
Multi-Allelic Haplotype-Based Association Analysis Identifies Genomic Regions Controlling Domestication Traits in Intermediate Wheatgrass


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Population
2.2. Genotyping
2.3. Phenotyping and Statistical Analysis
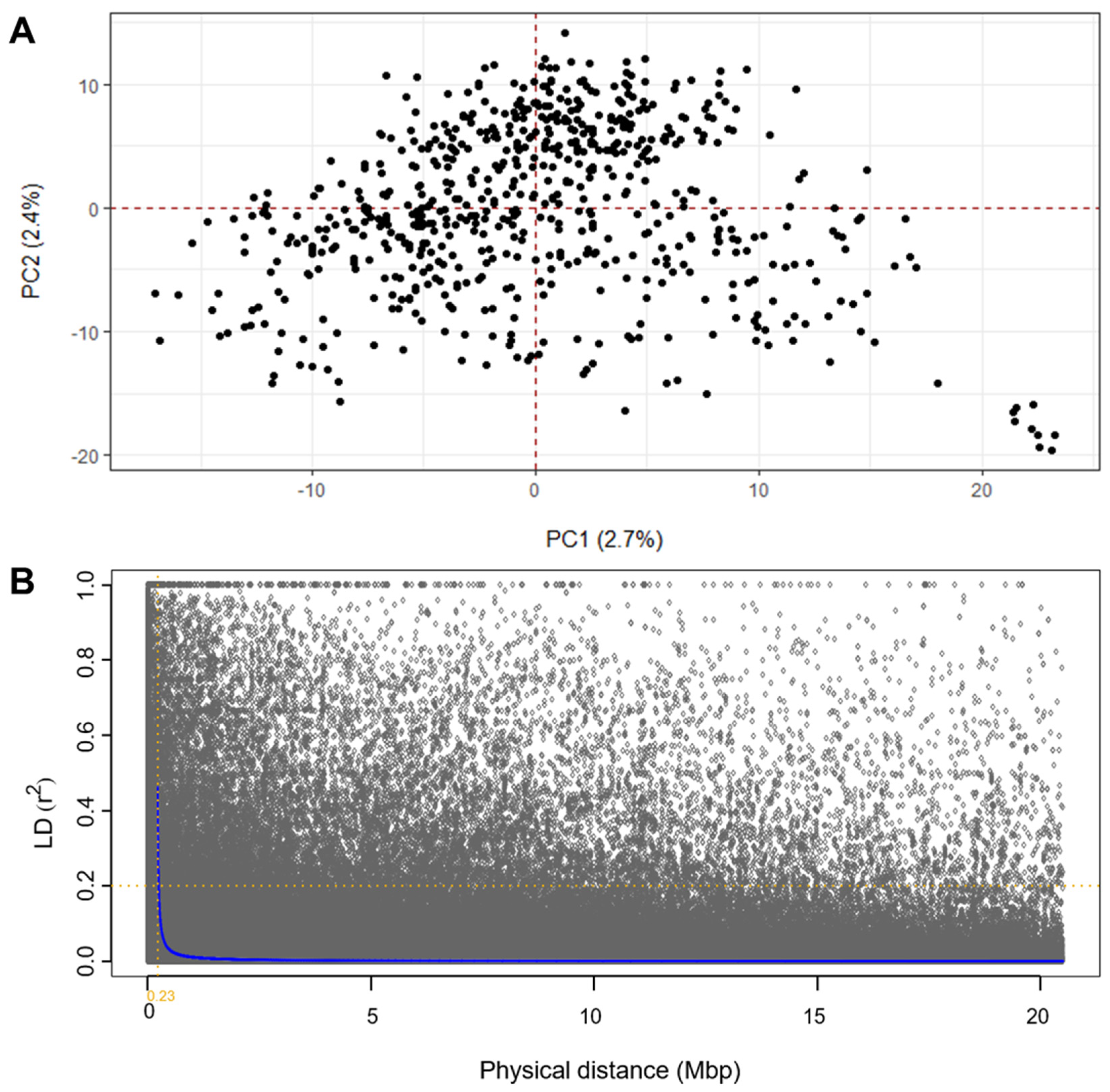
2.4. Linkage Disequilibrium and Population Relatedness
2.5. Haplotype Construction
2.6. Association Analysis
- For single SNP markers: at α/no. of observations = 0.05/25909 = p value of 1.93 × 10−06 or LOD equivalent of 5.71, and
- For multi-allelic haplotype blocks: at α/no. of observations = 0.05/5379 = p value of 9.30 × 10−06 or LOD equivalent of 5.03
3. Results
3.1. Genotyping and Population Properties
3.2. Haplotype Distribution
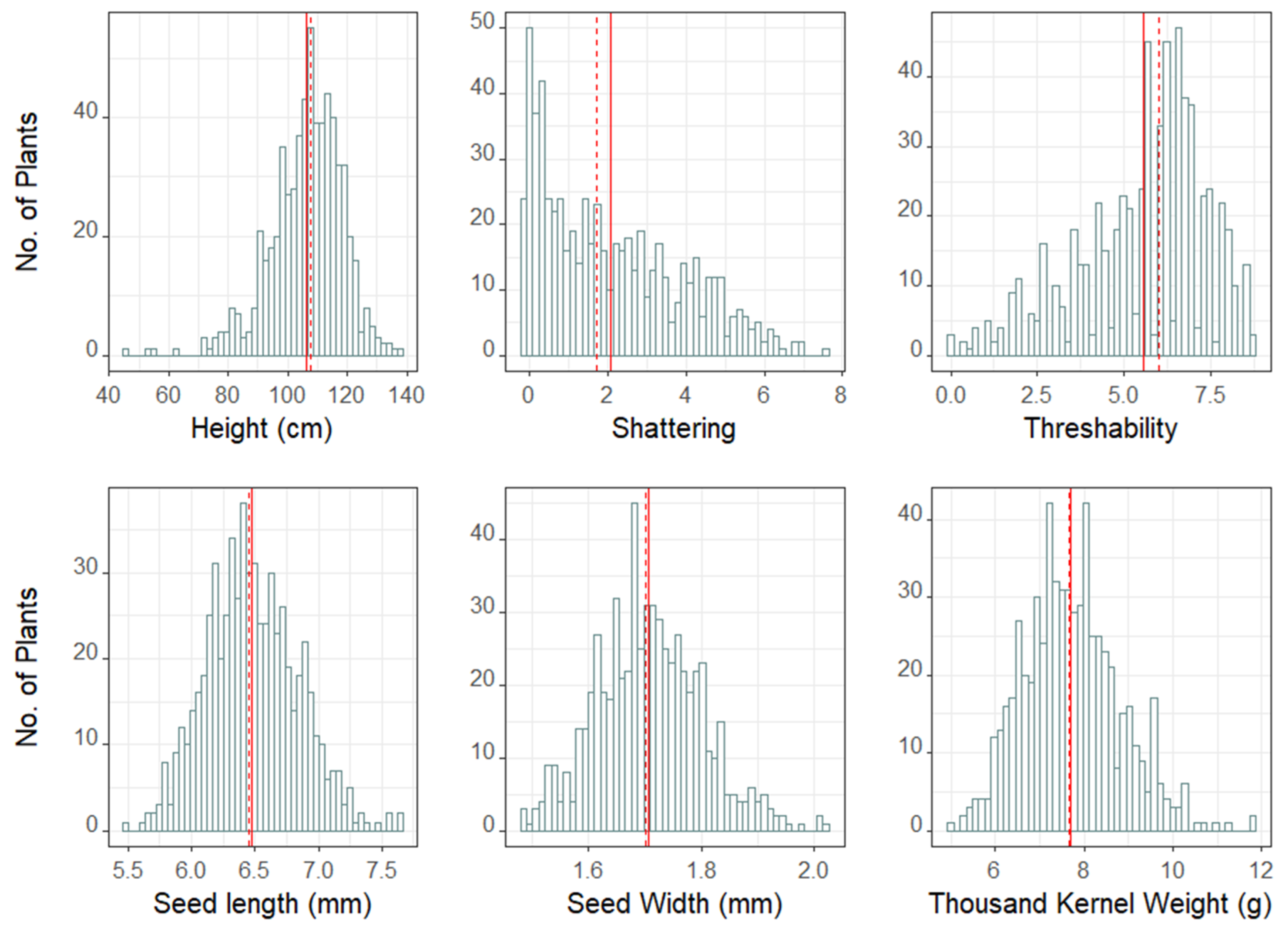
3.3. Trait Distribution, Correlation, Heritability
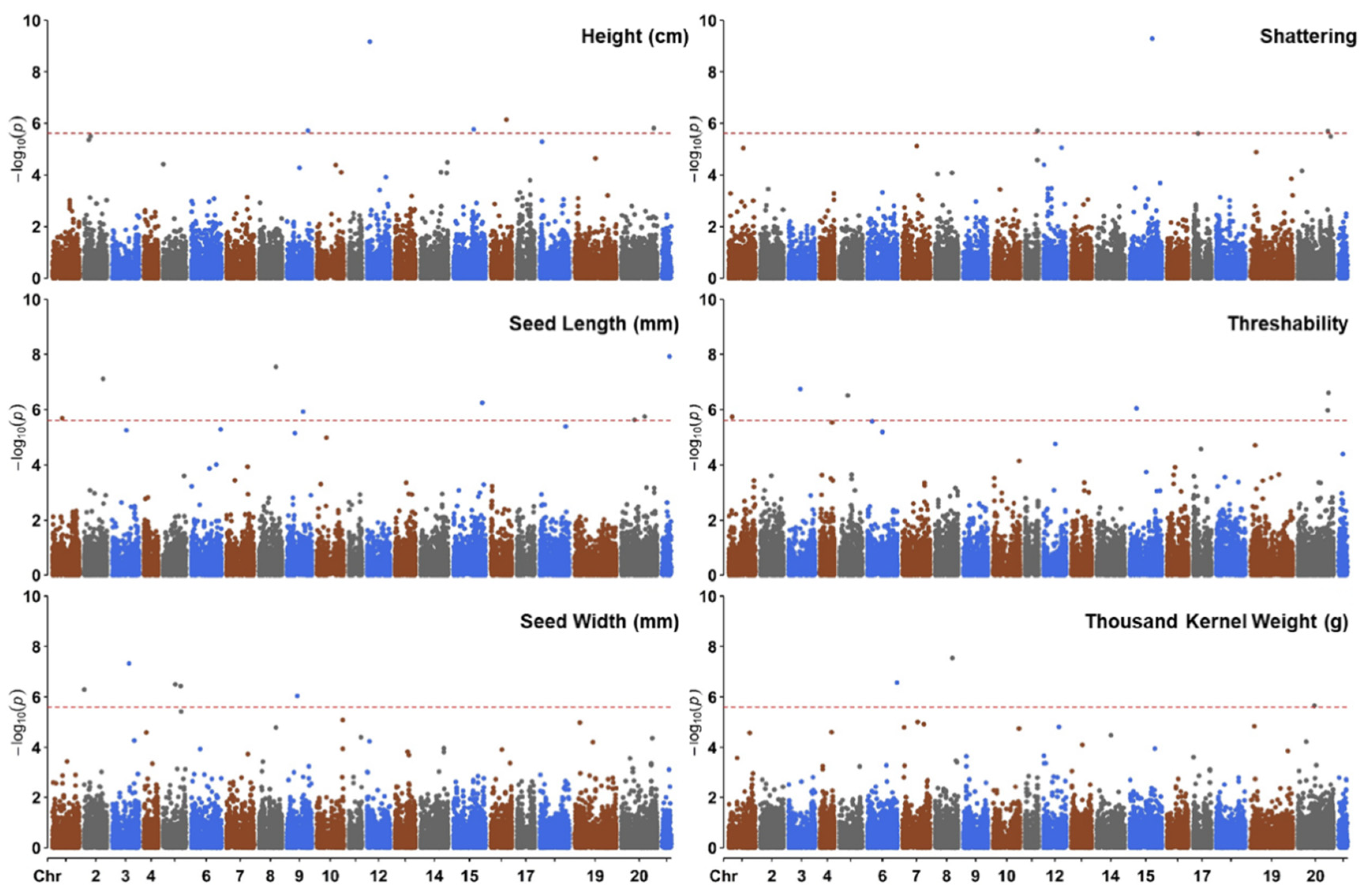
3.4. Association Analysis
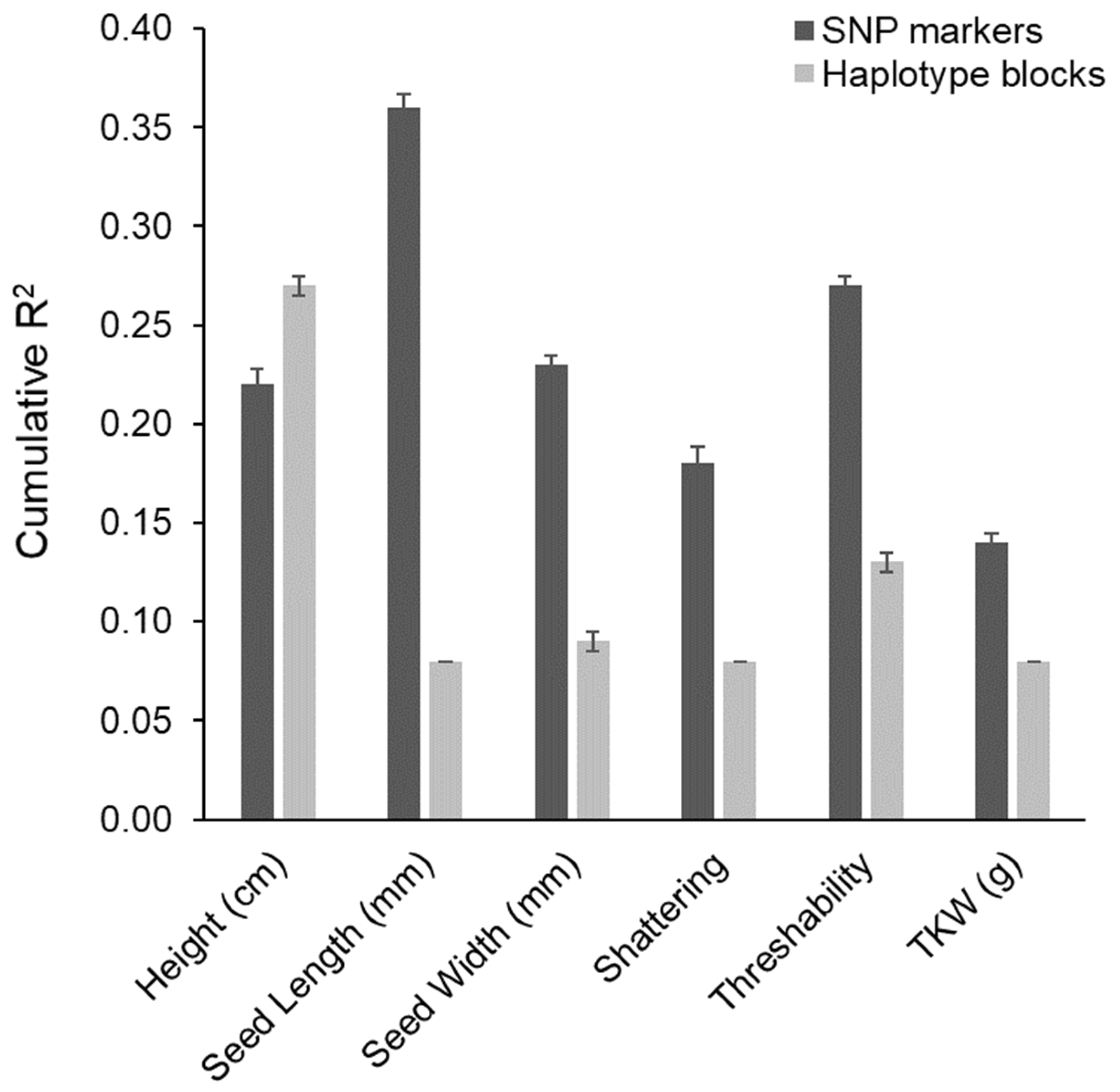
3.5. Comparison of Mapping Results between Marker Types
4. Discussion
4.1. IWG Population, Trait Properties, and Association Mapping
4.2. Comparison with Existing IWG QTL
4.3. QTL Size and Implications in Intermediate Wheatgrass Breeding
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wagoner, P. Perennial Grain New Use for Intermediate Wheatgrass. J. Soil Water Conserv. 1990, 45, 81–82. [Google Scholar]
- DeHaan, L.R.; Ismail, B.P. Perennial Cereals Provide Ecosystem Benefits. Cereal Foods World 2017, 62, 278–281. [Google Scholar] [CrossRef]
- Culman, S.W.; Snapp, S.S.; Ollenburger, M.; Basso, B.; DeHaan, L.R. Soil and Water Quality Rapidly Responds to the Perennial Grain Kernza Wheatgrass. Agron. J. 2013, 105, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Bergquist, G.E. Biomass Yield and Soil Microbial Response to Management of Perennial Intermediate Wheatgrass (Thinopyrum Intermedium) as Grain Crop and Carbon Sink; University of Minnesota: Minneapolis, MN, USA, 2019. [Google Scholar]
- Jungers, J.M.; DeHaan, L.H.; Mulla, D.J.; Sheaffer, C.C.; Wyse, D.L. Reduced Nitrate Leaching in a Perennial Grain Crop Compared to Maize in the Upper Midwest, USA. Agric. Ecosyst. Environ. 2019, 272, 63–73. [Google Scholar] [CrossRef]
- Vogel, K.P.; Jensen, K.J. Adaptation of Perennial Triticeae to the Eastern Central Great Plains. J. Range Manag. 2001, 54, 674–679. [Google Scholar] [CrossRef]
- Zhang, X.; Sallam, A.; Gao, L.; Kantarski, T.; Poland, J.; DeHaan, L.R.; Wyse, D.L.; Anderson, J.A. Establishment and Optimization of Genomic Selection to Accelerate the Domestication and Improvement of Intermediate Wheatgrass. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajgain, P.; Zhang, X.; Jungers, J.M.; DeHaan, L.R.; Heim, B.; Sheaffer, C.C.; Wyse, D.L.; Anderson, J.A. ‘MN-Clearwater’, the First Food-Grade Intermediate Wheatgrass (Kernza Perennial Grain) Cultivar. J. Plant Regist. 2020, 14, 288–297. [Google Scholar] [CrossRef]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The Molecular Genetics of Crop Domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [Green Version]
- Purugganan, M.D.; Fuller, D.Q. The Nature of Selection during Plant Domestication. Nature 2009, 457, 843–848. [Google Scholar] [CrossRef]
- Solberg, T.R.; Sonesson, A.K.; Woolliams, J.A.; Meuwissen, T.H.E. Genomic Selection Using Different Marker Types and Densities. J. Anim. Sci. 2008, 86, 2447–2454. [Google Scholar] [CrossRef] [Green Version]
- Korte, A.; Farlow, A. The Advantages and Limitations of Trait Analysis with GWAS: A Review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilhjálmsson, B.J.; Nordborg, M. The Nature of Confounding in Genome-Wide Association Studies. Nat. Rev. Genet. 2013, 14, 1–2. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A Unified Mixed-Model Method for Association Mapping That Accounts for Multiple Levels of Relatedness. Nat. Genet. 2005, 38, 203. [Google Scholar] [CrossRef]
- Jannink, J.-L.; Bink, M.C.A.M.; Jansen, R.C. Using Complex Plant Pedigrees to Map Valuable Genes. Trends Plant Sci. 2001, 6, 337–342. [Google Scholar] [CrossRef]
- Buckler, E.S.; Thornsberry, J.M. Plant Molecular Diversity and Applications to Genomics. Curr. Opin. Plant Biol. 2002, 5, 107–111. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP Markers and Their Impact on Plant Breeding. Int. J. Plant Genomics 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Albrechtsen, A.; Nielsen, F.C.; Nielsen, R. Ascertainment Biases in SNP Chips Affect Measures of Population Divergence. Mol. Biol. Evol. 2010, 27, 2534–2547. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Bajgain, P.; Rouse, M.N.; Anderson, J.A. Comparing Genotyping-by-Sequencing and Single Nucleotide Polymorphism Chip Genotyping for Quantitative Trait Loci Mapping in Wheat. Crop Sci. 2016, 56, 232–248. [Google Scholar] [CrossRef]
- Gupta, P.K.; Kulwal, P.L.; Jaiswal, V. Chapter Two-Association Mapping in Crop Plants: Opportunities and Challenges. In Advances in Genetics; Friedmann, T., Dunlap, J.C., Goodwin, S.F., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 85, pp. 109–147. ISBN 0065-2660. [Google Scholar]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological Implementation of Mixed Linear Models in Multi-Locus Genome-Wide Association Studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Shah, T.; Hao, Z.; Taba, S.; Zhang, S.; Gao, S.; Liu, J.; Cao, M.; Wang, J.; Prakash, A.B.; et al. Comparative SNP and Haplotype Analysis Reveals a Higher Genetic Diversity and Rapider LD Decay in Tropical than Temperate Germplasm in Maize. PLoS ONE 2011, 6, e24861. [Google Scholar] [CrossRef] [Green Version]
- Da, Y. Multi-Allelic Haplotype Model Based on Genetic Partition for Genomic Prediction and Variance Component Estimation Using SNP Markers. BMC Genet. 2015, 16, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving Power and Accuracy of Genome-Wide Association Studies via a Multi-Locus Mixed Linear Model Methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [Green Version]
- Thinopyrum Intermedium Genome Sequencing Consortium Thinopyrum Intermedium v2.1 DOE-JGI. Available online: http://Phytozome.Jgi.Doe.Gov/ (accessed on 14 July 2021).
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinforma. Oxf. Engl. 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Money, D.; Gardner, K.; Migicovsky, Z.; Schwaninger, H.; Zhong, G.-Y.; Myles, S. LinkImpute: Fast and Accurate Genotype Imputation for Nonmodel Organisms. G3 Genes Genomes Genet. 2015, 5, 2383–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, W.G.; Weir, B.S. Variances and Covariances of Squared Linkage Disequilibria in Finite Populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Vos, P.G.; Paulo, M.J.; Voorrips, R.E.; Visser, R.G.F.; van Eck, H.J.; van Eeuwijk, F.A. Evaluation of LD Decay and Various LD-Decay Estimators in Simulated and SNP-Array Data of Tetraploid Potato. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2017, 130, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The Structure of Haplotype Blocks in the Human Genome. Science 2002, 296, 2225. [Google Scholar] [CrossRef] [Green Version]
- Abed, A.; Belzile, F. Comparing Single-SNP, Multi-SNP, and Haplotype-Based Approaches in Association Studies for Major Traits in Barley. Plant Genome 2019, 12, 190036. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome Association and Prediction Integrated Tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Sen, Ś.; Churchill, G.A. A Statistical Framework for Quantitative Trait Mapping. Genetics 2001, 159, 371. [Google Scholar] [CrossRef]
- Broman, K.W.; Wu, H.; Sen, Ś.; Churchill, G.A. R/Qtl: QTL Mapping in Experimental Crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajgain, P.; Zhang, X.; Anderson, J.A. Genome-Wide Association Study of Yield Component Traits in Intermediate Wheatgrass and Implications in Genomic Selection and Breeding. G3 Genes Genomes Genet. 2019, 9, 2429–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crain, J.; Bajgain, P.; Anderson, J.; Zhang, X.; Poland, J.; DeHaan, L. Enhancing Crop Domestication Through Genomic Selection, a Case Study of Intermediate Wheatgrass. Front. Plant Sci. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, S.; DeHaan, L.; Poland, J.; Zhang, X.; Dorn, K.; Kantarski, T.; Anderson, J.; Schmutz, J.; Grimwood, J.; Jenkins, J.; et al. Genome Mapping of Quantitative Trait Loci (QTL) Controlling Domestication Traits of Intermediate Wheatgrass (Thinopyrum Intermedium). Theor. Appl. Genet. 2019, 132, 2325–2351. [Google Scholar] [CrossRef]
- Altendorf, K.R.; DeHaan, L.R.; Heineck, G.C.; Zhang, X.; Anderson, J.A. Floret Site Utilization and Reproductive Tiller Number Are Primary Components of Grain Yield in Intermediate Wheatgrass Spaced Plants. Crop Sci. 2021, 61, 1073–1088. [Google Scholar] [CrossRef]
- Altendorf, K. Phenotypic and Genetic Characterization of Domestication and Yield Component Traits in the Perennial Grain Crop Intermediate Wheatgrass; University of Minnesota: Minneapolis, MN, USA, 2020. [Google Scholar]
- Lorenz, A.J.; Hamblin, M.T.; Jannink, J.-L. Performance of Single Nucleotide Polymorphisms versus Haplotypes for Genome-Wide Association Analysis in Barley. PLoS ONE 2010, 5, e14079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luján Basile, S.M.; Ramírez, I.A.; Crescente, J.M.; Conde, M.B.; Demichelis, M.; Abbate, P.; Rogers, W.J.; Pontaroli, A.C.; Helguera, M.; Vanzetti, L.S. Haplotype Block Analysis of an Argentinean Hexaploid Wheat Collection and GWAS for Yield Components and Adaptation. BMC Plant Biol. 2019, 19, 553. [Google Scholar] [CrossRef]
- Slatkin, M. Linkage Disequilibrium—Understanding the Evolutionary Past and Mapping the Medical Future. Nat. Rev. Genet. 2008, 9, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblin, M.T.; Jannink, J.-L. Factors Affecting the Power of Haplotype Markers in Association Studies. Plant Genome 2011, 4. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Soto, R.I.; Mora, F.; de Oliveira, M.A.R.; Higashi, W.; Scapim, C.A.; Schuster, I. A Genome-Wide Association Study for Agronomic Traits in Soybean Using SNP Markers and SNP-Based Haplotype Analysis. PLoS ONE 2017, 12, e0171105. [Google Scholar] [CrossRef]
- Jia, Q.; Zhang, J.; Westcott, S.; Zhang, X.-Q.; Bellgard, M.; Lance, R.; Li, C. GA-20 Oxidase as a Candidate for the Semidwarf Gene Sdw1/Denso in Barley. Funct. Integr. Genom. 2009, 9, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Larson, S.R.; Gao, L.; Teh, S.L.; DeHaan, L.R.; Fraser, M.; Sallam, A.; Kantarski, T.; Frels, K.; Poland, J.; et al. Uncovering the Genetic Architecture of Seed Weight and Size in Intermediate Wheatgrass through Linkage and Association Mapping. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, R.W. History of Plant Population Genetics. Annu. Rev. Genet. 1999, 33, 1–27. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Sarinelli, J.M.; Murphy, J.P.; Tyagi, P.; Holland, J.B.; Johnson, J.W.; Mergoum, M.; Mason, R.E.; Babar, A.; Harrison, S.; Sutton, R.; et al. Training Population Selection and Use of Fixed Effects to Optimize Genomic Predictions in a Historical USA Winter Wheat Panel. Theor. Appl. Genet. 2019, 132, 1247–1261. [Google Scholar] [CrossRef] [Green Version]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic Selection and Association Mapping in Rice (Oryza Sativa): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar] [CrossRef] [Green Version]
- Minamikawa, M.F.; Nonaka, K.; Kaminuma, E.; Kajiya-Kanegae, H.; Onogi, A.; Goto, S.; Yoshioka, T.; Imai, A.; Hamada, H.; Hayashi, T.; et al. Genome-Wide Association Study and Genomic Prediction in Citrus: Potential of Genomics-Assisted Breeding for Fruit Quality Traits. Sci. Rep. 2017, 7, 4721. [Google Scholar] [CrossRef] [PubMed]
- Bajgain, P.; Zhang, X.; Turner, K.M.; Curland, D.R.; Heim, B.; Dill-Macky, R.; Ishimaru, A.C.; Anderson, A.J. Characterization of Genetic Resistance to Fusarium Head Blight and Bacterial Leaf Streak in Intermediate Wheatgrass (Thinopyrum Intermedium). Agronomy 2019, 9, 429. [Google Scholar] [CrossRef] [Green Version]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package RrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype Blocks | ||||
|---|---|---|---|---|
| Chromosome | No. of SNPs | SNP Position Range (bp) | Total | No. of SNPs |
| 1 | 1400 | 1,326,898–507,102,603 | 294 | 825 |
| 2 | 1617 | 300,679–433,297,397 | 325 | 928 |
| 3 | 1065 | 2,940,432–513,687,187 | 224 | 626 |
| 4 | 1133 | 115,039–267,218,805 | 234 | 626 |
| 5 | 850 | 1,020,684–434,249,891 | 197 | 515 |
| 6 | 1377 | 1,845,863–570,323,785 | 267 | 747 |
| 7 | 1173 | 364,376–520,161,072 | 231 | 645 |
| 8 | 1472 | 678,201–432,546,992 | 325 | 910 |
| 9 | 1111 | 42,820–473,689,827 | 240 | 647 |
| 10 | 961 | 811,258–512,842,958 | 185 | 514 |
| 11 | 789 | 70,253–250,601,048 | 163 | 457 |
| 12 | 819 | 1,201,293–434,811,611 | 175 | 469 |
| 13 | 1311 | 1,606,077–385,563,675 | 274 | 777 |
| 14 | 1252 | 265,147–524,672,644 | 246 | 695 |
| 15 | 1573 | 359,226–611,692,208 | 329 | 928 |
| 16 | 985 | 949,133–400,595,591 | 205 | 584 |
| 17 | 979 | 1,637–337,127,221 | 205 | 531 |
| 18 | 1372 | 49,467–560,298,643 | 283 | 776 |
| 19 | 1905 | 2,330,783–801,761,017 | 397 | 1117 |
| 20 | 2177 | 276,951–676,545,699 | 453 | 1290 |
| 21 | 588 | 134,660–169,591,600 | 127 | 344 |
| Mean | 1234 | -- | 256 | 712 |
| Total | 25,909 | -- | 5379 | 14,951 |
| Trait | Height (cm) | Shattering | Threshability | Seed Length (mm) | Seed Width (mm) |
|---|---|---|---|---|---|
| Shattering | 0.13 * | ||||
| Threshability | −0.07 | −0.37 * | |||
| Seed Length (mm) | 0.20 * | 0.15 * | −0.05 | ||
| Seed Width (mm) | 0.11 * | 0.34 * | −0.42 * | 0.30 * | |
| TKW (g) | 0.20 * | 0.19 * | −0.06 | 0.67 * | 0.66 * |
| Trait | SNP Marker | Chr | Pos (Mbp) | Alleles | MAF | LOD | R2 | Allelic Effect | Hb |
|---|---|---|---|---|---|---|---|---|---|
| Height (cm) | S09_398417599 | 9 | 398.42 | G/T | 0.15 | 5.71 | 0.04 | −3.43 | NA |
| Height (cm) | S12_54085941 | 12 | 54.09 | G/C | 0.30 | 9.18 | 0.06 | 3.58 | Chr12-Hb.032 |
| Height (cm) | S15_384414403 | 15 | 384.41 | C/T | 0.10 | 5.76 | 0.04 | −3.58 | Chr15-Hb.150 |
| Height (cm) | S16_293220465 | 16 | 293.22 | G/C | 0.27 | 6.14 | 0.04 | −2.70 | Chr16-Hb.110 |
| Height (cm) | S20_613713856 | 20 | 613.71 | A/G | 0.11 | 5.81 | 0.04 | 4.13 | Chr20-Hb.395 |
| Seed Length (mm) | S01_184375481 | 1 | 184.38 | C/T | 0.09 | 5.69 | 0.04 | −0.10 | NA |
| Seed Length (mm) | S02_360222683 | 2 | 360.22 | C/G | 0.09 | 7.11 | 0.05 | 0.13 | Chr02-Hb.254 |
| Seed Length (mm) | S08_323540788 | 8 | 323.54 | C/A | 0.30 | 7.54 | 0.05 | −0.08 | Chr08-Hb.228 |
| Seed Length (mm) | S09_306672331 | 9 | 306.67 | C/A | 0.42 | 5.92 | 0.04 | 0.06 | NA |
| Seed Length (mm) | S15_550008611 | 15 | 550.01 | T/A | 0.14 | 6.25 | 0.04 | −0.09 | Chr15-Hb.298 |
| Seed Length (mm) | S20_248288758 | 20 | 248.29 | C/T | 0.17 | 5.63 | 0.04 | −0.09 | NA |
| Seed Length (mm) | S20_440036613 | 20 | 440.04 | G/A | 0.09 | 5.75 | 0.04 | 0.13 | NA |
| Seed Length (mm) | S21_139054606 | 21 | 139.05 | G/A | 0.19 | 7.93 | 0.06 | −0.10 | NA |
| Seed Width (mm) | S02_627054 | 2 | 0.63 | A/C | 0.22 | 6.30 | 0.04 | −0.02 | Chr02-Hb.001 |
| Seed Width (mm) | S03_324339821 | 3 | 324.34 | G/C | 0.26 | 7.33 | 0.05 | 0.02 | NA |
| Seed Width (mm) | S05_229107942 | 5 | 229.11 | A/G | 0.14 | 6.50 | 0.05 | 0.03 | Chr05-Hb.076 |
| Seed Width (mm) | S05_330687025 | 5 | 330.69 | T/C | 0.30 | 6.44 | 0.05 | 0.02 | NA |
| Seed Width (mm) | S09_194463841 | 9 | 194.46 | G/A | 0.12 | 6.05 | 0.04 | −0.03 | NA |
| Shattering | S11_233685188 | 11 | 233.69 | C/A | 0.40 | 5.71 | 0.04 | 0.15 | NA |
| Shattering | S15_427773440 | 15 | 427.77 | C/A | 0.44 | 9.30 | 0.06 | −0.20 | Chr15-Hb.183 |
| Shattering | S17_89388699 | 17 | 89.39 | C/T | 0.30 | 5.61 | 0.04 | −0.19 | NA |
| Shattering | S20_569949391 | 20 | 569.95 | A/G | 0.18 | 5.70 | 0.04 | −0.26 | NA |
| Threshability | S01_67160311 | 1 | 67.16 | G/T | 0.24 | 5.74 | 0.04 | −0.40 | Chr01-Hb.041 |
| Threshability | S03_231083092 | 3 | 231.08 | A/G | 0.13 | 6.74 | 0.05 | 0.51 | NA |
| Threshability | S05_156358046 | 5 | 156.36 | G/T | 0.38 | 6.51 | 0.05 | 0.35 | NA |
| Threshability | S15_127556933 | 15 | 127.56 | G/A | 0.07 | 6.05 | 0.04 | −0.69 | NA |
| Threshability | S20_568849724 | 20 | 568.85 | T/C | 0.25 | 5.98 | 0.04 | 0.43 | Chr20-Hb.349 |
| Threshability | S20_583569893 | 20 | 583.57 | G/C | 0.08 | 6.61 | 0.05 | 0.61 | Chr20-Hb.361 |
| TKW (g) | S06_558612622 | 6 | 558.61 | T/C | 0.22 | 6.57 | 0.05 | −0.33 | Chr06-Hb.258 |
| TKW (g) | S08_323540788 | 8 | 323.54 | C/A | 0.30 | 7.55 | 0.05 | −0.26 | Chr08-Hb.228 |
| TKW (g) | S20_314990021 | 20 | 314.99 | A/G | 0.25 | 5.65 | 0.04 | 0.32 | NA |
| Trait | Hb | SNPs in Block | Chr | LOD | R2 | Allelic Effect | SigSNP |
|---|---|---|---|---|---|---|---|
| Height (cm) | Chr01-Hb.224 | 3 | Chr01 | 6.71 | 0.05 | 3.46 | NA |
| Height (cm) | Chr08-Hb.197 | 2 | Chr08 | 5.08 | 0.04 | −10.02 | NA |
| Height (cm) | Chr12-Hb.032 | 2 | Chr12 | 6.97 | 0.05 | 6.29 | S12_54085941 |
| Height (cm) | Chr14-Hb.107 | 4 | Chr14 | 6.08 | 0.04 | −24.63 | NA |
| Height (cm) | Chr15-Hb.150 | 3 | Chr15 | 5.82 | 0.04 | −8.44 | S15_384414403 |
| Height (cm) | Chr18-Hb.267 | 3 | Chr18 | 6.75 | 0.05 | 7.44 | NA |
| Seed Length (mm) | Chr02-Hb.254 | 2 | Chr02 | 5.85 | 0.04 | 0.02 | S02_360222683 |
| Seed Length (mm) | Chr08-Hb.228 | 4 | Chr08 | 5.18 | 0.04 | −0.14 | S08_323540788 |
| Seed Width (mm) | Chr02-Hb.001 | 2 | Chr02 | 5.34 | 0.04 | −0.10 | S02_627054 |
| Seed Width (mm) | Chr05-Hb.076 | 5 | Chr05 | 6.41 | 0.05 | 0.08 | S05_229107942 |
| Shattering | Chr11-Hb.134 | 7 | Chr11 | 5.95 | 0.04 | 0.42 | NA |
| Shattering | Chr15-Hb.183 | 4 | Chr15 | 5.90 | 0.04 | −0.22 | S15_427773440 |
| Threshability | Chr06-Hb.180 | 4 | Chr06 | 5.14 | 0.04 | −1.66 | NA |
| Threshability | Chr20-Hb.349 | 3 | Chr20 | 5.60 | 0.04 | 0.68 | S20_568849724 |
| Threshability | Chr20-Hb.361 | 2 | Chr20 | 6.69 | 0.05 | 0.84 | S20_583569893 |
| TKW (g) | Chr06-Hb.186 | 2 | Chr06 | 6.01 | 0.04 | −3.77 | NA |
| TKW (g) | Chr08-Hb.228 | 4 | Chr08 | 5.09 | 0.04 | −0.47 | S08_323540788 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajgain, P.; Anderson, J.A. Multi-Allelic Haplotype-Based Association Analysis Identifies Genomic Regions Controlling Domestication Traits in Intermediate Wheatgrass. Agriculture 2021, 11, 667. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture11070667
Bajgain P, Anderson JA. Multi-Allelic Haplotype-Based Association Analysis Identifies Genomic Regions Controlling Domestication Traits in Intermediate Wheatgrass. Agriculture. 2021; 11(7):667. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture11070667
Chicago/Turabian StyleBajgain, Prabin, and James A. Anderson. 2021. "Multi-Allelic Haplotype-Based Association Analysis Identifies Genomic Regions Controlling Domestication Traits in Intermediate Wheatgrass" Agriculture 11, no. 7: 667. https://0-doi-org.brum.beds.ac.uk/10.3390/agriculture11070667