
The Diversity, Composition, and Metabolic Pathways of Archaea in Pigs

 , , and
, , and 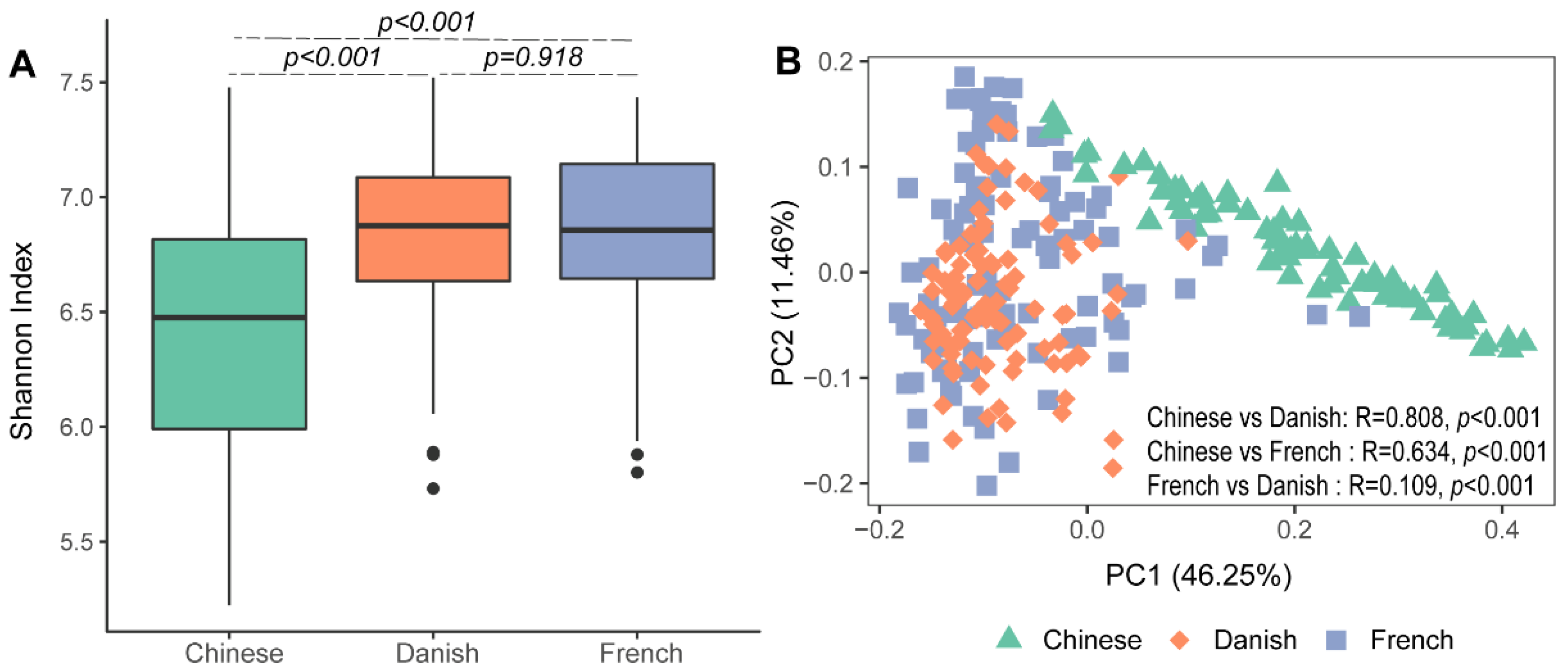{kind=link}
{kind=link}
{kind=link}
{kind=link}
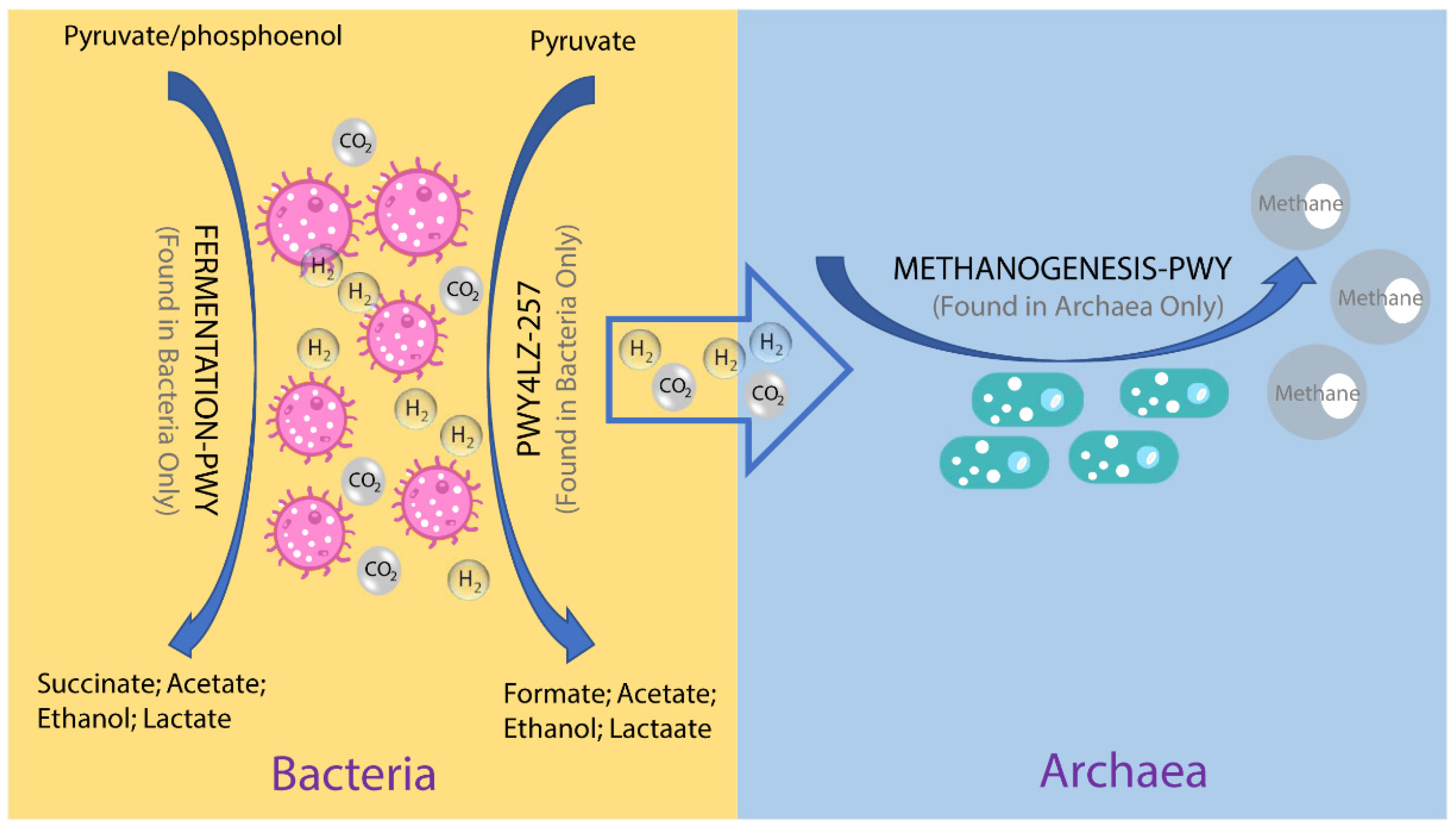{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Obtaining Data (Metadata (from Nature Microbiology))
2.2. Raw Reads Pre-Processing
2.3. Taxonomic Profiling and Diversity
2.4. CAZyme and ARG Analysis
2.5. Statistical Analysis
3. Results
3.1. The Diversity of Archaea in Pigs
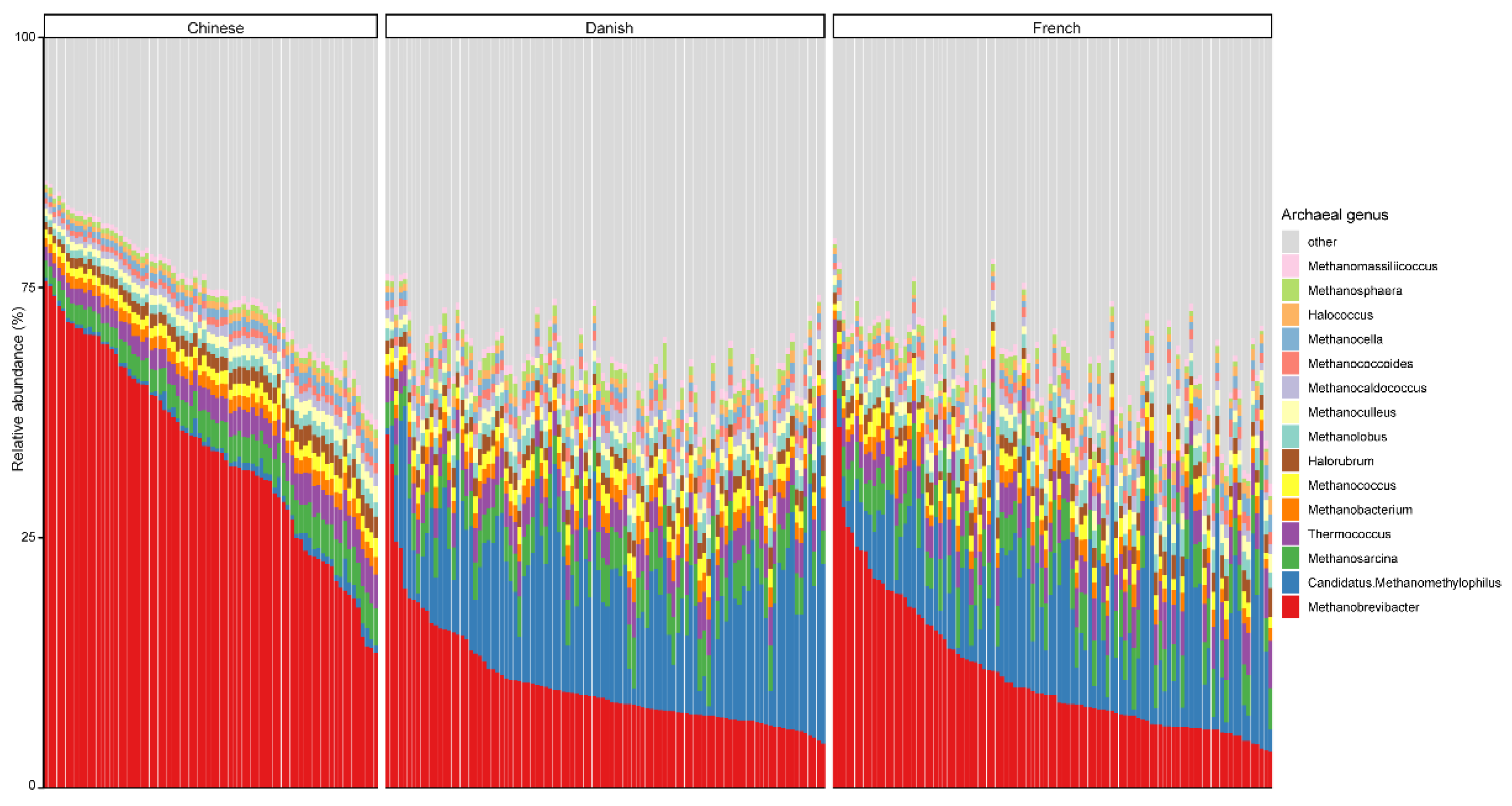
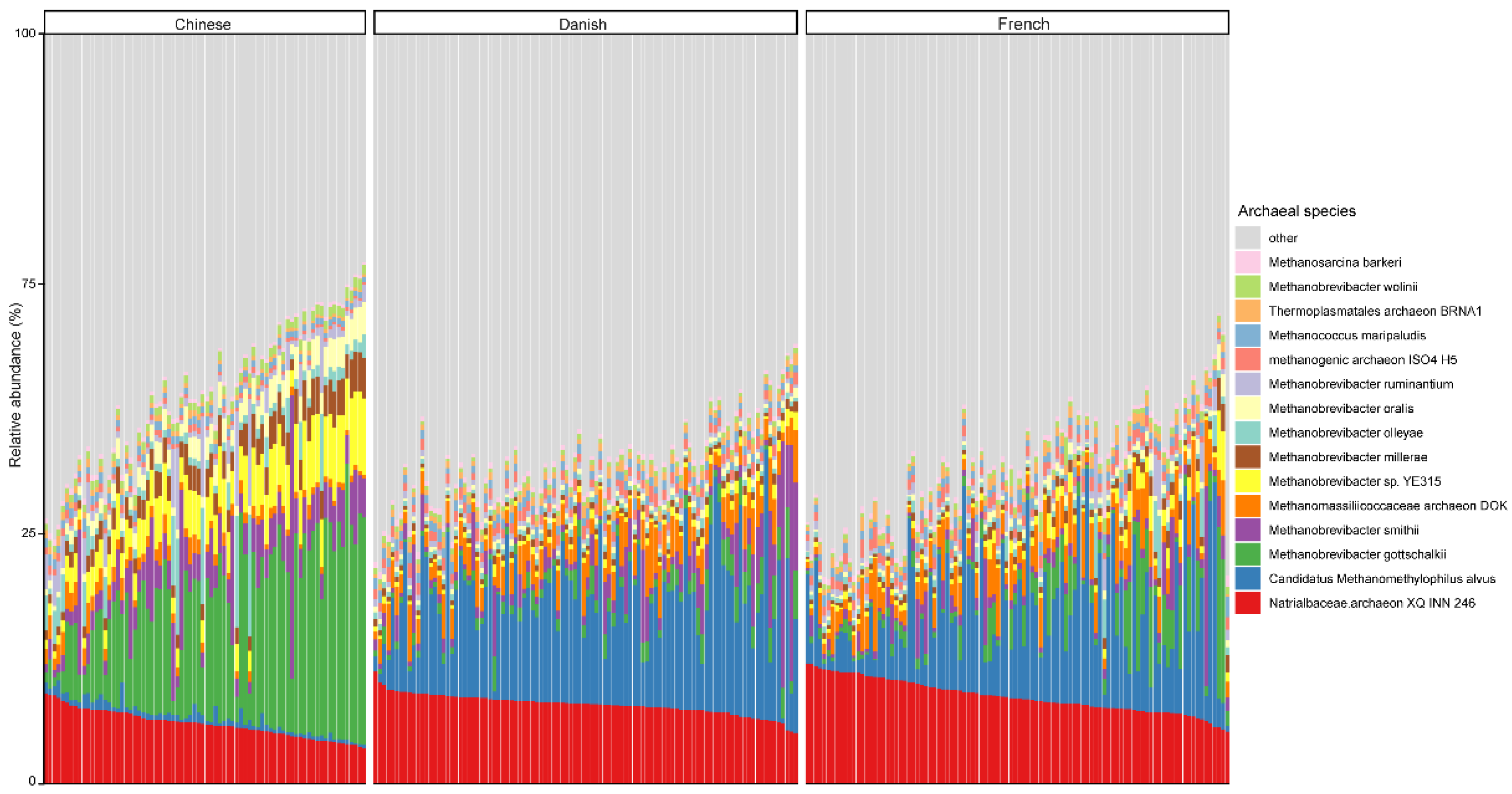
3.2. The Archaeal Composition in Pigs
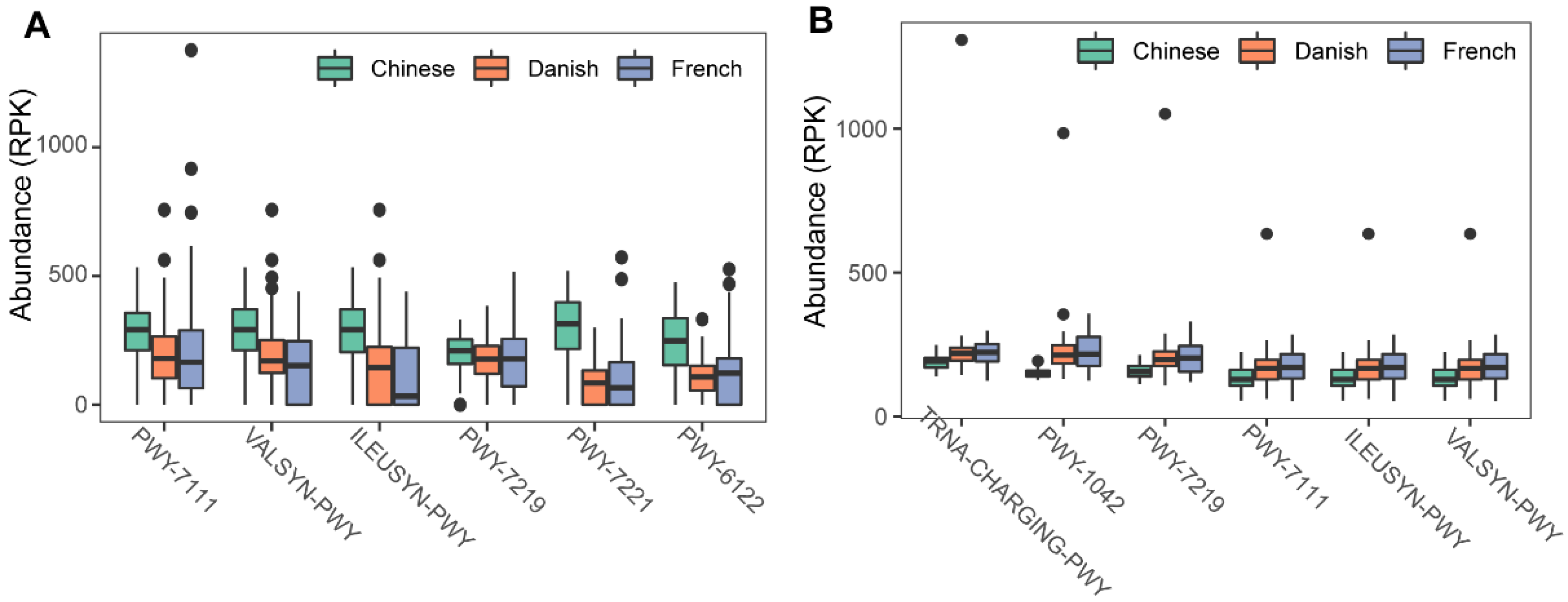
3.3. The metabolic Pathways of the Swine Archaea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [Green Version]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [Green Version]
- Cavicchioli, R. Cold-adapted archaea. Nat. Rev. Microbiol. 2006, 4, 331–343. [Google Scholar] [CrossRef]
- Andrei, A.-Ş.; Banciu, H.L.; Oren, A. Living with salt: Metabolic and phylogenetic diversity of archaea inhabiting saline ecosystems. FEMS Microbiol. Lett. 2012, 330, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.p.; Zhang, L.m.; Zhu, Y.g.; Zhang, J.b.; He, J.z. Abundance and composition of ammonia-oxidizing bacteria and ammonia-oxidizing archaea communities of an alkaline sandy loam. Environ. Microbiol. 2008, 10, 1601–1611. [Google Scholar] [CrossRef]
- Herbold, C.W.; Lehtovirta-Morley, L.E.; Jung, M.Y.; Jehmlich, N.; Hausmann, B.; Han, P.; Loy, A.; Pester, M.; Sayavedra-Soto, L.A.; Rhee, S.K. Ammonia-oxidising archaea living at low pH: Insights from comparative genomics. Environ. Microbiol. 2017, 19, 4939–4952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea are interactive components of complex microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Brugere, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The host-associated archaeome. Nat. Rev. Microbiol. 2020, 18, 622–636. [Google Scholar] [CrossRef]
- Kim, J.Y.; Whon, T.W.; Lim, M.Y.; Kim, Y.B.; Kim, N.; Kwon, M.-S.; Kim, J.; Lee, S.H.; Choi, H.-J.; Nam, I.-H. The human gut archaeome: Identification of diverse haloarchaea in Korean subjects. Microbiome 2020, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.A.; Faull, R.; Fornasini, G.; Milne, R.W.; Evans, A.M. Accumulation of trimethylamine and trimethylamine-N-oxide in end-stage renal disease patients undergoing haemodialysis. Nephrol. Dial. Transplant. 2006, 21, 1300–1304. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, M.; Gunsalus, R.P.; Rao, S.S.; Zhang, H. Methanogens in human health and disease. Am. J. Gastroenterol. Suppl. 2012, 1, 28. [Google Scholar] [CrossRef]
- Khelaifia, S.; Raoult, D. Haloferax massiliensis sp. nov., the first human-associated halophilic archaea. New Microbes New Infect. 2016, 12, 96. [Google Scholar] [CrossRef] [Green Version]
- Coker, O.O.; Wu, W.K.K.; Wong, S.H.; Sung, J.J.; Yu, J. Altered gut archaea composition and interaction with bacteria are associated with colorectal cancer. Gastroenterology 2020, 159, 1459–1470.e1455. [Google Scholar] [CrossRef] [PubMed]
- Gresse, R.; Chaucheyras Durand, F.; Dunière, L.; Blanquet-Diot, S.; Forano, E. Microbiota composition and functional profiling throughout the gastrointestinal tract of commercial weaning piglets. Microorganisms 2019, 7, 343. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Bian, G.; Zhu, Z.; Smidt, H.; Zhu, W. Early methanogenic colonisation in the faeces of Meishan and Yorkshire piglets as determined by pyrosequencing analysis. Archaea 2014, 2014, 547908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.-h.; Su, Y.; Wright, A.-D.G.; Zhang, L.-l.; Smidt, H.; Zhu, W.-Y. Lean breed Landrace pigs harbor fecal methanogens at higher diversity and density than obese breed Erhualian pigs. Archaea 2012, 2012, 605289. [Google Scholar] [CrossRef]
- Mi, J.; Peng, H.; Wu, Y.; Wang, Y.; Liao, X. Diversity and community of methanogens in the large intestine of finishing pigs. BMC Microbiol. 2019, 19, 83. [Google Scholar] [CrossRef]
- Federici, S.; Miragoli, F.; Pisacane, V.; Rebecchi, A.; Morelli, L.; Callegari, M.L. Archaeal microbiota population in piglet feces shifts in response to weaning: Methanobrevibacter smithii is replaced with Methanobrevibacter boviskoreani. FEMS Microbiol. Lett. 2015, 362, fnv064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Chen, H.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Tian, G.; Yu, J.; Huang, Z.; Luo, J. Dietary pea fiber increases diversity of colonic methanogens of pigs with a shift from Methanobrevibacter to Methanomassiliicoccus-like genus and change in numbers of three hydrogenotrophs. BMC Microbiol. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Estellé, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.Ø.; Kjeldsen, N.J.; Liu, C. A reference gene catalogue of the pig gut microbiome. Nat. Microbiol. 2016, 1, 16161. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BMTagger: Best Match Tagger for Removing Human Reads from Metagenomics Datasets. Available online: Ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/ (accessed on 1 July 2020).
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef]
- Kirstahler, P.; Bjerrum, S.S.; Friis-Møller, A.; La Cour, M.; Aarestrup, F.M.; Westh, H.; Pamp, S.J. Genomics-based identification of microorganisms in human ocular body fluid. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.L.; Wolin, M.; Kusel, E. Isolation and characterization of methanogens from animal feces. Syst. Appl. Microbiol. 1986, 8, 234–238. [Google Scholar] [CrossRef]
- Al-Mailem, D.; Sorkhoh, N.; Al-Awadhi, H.; Eliyas, M.; Radwan, S. Biodegradation of crude oil and pure hydrocarbons by extreme halophilic archaea from hypersaline coasts of the Arabian Gulf. Extremophiles 2010, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Plugge, C.M.; Stams, A.J. Anaerobic degradation of propionate by a mesophilic acetogenic bacterium in coculture and triculture with different methanogens. Appl. Environ. Microbiol. 1994, 60, 2834–2838. [Google Scholar] [CrossRef] [Green Version]
- Noel, S.J.; Højberg, O.; Urich, T.; Poulsen, M. Draft genome sequence of “Candidatus Methanomethylophilus” sp. 1R26, enriched from bovine rumen, a methanogenic archaeon belonging to the Methanomassiliicoccales order. Genome Announc. 2016, 4, e01734-15. [Google Scholar]
- Kennedy, K.; Hall, M.W.; Lynch, M.D.; Moreno-Hagelsieb, G.; Neufeld, J.D. Evaluating bias of Illumina-based bacterial 16S rRNA gene profiles. Appl. Environ. Microbiol. 2014, 80, 5717–5722. [Google Scholar] [CrossRef] [Green Version]
- Eloe-Fadrosh, E.A.; Ivanova, N.N.; Woyke, T.; Kyrpides, N.C. Metagenomics uncovers gaps in amplicon-based detection of microbial diversity. Nat. Microbiol. 2016, 1, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, B.S.; Gordon, J.I. A humanized gnotobiotic mouse model of host–archaeal–bacterial mutualism. Proc. Natl. Acad. Sci. USA 2006, 103, 10011–10016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavromichalis, I.; Kerr, B.; Parr, T.; Albin, D.; Gabert, V.; Baker, D. Valine requirement of nursery pigs. Anim. Sci. J. 2001, 79, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.; Lewis, A.; Miller, P.; Fischer, R.; Diedrichsen, R. Growth, carcass traits, and plasma amino acid concentrations of gilts fed low-protein diets supplemented with amino acids including histidine, isoleucine, and valine. Anim. Sci. J. 2003, 81, 1529–1537. [Google Scholar] [CrossRef] [Green Version]
- Alfaia, C.; Pestana, J.; Rodrigues, M.; Coelho, D.; Aires, M.; Ribeiro, D.; Major, V.; Martins, C.; Santos, H.; Lopes, P. Influence of dietary Chlorella vulgaris and carbohydrate-active enzymes on growth performance, meat quality and lipid composition of broiler chickens. Poult. Sci. 2021, 100, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, T.; Zhou, Y.; Li, F. Effects of enzyme+ bacteria treatment on growth performance, rumen bacterial diversity, KEGG pathways, and the CAZy spectrum of Tan sheep. Bioengineered 2020, 11, 1221–1232. [Google Scholar] [CrossRef]
- Fang, S.; Chen, X.; Ye, X.; Zhou, L.; Xue, S.; Gan, Q. Effects of gut microbiome and short-chain fatty acids (SCFAs) on finishing weight of meat rabbits. Front. Microbiol. 2020, 11, 1835. [Google Scholar] [CrossRef] [PubMed]
- White, B.A.; Lamed, R.; Bayer, E.A.; Flint, H.J. Biomass utilization by gut microbiomes. Annu. Rev. Microbiol. 2014, 68, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Kırtel, O.; Lescrinier, E.; Van den Ende, W.; Öner, E.T. Discovery of fructans in Archaea. Carbohydr. Polym. 2019, 220, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, M.; Krüger, A.; Antranikian, G. Biomass-degrading glycoside hydrolases of archaeal origin. Biotechnol. Biofuels 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D. XSEDE: Accelerating scientific discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, F.; Li, Y.; Peng, Y.; Wei, X.; Wang, X.; Howe, S.; Yang, H.; Xiao, Y.; Li, H.; Zhao, J.; et al. The Diversity, Composition, and Metabolic Pathways of Archaea in Pigs. Animals 2021, 11, 2139. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11072139
Deng F, Li Y, Peng Y, Wei X, Wang X, Howe S, Yang H, Xiao Y, Li H, Zhao J, et al. The Diversity, Composition, and Metabolic Pathways of Archaea in Pigs. Animals. 2021; 11(7):2139. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11072139
Chicago/Turabian StyleDeng, Feilong, Yushan Li, Yunjuan Peng, Xiaoyuan Wei, Xiaofan Wang, Samantha Howe, Hua Yang, Yingping Xiao, Hua Li, Jiangchao Zhao, and et al. 2021. "The Diversity, Composition, and Metabolic Pathways of Archaea in Pigs" Animals 11, no. 7: 2139. https://0-doi-org.brum.beds.ac.uk/10.3390/ani11072139