
Antibody Conjugates: From Heterogeneous Populations to Defined Reagents


Abstract
:
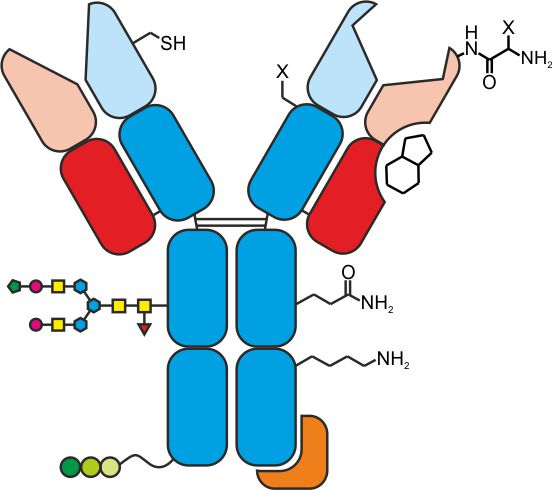{kind=link}
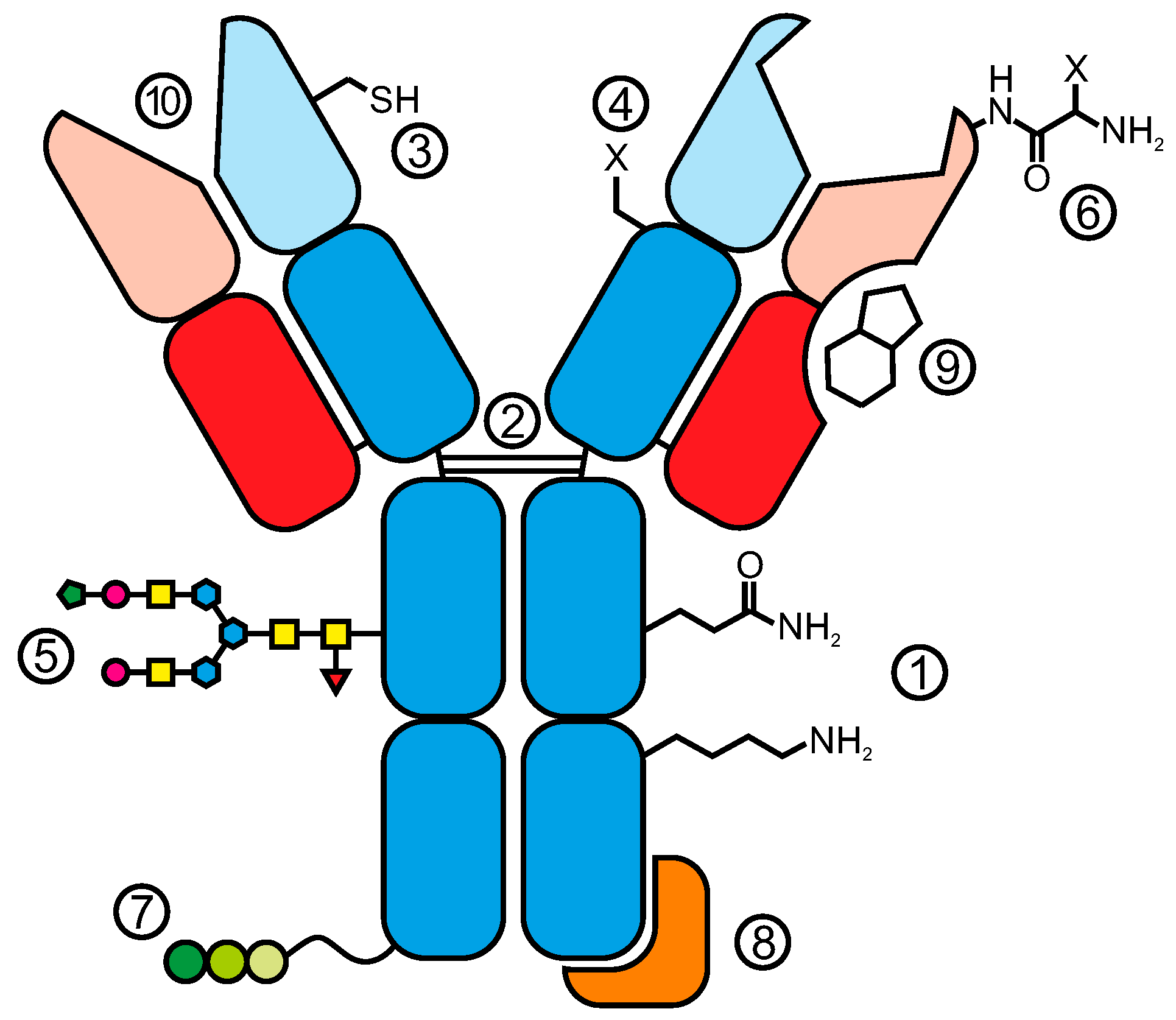{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Conjugation via Natural Amino Acid Residues
2.1. Conjugation via Lysine

2.2. Conjugation via Cysteines
2.2.1. Native Cysteines
2.2.2. Engineered Cysteines
2.3. Conjugation via Selenocysteine
2.4. Conjugation via Tyrosine
3. Conjugation via Bioorthogonal Amino Acid Residues

4. Conjugation via the Carbohydrate Moiety
4.1. Chemical Oxidation of Glycans

4.2. Enzymatic and Chemo-Enzymatic Modification of Glycans
4.3. Metabolic Engineering of the Carbohydrate Moiety
5. Conjugation via the N-Terminus

6. Conjugation via Tags
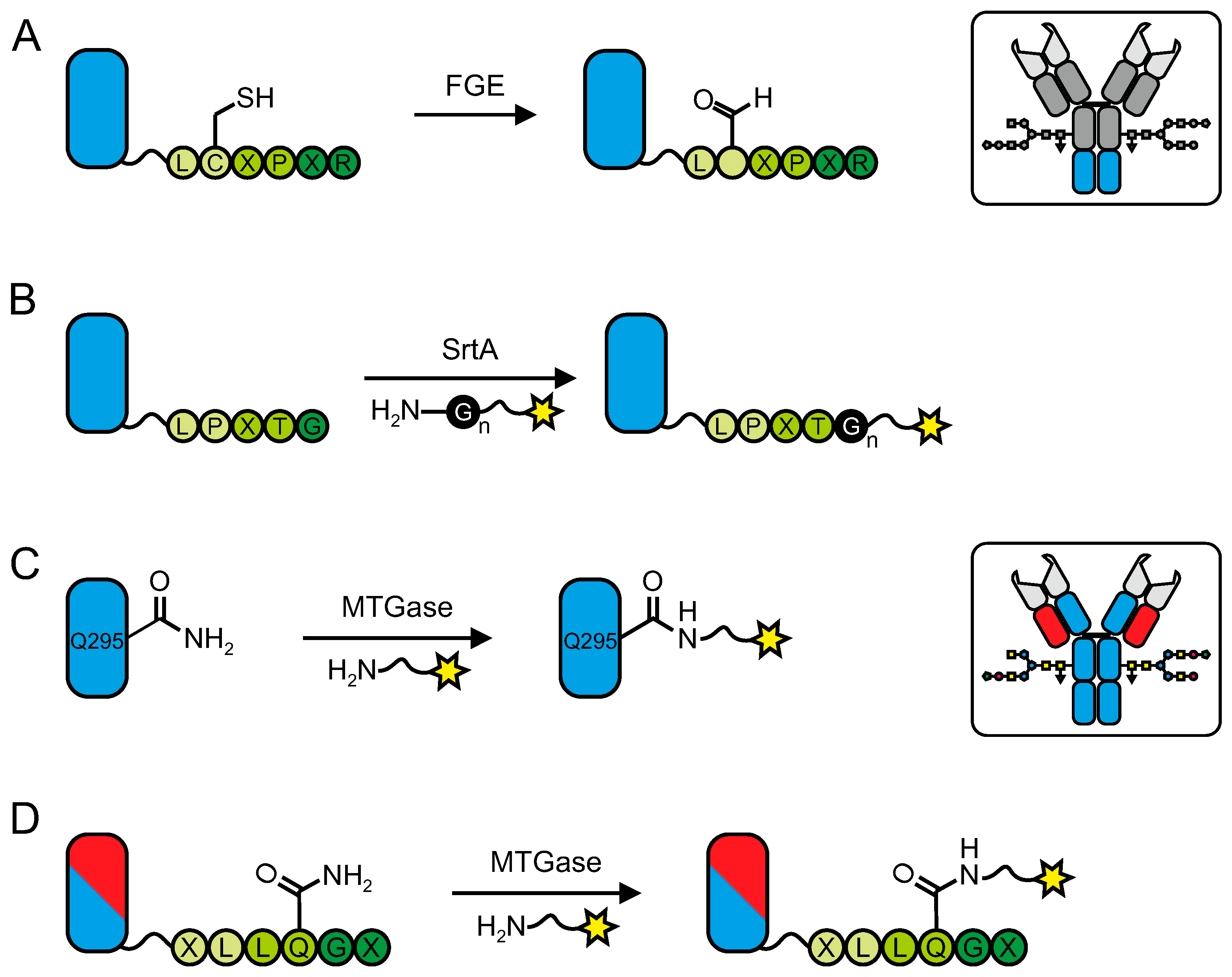
6.1. Formylglycin Generating Enzyme (FGE)

6.2. Sortase
6.3. Transglutaminases
7. Miscellaneous Conjugation Methods
7.1. Conjugation via Fc-Binding Domains (FcBD)
7.1.1. ZZ-Domain
7.1.2. Photoactivable FcBDs

7.2. Bioconjugation via the Nucleotide Binding Site
7.3. Catalytic Antibodies

8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautes-Fridman, C.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Tumor-targeting monoclonal antibodies in cancer therapy. Oncoimmunology 2014, 3, e27048. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Antibodies to watch in 2015. MAbs 2015, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Reichert, J.M. Antibody-drug conjugates: present and future. MAbs 2014, 6, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Papachristos, A.; Pippa, N.; Demetzos, C.; Sivolapenko, G. Antibody-drug conjugates: A mini-review. The synopsis of two approved medicines. The synopsis of two approved medicines. Drug Deliv. 2015, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kraeber-Bodere, F.; Bodet-Milin, C.; Rousseau, C.; Eugene, T.; Pallardy, A.; Frampas, E.; Carlier, T.; Ferrer, L.; Gaschet, J.; Davodeau, F.; et al. Radioimmunoconjugates for the treatment of cancer. Semin. Oncol. 2014, 41, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Hooper, A.; O'Donnell, C.; Gerber, H.-P. Investigational antibody drug conjugates for solid tumors. Expert Opin. Investig. Drugs 2011, 20, 1131–1180. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.; Pillow, T.; Aristoff, P. Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2013, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, N.; Francis, M.B. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011, 7, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer. Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Mundo, E.E.; Zhang, C.; Bumbaca, D.; Valle, N.R.; Kozak, K.R.; Fourie, A.; Chuh, J.; Koppada, N.; Saad, O.; et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug. Chem. 2011, 22, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Atkinson, J.; Guevara, C.I.; Zhang, C.; Kery, V.; Moon, S.-J.J.; Virata, C.; Yang, P.; Lowe, C.; Pinkstaff, J.; et al. In vitro and in vivo evaluation of cysteine and site-specific conjugated herceptin antibody-drug conjugates. PloS One 2014, 9, e83865:14. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.R.; Liu, B. Methods for site-specific drug conjugation to antibodies. mAbs 2013, 6, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kline, T.; Steiner, A.R.; Penta, K.; Sato, A.K.; Hallam, T.J.; Yin, G. Methods to make homogenous antibody drug conjugates. Pharm. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody-drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug. Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Sochaj, A.M.; Swiderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Kim, J. Advances in the development of site-specific antibody-drug conjugation. Anticancer Agents Med. Chem. 2015, 15, 828–836. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.N. Specific chemical modification of proteins. Annu. Rev. Biochem. 1970, 39, 101–130. [Google Scholar] [CrossRef] [PubMed]
- Ghose, T.I.; Blair, A.H.; Kulkarni, P.N. Preparation of antibody-linked cytotoxic agents. Methods Enzymol. 1983, 93, 280–333. [Google Scholar] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: New York, NY, USA, 2008; pp. 1041–1132. [Google Scholar]
- Gauthier, M.A.; Klok, H.-A. Peptide/protein-polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. (Cambridge, U. K.) 2008, 21, 2591–2611. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.B. New methods for protein bioconjugation. In Chemical Biology; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; pp. 593–634. [Google Scholar]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.M.; Wrasidlo, W.A.; Reisfeld, R.A. Determination of the number of e-amino groups available for conjugation of effector molecules to monoclonal antibodies. Hybridoma 1988, 7, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Feeney, M.B.; Rivera, J.; Chen, Y.; Kim, M.; Sharma, V.K.; Wang, Y.J. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: Changes due to modification and conjugation processes. Bioconjug. Chem. 2010, 21, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.M.; Tabei, K.; Kunz, A.; Hollander, I.J.; Hamann, R.R.; Bell, D.H.; Berkenkamp, S.; Hillenkamp, F. Calicheamicin derivatives conjugated to monoclonal antibodies: determination of loading values and distributions by infrared and UV matrix-assisted laser desorption/ionization mass spectrometry and electrospray ionization mass spectrometry. Anal. Chem. 1997, 69, 2716–2726. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, M.; Gebler, J.; Shreder, K.; Wu, J. Region-selective labeling of antibodies as determined by electrospray ionization-mass spectrometry (ESI-MS). Bioconjug. Chem. 2000, 11, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Amphlett, G.; Blattler, W.A.; Lambert, J.M.; Zhang, W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005, 14, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, Z.; Chen, W.; Prabakaran, P.; Lin, K.; Dimitrov, D. Conjugates of small molecule drugs with antibodies and other proteins. Biomedicines 2014, 2, 1–13. [Google Scholar] [CrossRef]
- Hess, C.; Venetz, D.; Neri, D. Emerging classes of armed antibody therapeutics against cancer. Med. Chem. Commun. 2014, 5, 408–431. [Google Scholar] [CrossRef]
- Kalkhof, S.; Sinz, A. Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal. Bioanal. Chem. 2008, 392, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Madler, S.; Bich, C.; Touboul, D.; Zenobi, R. Chemical cross-linking with NHS esters: A systematic study on amino acid reactivities. J. Mass Spectrom. 2009, 44, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Chih, H.W.; Gikanga, B.; Yang, Y.; Zhang, B. Identification of amino acid residues responsible for the release of free drug from an antibody-drug conjugate utilizing lysine-succinimidyl ester chemistry. J. Pharm. Sci. 2011, 100, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Hamaguchi, K. The role of the intrachain disulfide bond in the conformation and stability of the constant fragment of the immunoglobulin light chain. J. Biochem. 1979, 86, 1433–1441. [Google Scholar] [PubMed]
- Proba, K.; Honegger, A.; Pluckthun, A. A natural antibody missing a cysteine in VH: consequences for thermodynamic stability and folding. J. Mol. Biol. 1997, 265, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Thies, M.J.; Talamo, F.; Mayer, M.; Bell, S.; Ruoppolo, M.; Marino, G.; Buchner, J. Folding and oxidation of the antibody domain CH3. J. Mol. Biol. 2002, 319, 1267–1277. [Google Scholar] [CrossRef]
- McAuley, A.; Jacob, J.; Kolvenbach, C.G.; Westland, K.; Lee, H.J.; Brych, S.R.; Rehder, D.; Kleemann, G.R.; Brems, D.N.; Matsumura, M. Contributions of a disulfide bond to the structure, stability, and dimerization of human IgG1 antibody CH3 domain. Protein Sci. 2008, 17, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chumsae, C.; Gaza-Bulseco, G.; Hurkmans, K.; Radziejewski, C.H. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal. Chem. 2010, 82, 5219–5226. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Cal, P.M.; Bernardes, G.J.; Gois, P.M. Cysteine-selective reactions for antibody conjugation. Angew. Chem. Int. Ed. Engl. 2014, 53, 10585–10587. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.T.; Asano, S.; Li, X.; Rader, C.; Barbas, C.F. Improving the serum stability of site-specific antibody conjugates with sulfone linkers. Bioconjug. Chem. 2014, 25, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, K.; Tommasi, R.; Henseleit, K.; et al. In vitro and In vivo evaluation of cysteine rebridged trastuzumab-MMAE antibody drug conjugates with defined drug-to-antibody ratios. Mol. Pharm. 2015, 12, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.F.; Nunes, J.P.; Maruani, A.; Chudasama, V.; Smith, M.E.; Chester, K.A.; Baker, J.R.; Caddick, S. Next generation maleimides enable the controlled assembly of antibody-drug conjugates via native disulfide bond bridging. Org. Biomol. Chem. 2014, 12, 7261–7269. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.P.; Morais, M.; Vassileva, V.; Robinson, E.; Rajkumar, V.S.; Smith, M.E.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody-drug conjugate (ADC). Chem. Commun. (Camb.) 2015, 51, 10624–10627. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Smith, M.E.; Miranda, E.; Chester, K.A.; Chudasama, V.; Caddick, S. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 2015, 6, 6645. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Li, F.; et al. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Charati, M.; He, T.; Sousa, E.; Ma, D.; Han, X.; Clark, T.; Casavant, J.; Loganzo, F.; Barletta, F.; et al. Mild method for succinimide hydrolysis on ADCs: impact on ADC potency, stability, exposure, and efficacy. Bioconjug. Chem. 2014, 25, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Stimmel, J.B.; Merrill, B.M.; Kuyper, L.F.; Moxham, C.P.; Hutchins, J.T.; Fling, M.E.; Kull, F.C., Jr. Site-specific conjugation on serine right-arrow cysteine variant monoclonal antibodies. J. Biol. Chem. 2000, 275, 30445–30450. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Raab, H.; Junutula, J. Engineering THIOMABs for site-specific conjugation of thiol-reactive linkers. In Antibody-Drug Conjugates; Ducry, L., Ed.; Humana Press: New York, NJ, USA, 2013; Volume 1045, pp. 189–203. [Google Scholar]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Caron, P.C.; Laird, W.; Co, M.S.; Avdalovic, N.M.; Queen, C.; Scheinberg, D.A. Engineered humanized dimeric forms of IgG are more effective antibodies. J. Exp. Med. 1992, 176, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Shopes, B. A genetically engineered human IgG with limited flexibility fully initiates cytolysis via complement. Mol. Immunol. 1993, 30, 603–609. [Google Scholar] [CrossRef]
- Baldwin, A.D.; Kiick, K.L. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.; Chen, C.; Thorell, J.-O.; Fredriksson, A.; Stone-Elander, S.; Gafvelin, G.; Arnér, E. Exploiting the 21st amino acid-purifying and labeling proteins by selenolate targeting. Nat. Methods 2004, 1, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Skeffington, L.R.; Chapman, C.M.; Rader, C. Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry 2009, 48, 12047–12057. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Thomas, J.D.; Burke, T.R., Jr.; Rader, C. An engineered selenocysteine defines a unique class of antibody derivatives. Proc. Natl. Acad. Sci. USA 2008, 105, 12451–12456. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.; Rader, C. Antibody conjugation via one and two C-terminal selenocysteines. Methods (San Diego, Calif.) 2014, 65, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Gavrilyuk, J.; Barbas, C.F., 3rd. Tyrosine bioconjugation through aqueous ene-type reactions: a click-like reaction for tyrosine. J. Am. Chem. Soc. 2010, 132, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nagano, M.; Gavrilyuk, J.; Hakamata, W.; Inokuma, T.; Barbas, C. Facile and stabile linkages through tyrosine: Bioconjugation strategies with the tyrosine-click reaction. Bioconjug. Chem. 2013, 24, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Gavrilyuk, J.; Ban, H.; Nagano, M.; Hakamata, W.; Barbas, C. Formylbenzene diazonium hexafluorophosphate reagent for tyrosine-selective modification of proteins and the introduction of a bioorthogonal aldehyde. Bioconjug. Chem. 2012, 23, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Gavrilyuk, J.; Ban, H.; Uehara, H.; Sirk, S.J.; Saye-Francisco, K.; Cuevas, A.; Zablowsky, E.; Oza, A.; Seaman, M.S.; Burton, D.R.; et al. Antibody conjugation approach enhances breadth and potency of neutralization of anti-HIV-1 antibodies and CD4-IgG. J. Virol. 2013, 87, 4985–4993. [Google Scholar] [CrossRef] [PubMed]
- Hallam, T.J.; Smider, V.V. Unnatural amino acids in novel antibody conjugates. Future Med. Chem. 2014, 6, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Parrish, A.R.; Wang, L. Expanding the genetic code for biological studies. Chem. Biol. 2009, 16, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Axup, J.; Bajjuri, K.; Ritland, M.; Hutchins, B.; Kim, C.; Kazane, S.; Halder, R.; Forsyth, J.; Santidrian, A.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Chatterjee, A.; Choi, S.-H.; Bajjuri, K.M.; Sinha, S.C.; Schultz, P.G. Genetic incorporation of multiple unnatural amino acids into proteins in mammalian cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 14080–14083. [Google Scholar] [CrossRef] [PubMed]
- Abaturov, L.V.; Nezlin, R.S.; Vengerova, T.I.; Varshavsky, J.M. Conformational studies of immunoglobulin G and its subunits by the methods of hydrogen-deuterium exchange and infrared spectroscopy. Biochim. Biophys. Acta 1969, 194, 386–396. [Google Scholar] [CrossRef]
- Calmettes, P.; Cser, L.; Rajnavolgyi, E. Temperature and pH dependence of immunoglobulin G conformation. Arch. Biochem. Biophys. 1991, 291, 277–283. [Google Scholar] [CrossRef]
- Ejima, D.; Tsumoto, K.; Fukada, H.; Yumioka, R.; Nagase, K.; Arakawa, T.; Philo, J.S. Effects of acid exposure on the conformation, stability, and aggregation of monoclonal antibodies. Proteins 2007, 66, 954–962. [Google Scholar] [CrossRef] [PubMed]
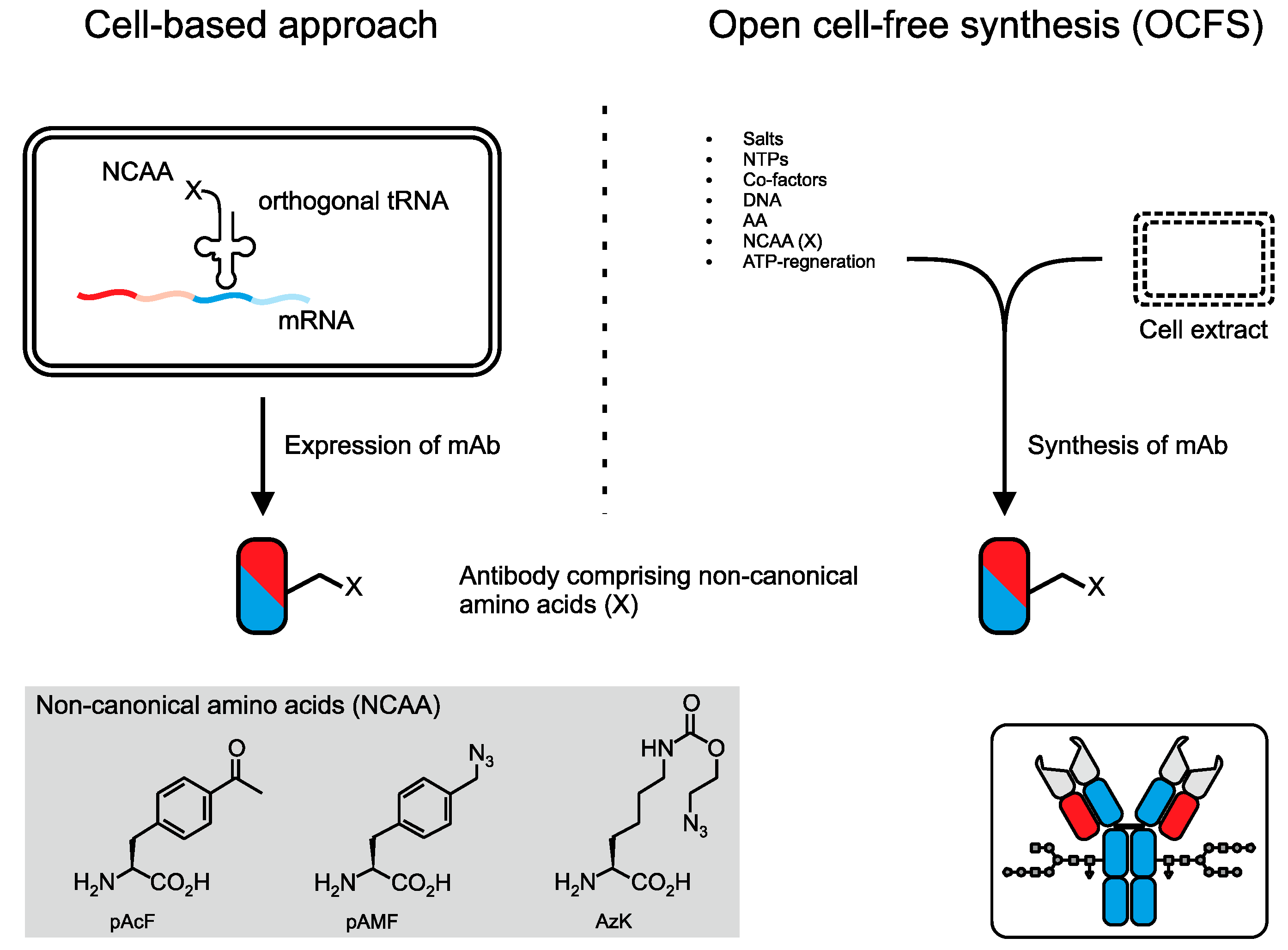
- Yin, G.; Garces, E.D.; Yang, J.; Zhang, J.; Tran, C.; Steiner, A.R.; Roos, C.; Bajad, S.; Hudak, S.; Penta, K.; et al. Aglycosylated antibodies and antibody fragments produced in a scalable in vitro transcription-translation system. MAbs 2012, 4, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Loscha, K.V.; Kuppan, K.V.; Loh, C.T.; Dixon, N.E.; Otting, G. High-yield cell-free protein synthesis for site-specific incorporation of unnatural amino acids at two sites. Biochem. Biophys. Res. Commun. 2012, 418, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Caschera, F.; Noireaux, V. Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 2014, 99, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Kwon, Y.-C.C.; Jewett, M.C. Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front. Chem. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tran, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug. Chem. 2014, 25, 351–361. [Google Scholar] [CrossRef] [PubMed]
- O'Shannessy, D.J.; Dobersen, M.J.; Quarles, R.H. A novel procedure for labeling immunoglobulins by conjugation to oligosaccharide moieties. Immunol. Lett. 1984, 8, 273–277. [Google Scholar] [CrossRef]
- Kralovec, J.; Spencer, G.; Blair, A.H.; Mammen, M.; Singh, M.; Ghose, T. Synthesis of methotrexate-antibody conjugates by regiospecific coupling and assessment of drug and antitumor activities. J. Med. Chem. 1989, 32, 2426–2431. [Google Scholar] [CrossRef] [PubMed]
- Laguzza, B.C.; Nichols, C.L.; Briggs, S.L.; Cullinan, G.J.; Johnson, D.A.; Starling, J.J.; Baker, A.L.; Bumol, T.F.; Corvalan, J.R. New antitumor monoclonal antibody-vinca conjugates LY203725 and related compounds: Design, preparation, and representative in vivo activity. J. Med. Chem. 1989, 32, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Baker, A.L.; Laguzza, B.C.; Fix, D.V.; Gutowski, M.C. Antitumor activity of L/1C2-4-desacetylvinblastine-3-carboxhydrazide immunoconjugate in xenografts. Cancer Res. 1990, 50, 1790–1794. [Google Scholar] [PubMed]
- Zuberbühler, K.; Casi, G.; Bernardes, G.J.; Neri, D. Fucose-specific conjugation of hydrazide derivatives to a vascular-targeting monoclonal antibody in IgG format. Chem. Commun. (Camb.) 2012, 48, 7100–7102. [Google Scholar] [CrossRef] [PubMed]
- Ranadive, G.N.; Rosenzweig, H.S.; Epperly, M.W.; Seskey, T.; Bloomer, W.D. A new method of technetium-99m labeling of monoclonal antibodies through sugar residues. A study with TAG-72 specific CC-49 antibody. Nucl. Med. Biol. 1993, 20, 719–726. [Google Scholar] [CrossRef]
- Rodwell, J.D.; Alvarez, V.L.; Lee, C.; Lopes, A.D.; Goers, J.W.; King, H.D.; Powsner, H.J.; McKearn, T.J. Site-specific covalent modification of monoclonal antibodies: In vitro and in vivo evaluations. Proc. Natl. Acad. Sci. USA 1986, 83, 2632–2636. [Google Scholar] [CrossRef] [PubMed]
- Husain, M.; Bieniarz, C. Fc site-specific labeling of immunoglobulins with calf intestinal alkaline phosphatase. Bioconjug. Chem. 1994, 5, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.F.; Li, Y.T.; Connell, M.E.; Yang, V.C. Synthesis and characterization of positively charged tPA as a prodrug using heparin/protamine-based drug delivery system. AAPS PharmSci 2000, 2, E7. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.F.; Park, Y.J.; Song, H.; Li, Y.T.; Yang, V.C. ATTEMPTS: a heparin/protamine-based prodrug approach for delivery of thrombolytic drugs. J. Control. Release 2001, 72, 145–156. [Google Scholar] [CrossRef]
- Park, Y.-J.; Liang, J.-F.; Song, H.; Li, Y.T.; Naik, S.; Yang, V.C. ATTEMPTS: A heparin/protamine-based triggered release system for the delivery of enzyme drugs without associated side-effects. Adv. Drug Deliv. Rev. 2003, 55, 251–265. [Google Scholar] [CrossRef]
- Raju, T.S.; Jordan, R.E. Galactosylation variations in marketed therapeutic antibodies. MAbs 2012, 4, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Zauner, G.; Selman, M.H.; Bondt, A.; Rombouts, Y.; Blank, D.; Deelder, A.M.; Wuhrer, M. Glycoproteomic analysis of antibodies. Mol. Cell. Proteomics 2013, 12, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Prog. 2005, 21, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Moller, D.; Gabel, D.; Senter, P.; Hellstrom, I.; Hellstrom, K.E. The influence of periodate oxidation on monoclonal antibody avidity and immunoreactivity. J. Immunol. Methods 1991, 144, 77–86. [Google Scholar] [CrossRef]
- Wolfe, C.A.; Hage, D.S. Studies on the rate and control of antibody oxidation by periodate. Anal. Biochem. 1995, 231, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hage, D.S.; Wolfe, C.A.; Oates, M.R. Development of a kinetic model to describe the effective rate of antibody oxidation by periodate. Bioconjug. Chem. 1997, 8, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.; Koppel, R.; Schwartz, F.; Fleminger, G. Enzymic oxidation of monoclonal antibodies by soluble and immobilized bifunctional enzyme complexes. J. Chromatogr. A 1990, 510, 321–329. [Google Scholar] [CrossRef]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Boons, G.J. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem. Int. Ed. Engl. 2014, 53, 7179–7182. [Google Scholar] [CrossRef] [PubMed]
- Boeggeman, E.; Ramakrishnan, B.; Kilgore, C.; Khidekel, N.; Hsieh-Wilson, L.C.; Simpson, J.T.; Qasba, P.K. Direct identification of nonreducing GlcNAc residues on N-glycans of glycoproteins using a novel chemoenzymatic method. Bioconjug. Chem. 2007, 18, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Boeggeman, E.; Ramakrishnan, B.; Pasek, M.; Manzoni, M.; Puri, A.; Loomis, K.H.; Waybright, T.J.; Qasba, P.K. Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: Application for cell surface antigen detection. Bioconjug. Chem. 2009, 20, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Davis, C.B.; Aggeler, R.; Kang, H.-C.; Chen, A.; Agnew, B.; Lewis, J.S. An enzyme-mediated methodology for the site-specific radiolabeling of antibodies based on catalyst-free click chemistry. Bioconjug. Chem. 2013, 42, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Davis, C.B.; Abdel-Atti, D.; Carlin, S.D.; Chen, A.; Aggeler, R.; Agnew, B.J.; Lewis, J.S. Chemoenzymatic strategy for the synthesis of site-specifically labeled immunoconjugates for multimodal PET and optical imaging. Bioconjug. Chem. 2014, 25, 2123–2128. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; Dyba, M.A.; Qasba, P.K.; Dimitrov, D.S. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs 2014, 6, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Okeley, N.M.; Toki, B.E.; Zhang, X.; Jeffrey, S.C.; Burke, P.J.; Alley, S.C.; Senter, P.D. Metabolic engineering of monoclonal antibody carbohydrates for antibody-drug conjugation. Bioconjug. Chem. 2013, 24, 1650–1655. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Bondt, A.; Rombouts, Y.; Selman, M.H.; Hensbergen, P.J.; Reiding, K.R.; Hazes, J.M.; Dolhain, R.J.; Wuhrer, M. Immunoglobulin G (IgG) Fab glycosylation analysis using a new mass spectrometric high-throughput profiling method reveals pregnancy-associated changes. Mol. Cell. Proteomics 2014, 13, 3029–3039. [Google Scholar] [CrossRef] [PubMed]
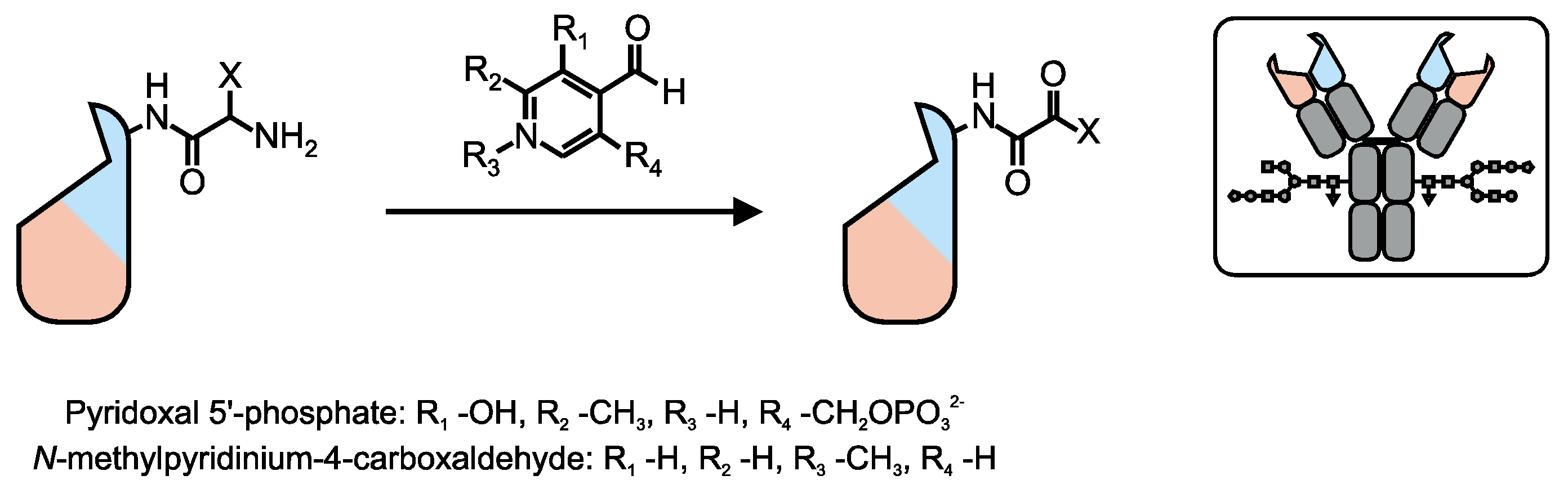
- Scheck, R.A.; Francis, M.B. Regioselective labeling of antibodies through N-terminal transamination. ACS Chem. Biol. 2007, 2, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Witus, L.S.; Netirojjanakul, C.; Palla, K.S.; Muehl, E.M.; Weng, C.H.; Iavarone, A.T.; Francis, M.B. Site-specific protein transamination using N-methylpyridinium-4-carboxaldehyde. J. Am. Chem. Soc. 2013, 135, 17223–17229. [Google Scholar] [CrossRef] [PubMed]
- Sack, J.T.; Stephanopoulos, N.; Austin, D.C.; Francis, M.B.; Trimmer, J.S. Antibody-guided photoablation of voltage-gated potassium currents. J. Gen. Physiol. 2013, 142, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Rabuka, D. Chemoenzymatic methods for site-specific protein modification. Curr. Opin. Chem. Biol. 2010, 14, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Dozier, J.; Distefano, M. Enzymatic labeling of proteins: techniques and approaches. Bioconjug. Chem. 2013, 24, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 2007, 3, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed]
- Rabuka, D.; Rush, J.; deHart, G.; Wu, P.; Bertozzi, C. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1119. [Google Scholar] [CrossRef] [PubMed]
- Hudak, J.; Barfield, R.; de Hart, G.; Grob, P.; Nogales, E.; Bertozzi, C.; Rabuka, D. Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew. Chem. Int. Ed. Engl. 2012, 51, 4161–4166. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; van der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A Pictet-Spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Subramanian, S.; Boder, E.T. Sortase A as a novel molecular "stapler" for sequence-specific protein conjugation. Bioconjug. Chem. 2007, 18, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Ritzefeld, M. Sortagging: A robust and efficient chemoenzymatic ligation strategy. Chemistry 2014, 20, 8516–8529. [Google Scholar] [CrossRef] [PubMed]
- Levary, D.A.; Parthasarathy, R.; Boder, E.T.; Ackerman, M.E. Protein-protein fusion catalyzed by sortase A. PLoS One 2011, 6, e18342:6. [Google Scholar] [CrossRef] [PubMed]
- Swee, L.K.; Guimaraes, C.P.; Sehrawat, S.; Spooner, E.; Barrasa, M.I.; Ploegh, H.L. Sortase-mediated modification of αDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc. Natl. Acad. Sci. USA 2013, 110, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Kwakkenbos, M.J.; Claassen, Y.B.; Maijoor, K.; Böhne, M.; van der Sluijs, K.F.; Witte, M.D.; van Zoelen, D.J.; Cornelissen, L.A.; Beaumont, T.; et al. Bispecific antibody generated with sortase and click chemistry has broad antiinfluenza virus activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16820–16825. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, J.J.; Bhattacharyya, J.; Chilkoti, A. A noncanonical function of sortase enables site-specific conjugation of small molecules to lysine residues in proteins. Angew. Chem. Int. Ed. Engl. 2014, 54, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Adachi, M.; Umeda, K.; Matsuura, A.; Nonaka, M.; Uchio, R.; Tanaka, H.; Motoki, M. Purification and characteristics of a novel transglutaminase derived from microorganisms. Agric. Biol. Chem. 1989, 53, 2613–2617. [Google Scholar] [CrossRef]
- Zhu, Y.; Tramper, J. Novel applications for microbial transglutaminase beyond food processing. Trends Biotechnol. 2008, 26, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Strop, P. Versatility of microbial transglutaminase. Bioconjug. Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Josten, A.; Haalck, L.; Spener, F.; Meusel, M. Use of microbial transglutaminase for the enzymatic biotinylation of antibodies. J. Immunol. Methods 2000, 240, 47–54. [Google Scholar] [CrossRef]
- Mindt, T.L.; Jungi, V.; Wyss, S.; Friedli, A.; Pla, G.; Novak-Hofer, I.; Grünberg, J.; Schibli, R. Modification of different IgG1 antibodies via glutamine and lysine using bacterial and human tissue transglutaminase. Bioconjug. Chem. 2008, 19, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Gruenberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Bregeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-specific conjugation of monomethyl auristatin E to anti-CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.-H.; Dorywalska, M.; Delaria, K.; Dushin, R.; Tran, T.-T.; Ho, W.-H.; Farias, S.; Casas, M.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C.; et al. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjug. Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsen, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlen, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Forsgren, A.; Sjöquist, J. Protein A from S. Aureus. J. Immunol. 1966, 97, 822–827. [Google Scholar] [PubMed]
- Iangone, J.J. Protein A of Staphylococcus aureus and related immunoglobulin receptors produced by Streptococci and Pneumonococci. In Advances in Immunology; Frank, J.D., Henry, G.K., Eds.; Academic Press: New York, NJ, USA, 1982; Volume 32, pp. 157–252. [Google Scholar]
- Kurata, N.; Shishido, T.; Muraoka, M.; Tanaka, T.; Ogino, C.; Fukuda, H.; Kondo, A. Specific protein delivery to target cells by antibody-displaying bionanocapsules. J. Biochem. 2008, 144, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Matsuzaki, T.; Yoshimoto, N.; Niimi, T.; Tanizawa, K.; Kuroda, S.I. Fluorophore-labeled nanocapsules displaying IgG Fc-binding domains for the simultaneous detection of multiple antigens. Biomaterials 2011, 32, 9011–9031. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Kadoya, H.; Hatahira, S.; Hiramatsu, S.; Jung, G.; Martin, A.; Quinn, J.; Jung, J.; Jeong, S.-Y.; Choi, E.; et al. Nanocapsules incorporating IgG Fc-binding domain derived from Staphylococcus aureus protein A for displaying IgGs on immunosensor chips. Biomaterials 2011, 32, 1455–1519. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Somiya, M.; Yoshimoto, N.; Niimi, T.; Kuroda, S.I. Nano-visualization of oriented-immobilized IgGs on immunosensors by high-speed atomic force microscopy. Sci. Rep. 2012, 2, 790. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, Y.; Tomizawa, K.; Nagita, M.; Michiue, H.; Nishiki, T.-I.; Ohmori, I.; Seno, M.; Matsui, H. Development of bionanocapsules targeting brain tumors. J. Control. Release 2007, 122, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Barnea, I.; Keydar, I.; Benhar, I. Antibody internalization studied using a novel IgG binding toxin fusion. J. Immunol. Methods 2007, 321, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Noy, R.; Wels, W.S.; Benhar, I. chFRP5-ZZ-PE38, a large IgG-toxin immunoconjugate outperforms the corresponding smaller FRP5(Fv)-ETA immunotoxin in eradicating ErbB2-expressing tumor xenografts. Cancer Lett. 2007, 257, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.; Shapira, A.; Starr, A.; Kazanov, D.; Kraus, S.; Benhar, I.; Arber, N. An immunoconjugate of anti-CD24 and Pseudomonas exotoxin selectively kills human colorectal tumors in mice. Gastroenterology 2011, 140, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Barnea, I.; Ben-Yosef, R.; Karaush, V.; Benhar, I.; Vexler, A. Targeting EGFR-positive cancer cells with cetuximab-ZZ-PE38: Results of in vitro and in vivo studies. Head Neck 2013, 35, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Sawamoto, S.; Tanaka, T.; Fukuda, H.; Kondo, A. Enzyme-mediated site-specific antibody-protein modification using a ZZ domain as a linker. Bioconjug. Chem. 2010, 21, 2227–2233. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Karlstrom, A.E.; Hober, S. Covalent immunoglobulin labeling through a photoactivable synthetic Z domain. Bioconjug. Chem. 2011, 22, 2395–2403. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Järver, P.; Nygren, P.-Å.Å. Tailor-making a protein a-derived domain for efficient site-specific photocoupling to Fc of mouse IgG1. PLoS One 2013, 8, e56597:11. [Google Scholar]
- Perols, A.; Karlström, A.E. Site-specific photoconjugation of antibodies using chemically synthesized IgG-binding domains. Bioconjug. Chem. 2014, 25, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.Z.; Tsourkas, A. Optimization of photoactive protein Z for fast and efficient site-specific conjugation of native IgG. Bioconjug. Chem. 2014, 25, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Lee, J.M.; Kim, J.W.; Yoon, J.; Cho, H.; Chung, B.H. Photoactivable antibody binding protein: site-selective and covalent coupling of antibody. Anal. Chem. 2009, 81, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, K.; Pavlinkova, G.; Levy, S.; Pokkuluri, P.R.; Schiffer, M.; Haley, B.E.; Kohler, H. Novel unconventional binding site in the variable region of immunoglobulins. Proc. Natl. Acad. Sci. USA 1996, 93, 6019–6024. [Google Scholar] [CrossRef] [PubMed]
- Pavlinkova, G.; Rajagopalan, K.; Muller, S.; Chavan, A.; Sievert, G.; Lou, D.; O'Toole, C.; Haley, B.; Kohler, H. Site-specific photobiotinylation of immunoglobulins, fragments and light chain dimers. J. Immunol. Methods 1997, 201, 77–88. [Google Scholar] [CrossRef]
- Pavlinkova, G.; Lou, D.; Kohler, H. Site-specific photobiotinylation of antibodies, light chains, and immunoglobulin fragments. Methods 2000, 22, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Alves, N.J.; Champion, M.M.; Stefanick, J.F.; Handlogten, M.W.; Moustakas, D.T.; Shi, Y.; Shaw, B.F.; Navari, R.M.; Kiziltepe, T.; Bilgicer, B. Selective photocrosslinking of functional ligands to antibodies via the conserved nucleotide binding site. Biomaterials 2013, 34, 5700–5710. [Google Scholar] [CrossRef] [PubMed]
- Alves, N.J.; Mustafaoglu, N.; Bilgicer, B. Conjugation of a reactive thiol at the nucleotide binding site for site-specific antibody functionalization. Bioconjug. Chem. 2014, 25, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Lerner, R.; Barbas, C. Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995, 270, 1797–1800. [Google Scholar] [CrossRef] [PubMed]
- Barbas, C.; Heine, A.; Zhong, G.; Hoffmann, T.; Gramatikova, S.; Björnestedt, R.; List, B.; Anderson, J.; Stura, E.; Wilson, I.; et al. Immune versus natural selection: antibody aldolases with enzymic rates but broader scope. Science 1997, 278, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Almer, H.; Lerner, R.A.; Barbas, C.F. Catalytic single-chain antibodies possessing β-lactamase activity selected from a phage displayed combinatorial library using a mechanism-based inhibitor. Tetrahedron Lett. 1999, 40, 8063–8066. [Google Scholar] [CrossRef]
- Tanaka, F.; Barbas, C. Reactive immunization: A unique approach to catalytic antibodies. J. Immunol. Methods 2002, 269, 67–79. [Google Scholar] [CrossRef]
- Sinha, S.C.; Das, S.; Li, L.S.; Lerner, R.A.; Barbas, C.F., 3rd. Preparation of integrin alpha(v)beta3-targeting Ab 38C2 constructs. Nat. Protoc. 2007, 2, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Laurent, O.; Lappe, R. CovX-Bodies. In Fusion Protein Technologies for Biopharmaceuticals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 571–582. [Google Scholar]
- Rader, C. Chemically programmed antibodies. Trends Biotechnol. 2014, 32, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Rader, C.; Sinha, S.; Popkov, M.; Lerner, R.; Barbas, C. Chemically programmed monoclonal antibodies for cancer therapy: adaptor immunotherapy based on a covalent antibody catalyst. Proc. Natl. Acad. Sci. USA 2003, 100, 5396–5400. [Google Scholar] [CrossRef] [PubMed]
- Doppalapudi, V.; Tryder, N.; Li, L.; Aja, T.; Griffith, D.; Liao, F.-F.; Roxas, G.; Ramprasad, M.; Bradshaw, C.; Barbas, C. Chemically programmed antibodies: Endothelin receptor targeting CovX-Bodies. Bioorg. Med. Chem. Lett. 2007, 17, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Coronella, J.; Li, L.; Johnson, K.; Pirie-Shepherd, S.; Roxas, G.; Levin, N. Selective activity against proliferating tumor endothelial cells by CVX-22, a thrombospondin-1 mimetic CovX-Body. Anticancer Res. 2009, 29, 2243–2252. [Google Scholar] [PubMed]
- Doppalapudi, V.; Huang, J.; Liu, D.; Jin, P.; Liu, B.; Li, L.; Desharnais, J.; Hagen, C.; Levin, N.; Shields, M.; et al. Chemical generation of bispecific antibodies. Proc. Natl. Acad. Sci. USA 2010, 107, 22611–22616. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Leedom, T.A.; Do, J.; Huang, H.; Lai, J.; Johnson, K.; Osothprarop, T.F.; Rizzo, J.D.; Doppalapudi, V.R.; Bradshaw, C.W.; et al. Antitumor efficacy of a thrombospondin 1 mimetic CovX-body. Transl. Oncol. 2011, 4, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.K.; Bajjuri, K.M.; Forsyth, J.S.; Das, S.; Hassenpflug, W.; Huang, Z.Z.; Lerner, R.A.; Felding-Habermann, B.; Sinha, S.C. Chemically programmed antibodies targeting multiple alpha(v) integrins and their effects on tumor-related functions in vitro. Bioconjug. Chem. 2011, 22, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.K.; Liu, Y.; Liu, C.; Lerner, R.A.; Sinha, S.C. Synthesis and evaluation of the aldolase antibody-derived chemical-antibodies targeting alpha5beta1 integrin. Mol. Pharm. 2013, 10, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Goswami, R.K.; Liu, C.; Sinha, S.C. Chemically programmed bispecific antibody targeting legumain protease and alphavbeta3 integrin mediates strong antitumor effects. Mol. Pharm. 2015, 12, 2544–2550. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Inokuma, T.; Otsubo, N.; Burton, D.R.; Barbas, C.F., 3rd. Chemically programmed antibodies as HIV-1 attachment inhibitors. ACS Med. Chem. Lett. 2013, 4, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Toda, N.; Carrillo, N.; Thornburg, N.J.; Crowe, J.E., Jr.; Barbas, C.F., 3rd. A chemically programmed antibody is a long-lasting and potent inhibitor of influenza neuraminidase. ChemBioChem 2012, 13, 2191–2195. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Brady, K.; Brown, A.; Davey, D.; Feng, L.; Huang, H.; Liu, D.; Malet, L.; McMurray, G.; Phelan, A.; et al. Kappa agonist CovX-Bodies. Bioorg. Med. Chem. Lett. 2012, 22, 4173–4178. [Google Scholar] [CrossRef] [PubMed]
- Palanki, M.S.S.; Bhat, A.; Bolanos, B.; Brunel, F.; Del Rosario, J.; Dettling, D.; Horn, M.; Lappe, R.; Preston, R.; Sievers, A.; et al. Development of a long acting human growth hormone analog suitable for once a week dosing. Bioorg. Med. Chem. Lett. 2013, 23, 402–406. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dennler, P.; Fischer, E.; Schibli, R. Antibody Conjugates: From Heterogeneous Populations to Defined Reagents. Antibodies 2015, 4, 197-224. https://0-doi-org.brum.beds.ac.uk/10.3390/antib4030197
Dennler P, Fischer E, Schibli R. Antibody Conjugates: From Heterogeneous Populations to Defined Reagents. Antibodies. 2015; 4(3):197-224. https://0-doi-org.brum.beds.ac.uk/10.3390/antib4030197
Chicago/Turabian StyleDennler, Patrick, Eliane Fischer, and Roger Schibli. 2015. "Antibody Conjugates: From Heterogeneous Populations to Defined Reagents" Antibodies 4, no. 3: 197-224. https://0-doi-org.brum.beds.ac.uk/10.3390/antib4030197