molecular biology techniques and bioinformatics accelerated the localization of genetic marker linked to trait of interest. Nowadays, DNA from hundreds of individuals can be whole-genome sequenced and simultaneously provides a big set of data for evaluating the frequency and position of single nucleotide polymorphism (SNP) after alignment to a reference genome. However, to identify the causative point mutation, it is essential to optimize experimental procedure to minimize the time and financial effort [4].
This is a preliminary study on the application of next-generation sequencing to the identification of pexophagy-related mutation in Arabidopsis thaliana. This method is currently developed in our laboratory and may replace the conventional fine mapping procedure for identification of other autophagy defective mutants. To achieve this goal, molecular methods are combined with bioinformatics approaches to identify the peup33 causal mutation.
2. Materials and Methods
2.1. Plant Material Preparation for Next-Generation Sequencing
Around 400 F2 seeds from the crossing between Arabidopsis the peup33 mutant (Columbia background, GFP-PTS1) and Landsberg erecta (Ler) and around 100 seeds from wild-type plants (Columbia background, expressing GFP-PTS1) were plated on growth media containing 0.5× strength Murashige and Skoog salts, 1% (w/v) sucrose, 0.5% (w/v) MES-KOH buffer (pH 5.7) and 0.4% Gellan Gum. Germination and growing of the seedlings were conducted under the condition of continuous light and 22 °C in the growth chamber. Six-day-old seedlings were stained with 4 µM FM4-64 fluorescence dye with addition of 5 µM E-64d (papain family protease inhibitor) for 24 h to visualize the phenotype of autophagy mutants. The screening of seedlings was performed with Zeiss Axio Imager 2 fluorescence microscope. The elongation zone near the root tip of the seedlings was assessed to isolate peup mutants [2]. In total, 72 peup33 mutants and 72 wild-type plants were chosen. After screening, seedlings were transferred to a fresh growth media for further growth. Two-week-old plants were collected and immediately frozen in liquid nitrogen until DNA extraction.
2.2. DNA Extraction and Next-Generation Sequencing
DNA was extracted from plant tissue using DNeasy Plant Mini Kit (QIAGEN; Venlo, The Netherlands) following the manufacturer’s protocol. To measure the concentration and purity of DNA NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific; Waltham, MA, U.S.A.) and Qubit 4 fluorometer (Thermo Fisher Scientific) were used. Subsequent steps, which were libraries preparation and sequencing of pooled DNA, were performed by the commercial company (Macrogen; Seoul, South Korea). Libraries were prepared using Illumina TruSeq DNA PCR-Free kit and sequenced on Illumina NovaSeq6000 sequencer with the conditions of 150 bp paired-end reads and 48× coverage.
2.3. Bioinformatics
Raw data was outputted as fastq reads. FastQC [5] was used for the quality control of the raw sequences. Flexbar [6] was used to remove adapters and to select reads with a quality score ≥ 20 and a fragment ≥ 36 bp. Filtered sequences were aligned to the A. thaliana genome (TAIR10 assembly, release48; [7]) with the BWA aligner [8]. SNPs were identified using SAMtools [9] and BCFtools [10] in the entire Arabidopsis genome with the high sample ploidy parameters (2n × 50 per pool) [11]. Then, rigorous variant filtering was applied to SNPs according to GATK [12]. The identified variants were analyzed by SnpEff [13] and the Ensembl Variant Effect Predictor [14] to determine the gene types associations and function. Mutations present in peup33 × Ler F2 individuals were identified as functional annotated using PANTHER [15], KEGG [16] and Kobas [17]. The final list of variants was evaluated manually to only retain the mutations in genes of our interest (i.e., involved in autophagy/pexophagy process).
3. Results and Discussion
3.1. peup33 Mutants Display Autophagy-Defective Phenotype after FM4-64/E-64d Treatment
A number of peup mutants have been isolated so far from a pool of EMS mutagenized seeds of Arabidopsis thaliana Columbia accession. These mutants display autophagy-defective phenotypes, i.e., early senescence, dark senescence and the absence of vesicle accumulation in the vacuole under the treatment of concanamycin A [1,2]. The new mutant peup33 also showed similar phenotype and was subjected to further examination.
A membrane fluorescent dye FM4-64 has been primarily utilized to monitor endocytosis in variety types of cells. The dye also has been used to stain vacuolar membrane and track its shape and fate in yeast and plants. The application of papain-family protease inhibitor E-64 induces accumulation of autophagy-related vesicles beside the vacuole [18]. In the previous study, we reported that the combination of FM4-64 and E-64d treatment is a useful tool to distinguish the autophagy mutants [2].
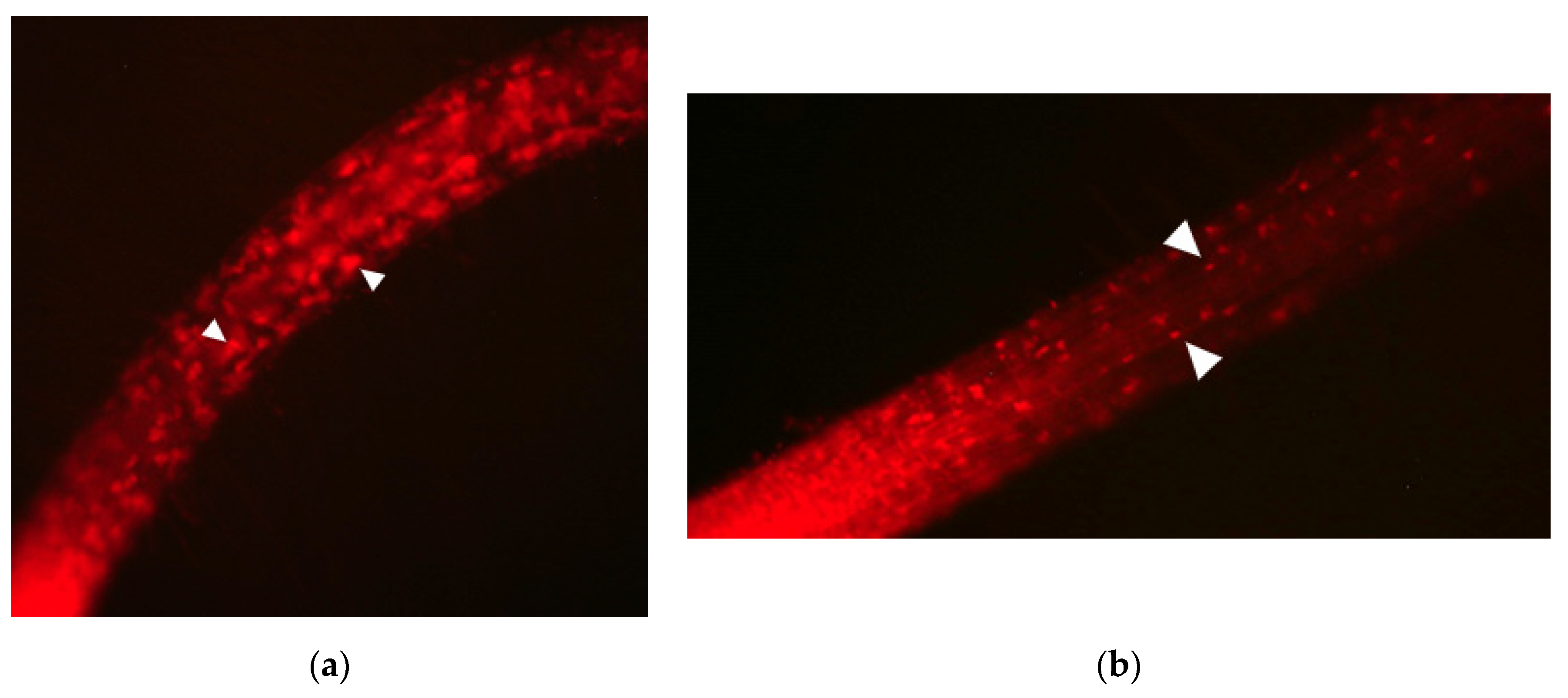
After 24 h of culturing in FM4-64/E-64d medium lacking sucrose, wild-type plants displayed a number of vessicle aggregates in the cells of the root area near the tip. In contrast, the observation of peup33 mutants after FM4-64/E-64d treatment revealed that the formation of vesicle aggregates was suppressed and the size of these aggregates decreased (Figure 1). Similar phenotype has been described by [2] for other peup mutants (peup1, peup2, peup4, peup17, peup22).
3.2. Next-Generation Sequencing for Identifaction of the peup33 Mutation
To identify the causative gene of the peup33 mutant, the F2 seedlings obtained from the crossing between peup33 and Ler were examined with the FM4-64/E64-d treatment, and the seedlings displaying mutant phenotype have been selected for next-generation sequencing. Whole-genome sequencing from the pool of F2 mutant seedlings revealed hundreds of thousands of mutations across the genome. After data pre-processing, more than 270 k of mutations have been displayed. Because peup33 shows autophagy-defective phenotype, it may possess the mutation in autophagy-related genes (ATG genes), but we want to consider also other genes that may be involved in autophagy process. First, we filtered out the variants with certain values of the primary parameters: alternative allele frequency ≥ 0.5, depth ≥ 50, mapping quality ≥ 60 and quality of reads ≥ 100. After the selection of the polymorphism, further verification was carried out using the genomic and gene annotation dataset of TAIR10; the variants that can affect protein function (the exonic missense, nonsense, stop-loss, frameshift and splice site variants) were selected [19]. Most of the variants were the type generating missense, several of others were splice site variants, and one was stop-gained mutation. After manual inquiring of functions of the candidate genes into common databases (TAIR, Ensembl, ThaleMine, UniProt) and publications, we focused on the genes and proteins that can be involved in autophagy process.
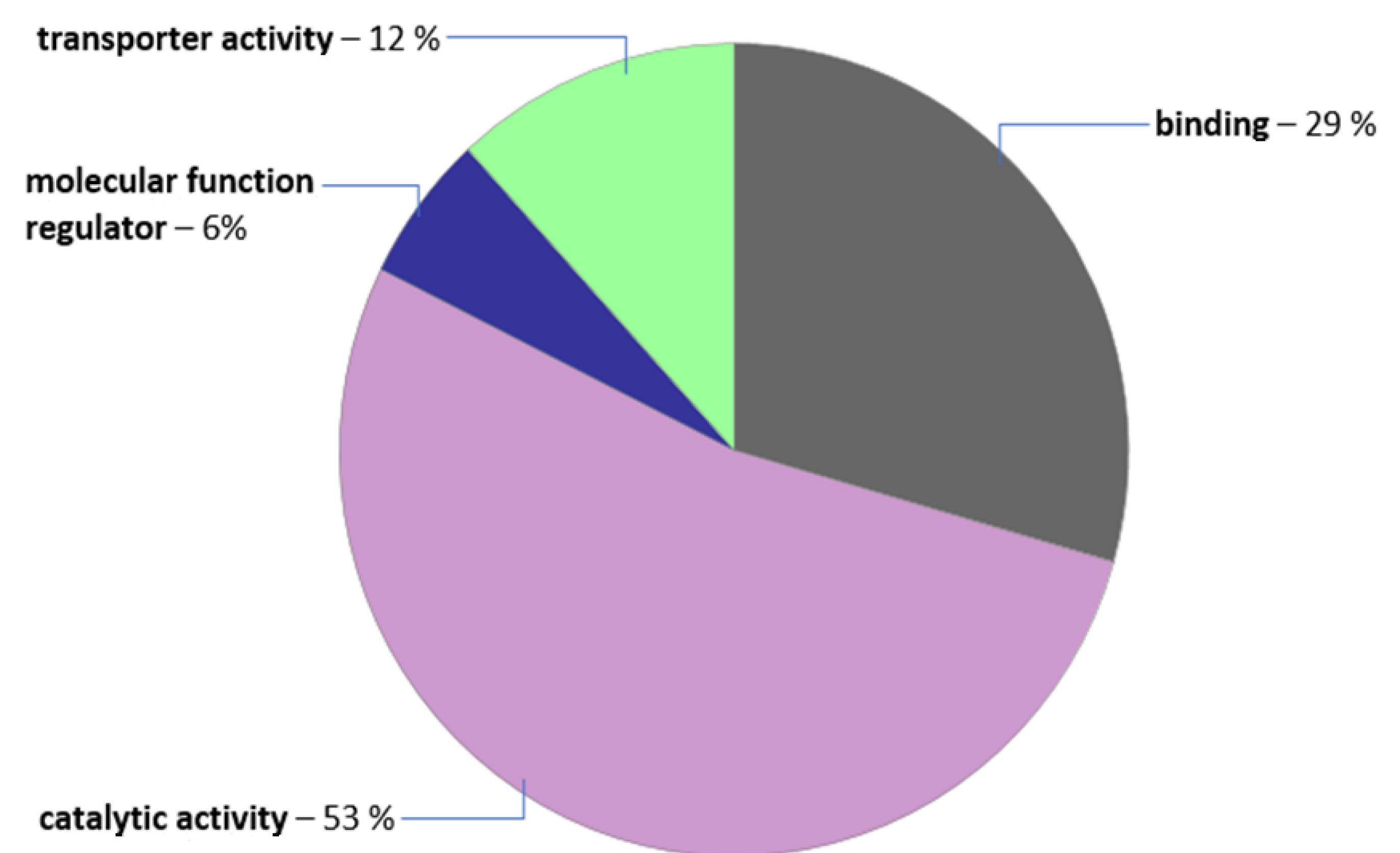
When all above-mentioned criteria have been applied to data filtration, we obtained 27 polymorphisms located mainly on chromosome 1 and 3 linked with 26 candidate genes that may be involved in autophagy/pexophagy process. PANTHER web-based tool gene ontology has been performed to verify the molecular function of candidate genes products (Figure 2). More than half of the variants identified were connected to catalytic activity.
A second big cluster included variants named as ‘binding’ activity, which was defined as ‘interaction of a molecule with one or more specific sites on another molecule’. A third was enrolled in transportation activity. These findings seems to be promising as the molecular function of these genes is likely connected to the pathways of our interest.
Once we have narrowed the range of potential peup33 causative mutation additional analyses are required for further verification e.g., direct sequencing, real-time PCR or performing allelism test to confirm the mutation and determine the causative gene.
4. Conclusions
Next-generation sequencing is a powerful tool to shorten the time of the identification of causative mutations and replacing the conventional mapping procedure. However, determination of a causative gene with whole-genome sequence requires an understanding of the genetic background of the biological process related to the mutant in order to link the phenotype with a proper SNP variant. Nowadays, it is getting easier as bioinformatics tools are being developed to be more user-friendly on NGS data analysis.
In future, we are going to analyze the function of PEUP33 protein by profiling gene expressions in the peup33 mutant and observing its phenotypes under a confocal laser-scanning microscope during inducing of pexophagy, as well as general autophagy. After establishing the series of gene identification processes using NGS, we aim to apply this technique to other autophagy/pexophagy mutants.
Author Contributions
Conceptualization, K.S. and S.G.-Y.; methodology, S.G.-Y. and K.S.; software, K.Ż.; formal analysis, K.Ż.; investigation, K.S., S.G.-Y.; data curation, K.S., K.Ż.; writing—original draft preparation, K.S.; writing—review and editing, S.G.-Y., K.Y.; visualization, K.S. and S.G.-Y.; supervision, S.G.-Y., K.Y.; funding acquisition, S.G.-Y. and K.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre, Poland: UMO-2019/34/NZ3/00299 to S.G.-Y. and UMO-2016/23/B/NZ1/01847 to K.Y. and K.S.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data available upon request from the authors.
Acknowledgments
This study was supported by the National Science Centre, Poland [UMO-2019/34/E/NZ3/00299 to SG-Y and UMO-2016/23/B/NZ1/01847 to K.Y. and K.S.] and in part by PLGrid Infrastructure.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Shibata, M.; Oikawa, K.; Yoshimoto, K.; Kondo, M.; Mano, S.; Yamada, K.; Hayashi, M.; Sakamoto, W.; Ohsumi, Y.; Nishimura, M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013, 25, 4967–4983. [Google Scholar] [CrossRef]
- Goto-Yamada, S.; Oikawa, K.; Bizan, J.; Shigenobu, S.; Yamaguchi, K.; Mano, S.; Hayashi, M.; Ueda, H.; Hara-Nishimura, I.; Nishimura, M.; et al. Sucrose Starvation Induces Microautophagy in Plant Root Cells. Front. Plant Sci. 2019, 10, 1604. [Google Scholar] [CrossRef]
- Allen, R.; Nakasugi, K.; Doran, R.L.; Millar, A.A.; Waterhouse, P.M. Facile mutant identification via a single parental backcross method and application of whole genome sequencing based mapping pipelines. Front. Plant Sci. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- James, G.V.; Patel, V.; Nordström, K.J.V.; Klasen, J.R.; Salomé, P.A.; Weigel, D.; Schneeberger, K. User guide for mapping-by-sequencing in Arabidopsis. Genome Biol. 2013, 14, R61. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. A quality control tool for high throughput sequence data. Babraham Bioinforma. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 November 2020).
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Moriyasu, Y.; Ohsumi, Y. Autophagy in Tobacco Suspension-Cultured Cells in Response to Sucrose Starvation. Plant Physiol. 1996, 111, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Dashti, M.J.S.; Gamieldien, J. A practical guide to filtering and prioritizing genetic variants. Biotechniques 2017, 62, 18–30. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The phenotypes of (a) wild-type plant (GFP-PTS1) and (b) peup33 mutant after the treatment with FM4-64 and E-64d for 24 h. White triangles indicate E-64d vesicle accumulation.
Figure 1.
The phenotypes of (a) wild-type plant (GFP-PTS1) and (b) peup33 mutant after the treatment with FM4-64 and E-64d for 24 h. White triangles indicate E-64d vesicle accumulation.

Figure 2.
Gene ontology molecular function classification of candidate genes for peup33 mutants. Pie chart was generated with PANTHER web-based tool.
Figure 2.
Gene ontology molecular function classification of candidate genes for peup33 mutants. Pie chart was generated with PANTHER web-based tool.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sieńko, K.; Żukowski, K.; Yamada, K.; Goto-Yamada, S. NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana. Proceedings 2021, 76, 7. https://0-doi-org.brum.beds.ac.uk/10.3390/IECGE-07155
AMA Style
Sieńko K, Żukowski K, Yamada K, Goto-Yamada S. NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana. Proceedings. 2021; 76(1):7. https://0-doi-org.brum.beds.ac.uk/10.3390/IECGE-07155
Chicago/Turabian StyleSieńko, Katarzyna, Kacper Żukowski, Kenji Yamada, and Shino Goto-Yamada. 2021. "NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana" Proceedings 76, no. 1: 7. https://0-doi-org.brum.beds.ac.uk/10.3390/IECGE-07155