Propylene Glycol and Non-Destructive DNA Extractions Enable Preservation and Isolation of Insect and Hosted Bacterial DNA

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Experiment 1: DNA Extraction Methods Comparison
2.3. qPCR
2.4. Experiment 2: Propylene Glycol DNA Preservation Time-Series Experiment
DNA Extraction Protocol and qPCR
2.5. Statistical Analysis of Cycle Threshold Data
3. Results
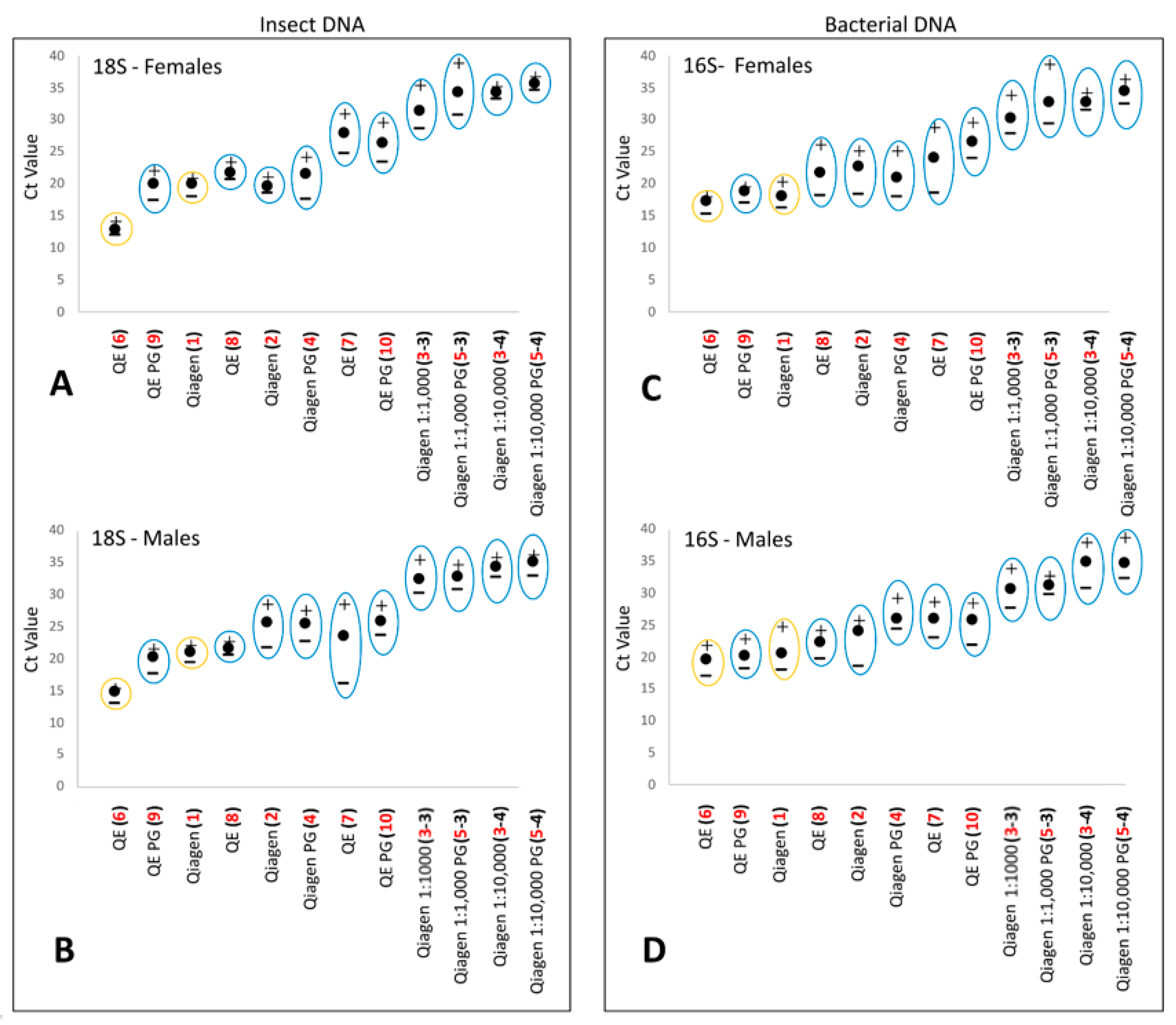
3.1. DNA Extraction Method Comparison
3.2. Controls
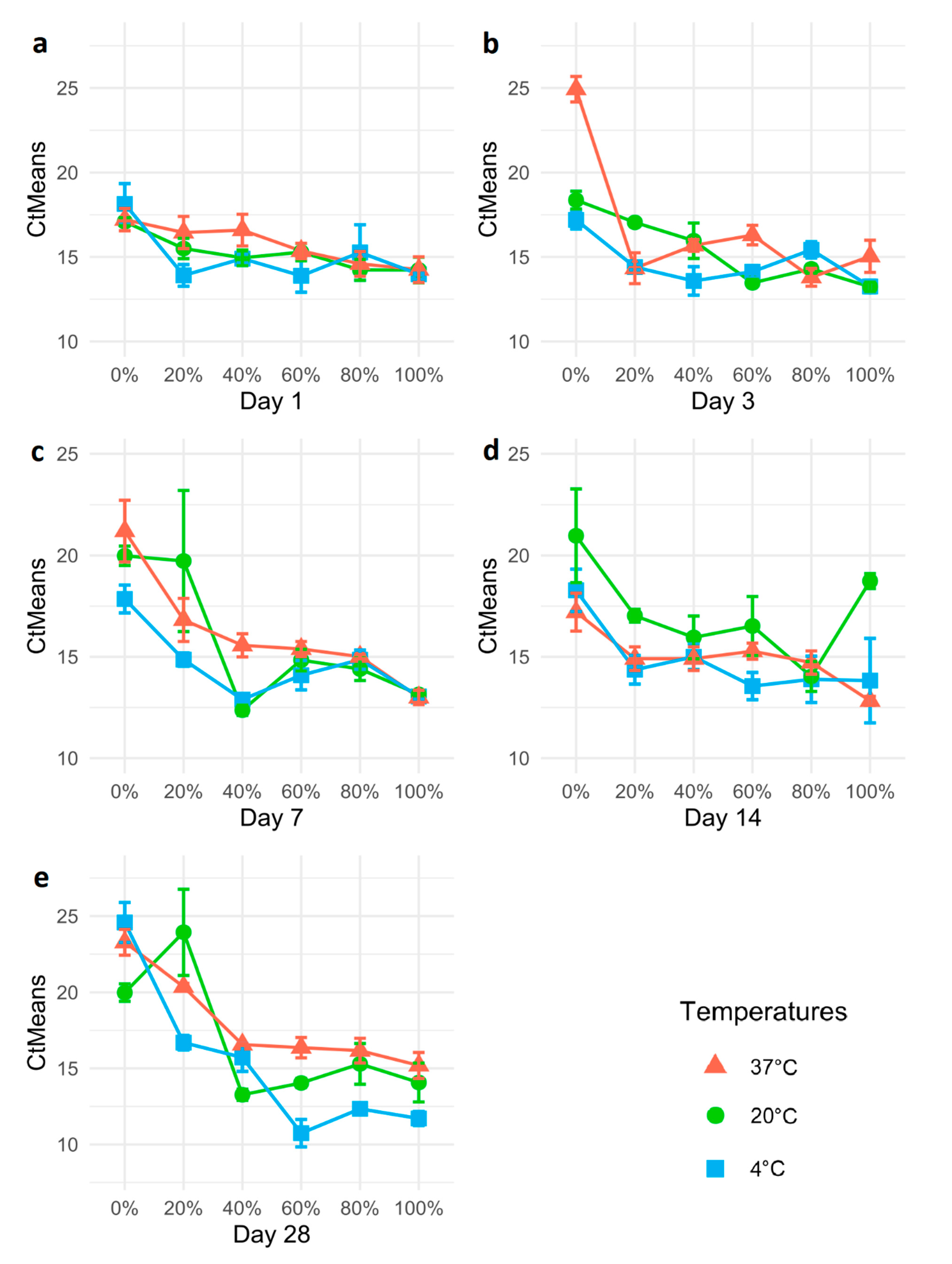
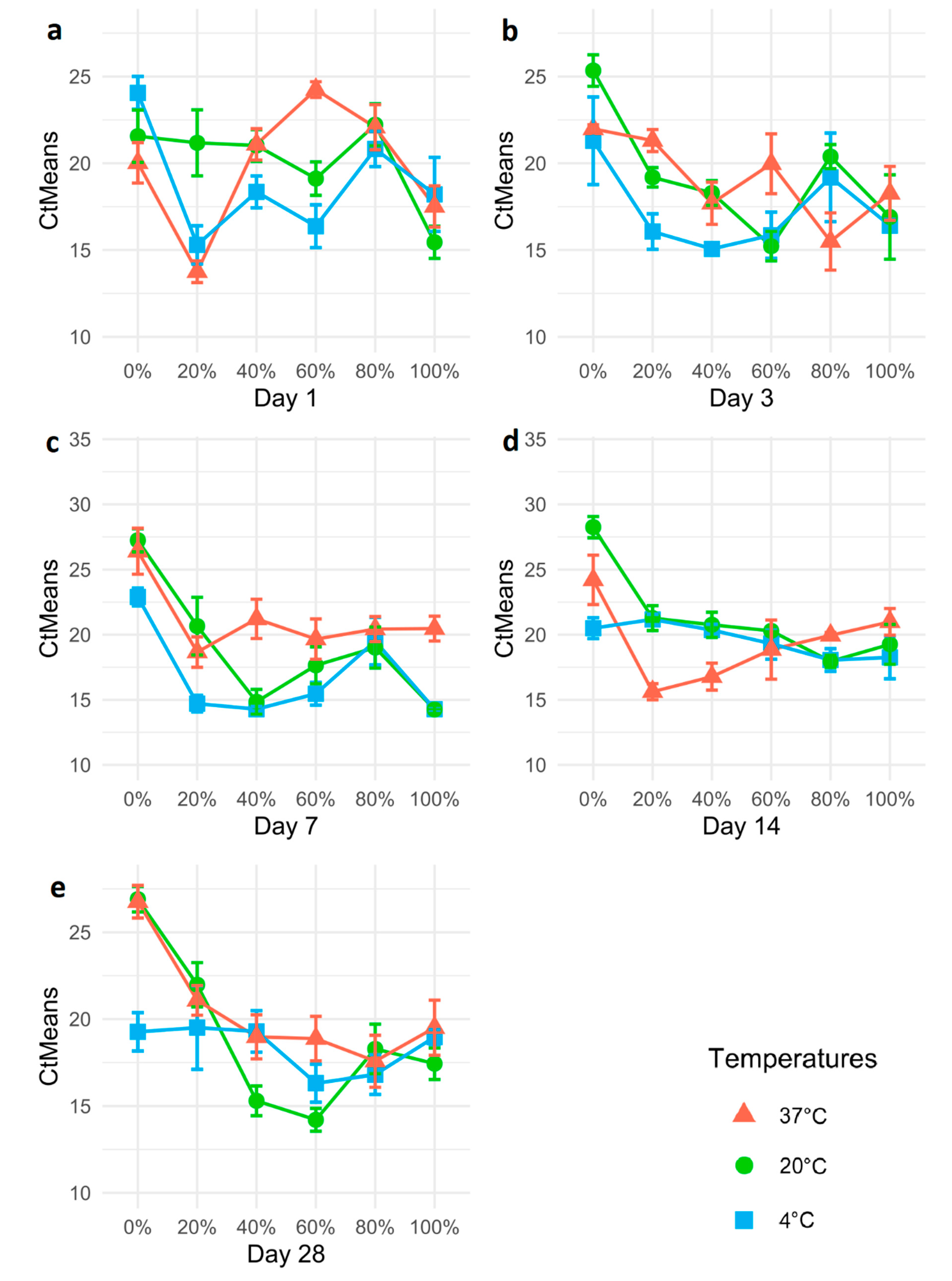
3.3. Propylene Glycol Preservation of Psyllids
3.4. Propylene Glycol Preservation of Ca. Liberibacter DNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liebhold, A.M.; Berec, L.; Brockerhoff, E.G.; Epanchin-Niell, R.S.; Hastings, A.; Herms, D.A.; Kean, J.M.; McCullough, D.G.; Suckling, D.M.; Tobin, P.C.; et al. Eradication of invading insect populations: From concepts to applications. Annu. Rev. Entomol. 2016, 61, 335–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebitz, A.S.; Hoffman, J.C.; Darling, J.A.; Pilgrim, E.M.; Kelly, J.R.; Brown, E.A.; Chadderton, W.L.; Egan, S.P.; Grey, E.K.; Hashsham, S.A.; et al. Early detection monitoring for aquatic non-indigenous species: Optimizing surveillance, incorporating advanced technologies, and identifying research needs. J. Environ. Manag. 2017, 202, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, P.G.; Beanland, L. Insect vectors of phytoplasmas. Annu. Rev. Entomol. 2006, 51, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Liefting, L.W.; Sutherland, P.W.; Ward, L.I.; Paice, K.L.; Weir, B.S.; Clover, G.R.G. A new “Candidatus Liberibacter” species associated with diseases of Solanaceous crops. Plant Dis. 2009, 93, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Overall, L.M.; Rebek, E.J. Insect vectors and current management strategies for diseases caused by Xylella fastidiosa in the Southern United States. J. Integr. Pest Manag. 2017, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Vereijssen, J.; Smith, G.R.; Weintraub, P.G. Bactericera cockerelli (Hemiptera: Triozidae) and Candidatus Liberibacter solanacearum in potatoes in New Zealand: Biology, transmission, and implications for management. J. Integr. Pest Manag. 2018, 9, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Lodge, D.M.; Williams, S.; MacIsaac, H.J.; Hayes, K.R.; Leung, B.; Reichard, S.; Mack, R.N.; Moyle, P.B.; Smith, M.; Andow, D.A.; et al. Biological invasions: Recommendations for U.S. policy and management. Ecol. Appl. 2006, 16, 2035–2054. [Google Scholar] [CrossRef] [Green Version]
- Piper, A.M.; Batovska, J.; Cogan, N.O.; Weiss, J.; Cunningham, J.P.; Rodoni, B.C.; Blacket, M.J. Prospects and challenges of implementing DNA metabarcoding for high throughput insect surveillance. GigaScience 2019, 8, giz092. [Google Scholar] [CrossRef]
- Martin, J.E.H. The Insects and Arachnids of Canada. Part 1: Collecting, Preparing, and Preserving Insects, Mites, and Spiders; Canada Department of Agriculture: Ottawa, ON, Canada, 1977; p. 182. [Google Scholar]
- Stein, E.D.; White, B.P.; Mazor, R.D.; Miller, P.E.; Pilgrim, E.M. Evaluating ethanol-based sample preservation to facilitate use of DNA barcoding in routine freshwater biomonitoring programs using benthic macroinvertebrates. PLoS ONE 2013, 8, e51273. [Google Scholar] [CrossRef] [Green Version]
- Hammer, T.J.; Dickerson, J.C.; Fierer, N. Evidence-based recommendations on storing and handling specimens for analyses of insect microbiota. PeerJ 2015, 3, e1190. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.V.; Porter, T.M.; Wright, M.T.G.; Hajibabaei, M. Propylene glycol-based antifreeze as an effective preservative for DNA metabarcoding of benthic arthropods. bioRxiv 2020. [Google Scholar] [CrossRef]
- Marquina, D.; Ronquist, F.; Łukasik, P. The effect of ethanol concentration on the preservation of insects for biodiversity studies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nakamura, S.; Tamura, S.; Taki, H.; Shoda-Kagaya, E. Propylene glycol: A promising preservative for insects, comparable to ethanol, from trapping to DNA analysis. Entomol. Exp. Appl. 2020, 168, 158–165. [Google Scholar] [CrossRef]
- Matos-Maraví, P.; Ritter, C.D.; Barnes, C.J.; Nielsen, M.; Olsson, U.; Wahlberg, N.; Marquina, D.; Sääksjärvi, I.; Antonelli, A. Biodiversity seen through the perspective of insects: 10 simple rules on methodological choices and experimental design for genomic studies. PeerJ 2019, 7, e6727. [Google Scholar] [CrossRef]
- Gurdebeke, S.; Maelfait, J.-P. Pitfall trapping in population genetics studies: Finding the right “solution”. J. Arachnol. 2002, 30, 255–261. [Google Scholar] [CrossRef]
- Moreau, C.S.; Wray, B.D.; Czekanski-Moir, J.E.; Rubin, B.E.R. DNA preservation: A test of commonly used preservatives for insects. Invertebr. Syst. 2013, 27, 81–86. [Google Scholar] [CrossRef]
- Pokluda, P.; Čižek, L.; Stříbrná, E.; Drag, L.; Lukeš, J.; Novotný, V. A goodbye letter to alcohol: An alternative method for field preservation of arthropod specimens and DNA suitable for mass collecting methods. Eur. J. Entomol. 2014, 111, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Ballare, K.M.; Pope, N.S.; Castilla, A.R.; Cusser, S.; Metz, R.P.; Jha, S. Utilizing field collected insects for next generation sequencing: Effects of sampling, storage, and DNA extraction methods. Ecol. Evol. 2019, 9, 13690–13705. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.T. A hands-on overview of tissue preservation methods for molecular genetic analyses. Org. Divers. Evol. 2010, 10, 91–105. [Google Scholar] [CrossRef]
- Aristophanous, M. Does your preservative preserve? A comparison of the efficacy of some pitfall trap solutions in preserving the internal reproductive organs of dung beetles. Zookeys 2010, 34, 1–16. [Google Scholar] [CrossRef]
- Rubink, W.L.; Murray, K.D.; Baum, K.A.; Pinto, M.A. Long term preservation of DNA from honey bees (Apis mellifera) collected in aerial pitfall traps. Texas J. Sci. 2003, 55, 159–168. [Google Scholar]
- Vink, C.J.; Thomas, S.M.; Paquin, P.; Hayashi, C.Y.; Hedin, M. The effects of preservatives and temperatures on arachnid DNA. Invertebr. Syst. 2005, 19, 99–104. [Google Scholar] [CrossRef]
- Stevens, M.M.; Warren, G.N.; Mo, J.; Schlipalius, D.I. Maintaining DNA quality in stored-grain beetles caught in Lindgren funnel traps. J. Stored Prod. Res. 2011, 47, 69–75. [Google Scholar] [CrossRef]
- Ferro, M.L.; Park, J.S. Effect of propylene glycol concentration on mid-term DNA preservation of Coleoptera. Coleopt. Bull. 2013, 67, 581–586. [Google Scholar] [CrossRef]
- Schutze, M.K. Propylene glycol as an effective preservation agent for subsequent genetic analysis of trap-caught fruit flies. Fruit Flies News 2012, 23, 1–2. [Google Scholar]
- Patrick, H.J.H.; Chomič, A.; Armstrong, K.F. Cooled propylene glycol as a pragmatic choice for preservation of DNA from remote field-collected Diptera for next-generation sequence analysis. J. Econ. Entomol. 2016, 109, 1469–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, Y.Y.; Sokolov, I.M.; Carlton, C.E. New microsporidia parasitizing bark lice (Insecta: Psocoptera). J. Invertebr. Pathol. 2010, 104, 186–194. [Google Scholar] [CrossRef]
- Martoni, F.; Valenzuela, I.; Blacket, M.J. On the complementarity of DNA barcoding and morphology to distinguish benign endemic insects from possible pests: The case of Dirioxa pornia and the tribe Acanthonevrini (Diptera: Tephritidae: Phytalmiinae) in Australia. Insect Sci. 2020. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.; Shiller, J.; Mann, R.; Smith, G.; Yen, A.; Rodoni, B. Novel ‘Candidatus Liberibacter’ species identified in the Australian eggplant psyllid, Acizzia solanicola”. Microb. Biotechnol. 2017, 10, 833–844. [Google Scholar] [CrossRef]
- Kent, D.S.; Taylor, G.S. Two new species of Acizzia Crawford (Hemiptera: Psyllidae) from the Solanaceae with a potential new economic pest of eggplant, Solanum melongena. Aust. J. Entomol. 2010, 49, 73–81. [Google Scholar] [CrossRef]
- Jagoueix, S.; Bove, J.M.; Garnier, M. The phloem-limited bacterium of greening disease of citrus is a member of the alpha subdivision of the Proteobacteria. Int. J. Syst. Evol. Microbiol. 1994, 44, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.; Livak, K. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Martoni, F.; Valenzuela, I.; Blacket, M.J. Non-destructive DNA extractions from fly larvae (Diptera: Muscidae) enable molecular identification of possible pests and enhance morphological features. Austral Entomol. 2019, 58, 848–856. [Google Scholar] [CrossRef]
- Morris, J.; Mann, R.; Sanka Perera, A.; Frampton, R.; Malipatil, M.; Norng, S.; Yen, A.; Smith, G.; Rodoni, B. ‘Candidatus Liberibacter brunswickensis’ colonization has no effect to the early development of Solanum melongena. Mol. Plant Microbe Interact. J. 2021. under review. [Google Scholar]
- Li, W.; Hartung, J.S.; Levy, L. Quantitative real-time PCR for detection and identification of Candidatus Liberibacter species associated with citrus huanglongbing. J. Microbiol. Methods 2006, 66, 104–115. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 1 December 2020).
- Tukey, J. Comparing individual means in the analysis of variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]
- Bretz, F.; Hothorn, T.; Westfall, P. Multiple Comparisons Using R; Chapman and Hall/CRC: Boca Raton, FL, USA, 2010; p. 208. [Google Scholar]
- Nakahama, N.; Isagi, Y.; Ito, M. Methods for retaining well-preserved DNA with dried specimens of insects. Eur. J. Entomol. 2019, 116, 486–491. [Google Scholar] [CrossRef] [Green Version]
- Favret, C. A new non-destructive DNA extraction and specimen clearing technique for aphids (Hemiptera). Proc. Entomol. Soc. Wash. 2005, 107, 469–470. [Google Scholar]
- Gilbert, M.T.P.; Moore, W.; Melchior, L.; Worobey, M. DNA extraction from dry museum beetles without conferring external morphological damage. PLoS ONE 2007, 2, e272. [Google Scholar] [CrossRef] [Green Version]
- Porco, D.; Rougerie, R.; Deharveng, L.; Hebert, P. Coupling non-destructive DNA extraction and voucher retrieval for small soft-bodied arthropods in a high-throughput context: The example of Collembola. Mol. Ecol. Resour. 2010, 10, 942–945. [Google Scholar] [CrossRef]
- Bahder, B.W.; Bollinger, M.L.; Sudarshana, M.R.; Zalom, F.G. Preparation of mealybugs (Hemiptera: Pseudococcidae) for genetic characterization and morphological examination. J. Insect Sci. 2015, 15, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munyaneza, J.E.; Sengoda, V.G.; Sundheim, L.; Meadow, R. Survey of “Candidatus Liberibacter solanacearum” in carrot crops affected by the psyllid Trioza apicalis (Hemiptera: Triozidae) in Norway. J. Plant Pathol. 2014, 96, 397–402. [Google Scholar] [CrossRef]
- Lucigen. Frequently asked questions—QuickExtract™ DNA Extraction Solution and QuickExtract™ FFPE DNA Extraction Kit. 2020. Available online: https://www.lucigen.com/docs/faqs/FAQ-QuickExtract-DNA-Solution.pdf (accessed on 1 December 2020).
- Sagdeev, D.I.; Fomina, M.G.; Abdulagatov, I.M. Density and viscosity of propylene glycol at high temperatures and high pressures. Fluid Phase Equilib. 2017, 450, 99–111. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Pre-Extraction | DNA Extraction | Post-Extraction | |||
|---|---|---|---|---|---|---|
| PG Incubation (Time, %, Temperature) | Kit | Destructive or Non-Destructive | Lysis Buffer Incubation (Time, Temperature) | Column Clean-Up | Lysate Dilution | |
| 1 | No | Qia | D | 1 h, 56 °C | Yes | No |
| 2 | No | Qia | ND | O/N, 56 °C | Yes | No |
| 3 | No | Qia | ND | O/N, 56 °C | No | No |
| 3–3 | No | Qia | ND | O/N, 56 °C | No | 1:1000 |
| 3–4 | No | Qia | ND | O/N, 56 °C | No | 1:10,000 |
| 4 | O/N, 50%, 23 °C | Qia | ND | O/N, 56 °C | Yes | No |
| 5 | O/N, 50%, 23 °C | Qia | ND | O/N, 56 °C | No | No |
| 5–3 | O/N, 50%, 23 °C | Qia | ND | O/N, 56 °C | No | 1:1000 |
| 5–4 | O/N, 50%, 23 °C | Qia | ND | O/N, 56 °C | No | 1:10,000 |
| 6 | No | QE | D | 6 min, 65 °C | No | No |
| 7 | No | QE | ND | 6 min, 65 °C | No | No |
| 8 | No | QE | ND | O/N, 65 °C | No | No |
| 9 | O/N, 50%, 23 °C | QE | ND | O/N, 65 °C | No | No |
| 10 | O/N, 50%, 23 °C | QE | ND | 6 min, 65 °C | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martoni, F.; Nogarotto, E.; Piper, A.M.; Mann, R.; Valenzuela, I.; Eow, L.; Rako, L.; Rodoni, B.C.; Blacket, M.J. Propylene Glycol and Non-Destructive DNA Extractions Enable Preservation and Isolation of Insect and Hosted Bacterial DNA. Agriculture 2021, 11, 77. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture11010077
Martoni F, Nogarotto E, Piper AM, Mann R, Valenzuela I, Eow L, Rako L, Rodoni BC, Blacket MJ. Propylene Glycol and Non-Destructive DNA Extractions Enable Preservation and Isolation of Insect and Hosted Bacterial DNA. Agriculture. 2021; 11(1):77. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture11010077
Chicago/Turabian StyleMartoni, Francesco, Elisse Nogarotto, Alexander M. Piper, Rachel Mann, Isabel Valenzuela, Lixin Eow, Lea Rako, Brendan C. Rodoni, and Mark J. Blacket. 2021. "Propylene Glycol and Non-Destructive DNA Extractions Enable Preservation and Isolation of Insect and Hosted Bacterial DNA" Agriculture 11, no. 1: 77. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture11010077