
What Makes A Bacterial Oral Vaccine a Strong Inducer of High-Affinity IgA Responses?

{kind=link}
Abstract
:1. Introduction
2. Models of Oral Immunogenicity
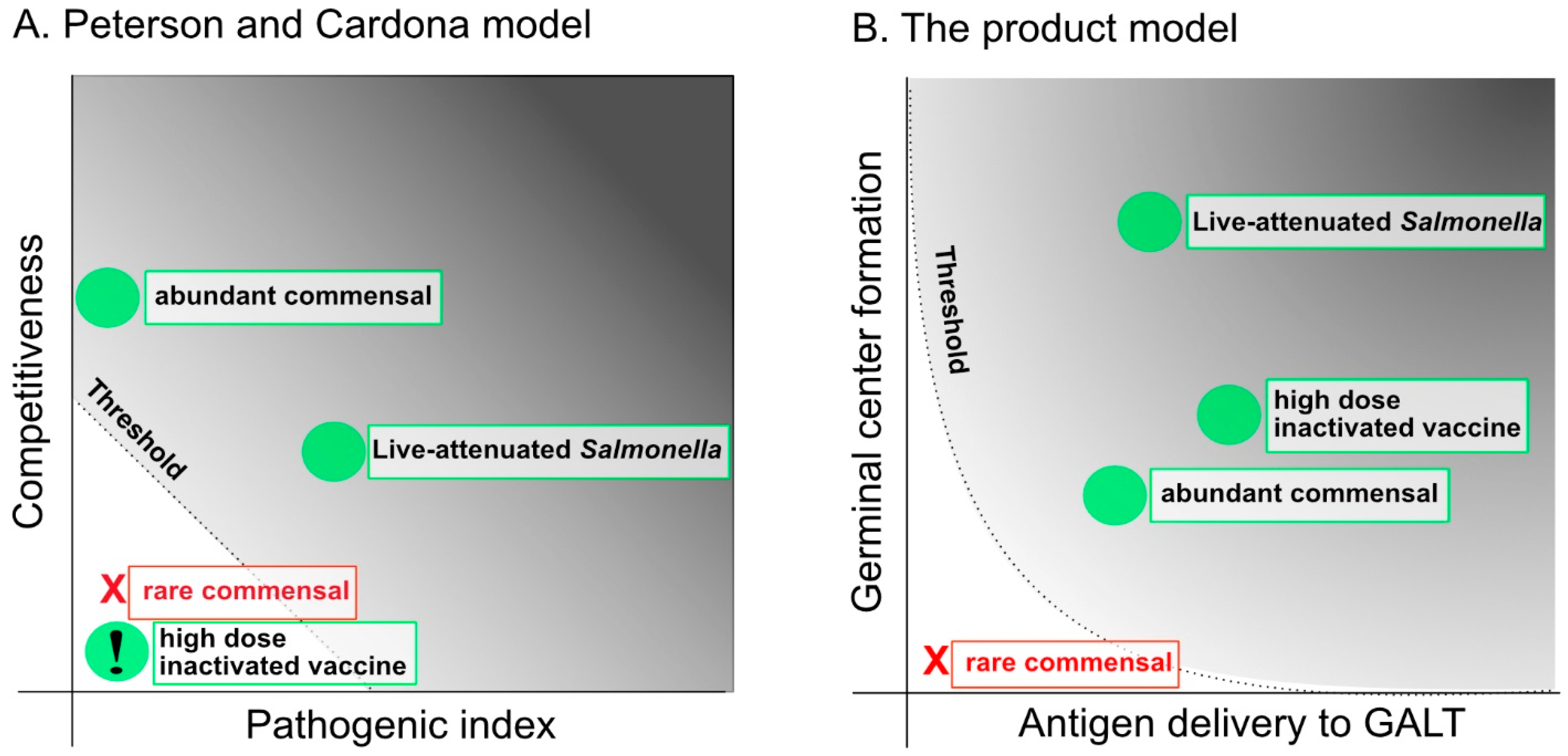
2.1. The Capacity to Compete with the Microbiota and the Pathogenic Index: Attenuation and Over-Attenuation
2.2. Novel Strategies to Uncouple Competitiveness from Pathology: Auxotrophy and Delayed Attenuation in Vivo
2.3. Can the “Pathogenic Index” Take a Zero Value?
2.4. The Tendency of Antigen to Enter the GALT: Importance of Being Particulate
2.5. The Requirement for Oral Adjuvants
2.6. The Tendency of Antigen to Reach GALT: The Role of Pre-existing Inflammation, Microbiota Composition, and Age
2.7. “Uniqueness”: Is There Evidence for Cross-Tolerance and Immunodominance in the Intestine?
3. Conclusions: The Perfect Oral Vaccine

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Macpherson, A.J.; Uhr, T. Compartmentalization of the mucosal immune responses to commensal intestinal bacteria. Ann. NY Acad. Sci. 2004, 1029, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Martinoli, C.; Chiavelli, A.; Rescigno, M. Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity 2007, 27, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Date, K.A.; Bentsi-Enchill, A.; Marks, F.; Fox, K. Typhoid fever vaccination strategies. Vaccine 2015, 33 (Suppl. 3), C55–C61. [Google Scholar] [CrossRef] [PubMed]
- Kabir, S. Critical analysis of compositions and protective efficacies of oral killed cholera vaccines. Clin. Vaccine Immunol. 2014, 21, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Cardona, R.A. Specificity of the adaptive immune response to the gut microbiota. Adv. Immunol. 2010, 107, 71–107. [Google Scholar]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, 2177–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef]
- Sztein, M.B.; Salerno-Goncalves, R.; McArthur, M.A. Complex adaptive immunity to enteric fevers in humans: lessons learned and the path forward. Front. Immunol. 2014, 5, 516. [Google Scholar] [PubMed]
- Bishop, A.L.; Camilli, A. Vibrio cholerae: Lessons for mucosal vaccine design. Expert Rev. Vaccines 2011, 10, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.A.; Angel, J.; Greenberg, H.B. Immunity and correlates of protection for rotavirus vaccines. Vaccine 2006, 24, 2718–2731. [Google Scholar] [CrossRef] [PubMed]
- Viret, J.F.; Dietrich, G.; Favre, D. Biosafety aspects of the recombinant live oral Vibrio cholerae vaccine strain CVD 103-HgR. Vaccine 2004, 22, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.T.; Calderwood, S.B.; Qadri, F. Live attenuated oral cholera vaccines. Expert Rev. Vaccines 2006, 5, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Hoiseth, S.K.; Stocker, B.A. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 1981, 291, 238–239. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, T.K.; Killar, L.M.; Stocker, B.A.; Sultzer, B.M. Cellular immunity induced by avirulent Salmonella in LPS-defective C3H/HeJ mice. J. Immunol. 1984, 133, 958–961. [Google Scholar] [PubMed]
- Killar, L.M.; Eisenstein, T.K. Immunity to Salmonella typhimurium infection in C3H/HeJ and C3H/HeNCrlBR mice: Studies with an aromatic-dependent live S. typhimurium strain as a vaccine. Infect. Immun. 1985, 47, 605–612. [Google Scholar] [PubMed]
- Harrison, J.A.; Villarreal-Ramos, B.; Mastroeni, P.; Demarco de Hormaeche, R.; Hormaeche, C.E. Correlates of protection induced by live Aro- Salmonella typhimurium vaccines in the murine typhoid model. Immunology 1997, 90, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Hormaeche, C.E.; Mastroeni, P.; Harrison, J.A.; Demarco de Hormaeche, R.; Svenson, S.; Stocker, B.A.D. Protection against oral challenge three months after i.v. immunization of BALB/c mice with live Aro Salmonella typhimurium and Salmonella enteritidis vaccines is serotype (species)-dependent and only partially determined by the main LPS O antigen. Vaccine 1996, 14, 251–259. [Google Scholar] [CrossRef]
- Chatfield, S.N.; Strahan, K.; Pickard, D.; Charles, I.G.; Hormaeche, C.E.; Dougan, G. Evaluation of Salmonella typhimurium strains harbouring defined mutations in htrA and aroA in the murine salmonellosis model. Microb. Pathog. 1992, 12, 145–151. [Google Scholar] [CrossRef]
- Lowe, D.C.; Savidge, T.C.; Pickard, D.; Eckmann, L.; Kagnoff, M.F.; Dougan, G.; Chatfield, S.N. Characterization of candidate live oral Salmonella typhi vaccine strains harboring defined mutations in aroA, aroC, and htrA. Infect. Immun. 1999, 67, 700–707. [Google Scholar] [PubMed]
- Tacket, C.O.; Sztein, M.B.; Losonsky, G.A.; Wasserman, S.S.; Nataro, J.P.; Edelman, R.; Pickard, D.; Dougan, G.; Chatfield, S.N.; Levine, M.M. Safety of live oral Salmonella typhi vaccine strains with deletions in htrA and aroC aroD and immune response in humans. Infect. Immun. 1997, 65, 452–456. [Google Scholar] [PubMed]
- Simon, R.; Tennant, S.M.; Galen, J.E.; Levine, M.M. Mouse models to assess the efficacy of non-typhoidal Salmonella vaccines: Revisiting the role of host innate susceptibility and routes of challenge. Vaccine 2011, 29, 5094–5106. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, R., 3rd; Kelly, S.M. Salmonella typhimurium deletion mutants lacking adenylate cyclase and cyclic AMP receptor protein are avirulent and immunogenic. Infect. Immun. 1987, 55, 3035–3043. [Google Scholar] [PubMed]
- Coe, N.E.; Wood, R.L. The effect of exposure to a delta cya/delta crp mutant of Salmonella typhimurium on the subsequent colonization of swine by the wild-type parent strain. Vet. Microbiol. 1992, 31, 207–220. [Google Scholar] [CrossRef]
- Hassan, J.O.; Curtiss, R., 3rd. Control of colonization by virulent Salmonella typhimurium by oral immunization of chickens with avirulent delta cya delta crp S. typhimurium. Res. Microbiol. 1990, 141, 839–850. [Google Scholar] [CrossRef]
- Hassan, J.O.; Curtiss, R., 3rd. Effect of vaccination of hens with an avirulent strain of Salmonella typhimurium on immunity of progeny challenged with wild-Type Salmonella strains. Infect. Immun. 1996, 64, 938–944. [Google Scholar]
- Galan, J.E.; Curtiss, R., 3rd. Virulence and vaccine potential of phoP mutants of Salmonella typhimurium. Microb. Pathog. 1989, 6, 433–443. [Google Scholar] [CrossRef]
- Hohmann, E.L.; Oletta, C.A.; Miller, S.I. Evaluation of a phoP/phoQ-deleted, aroA-deleted live oral Salmonella typhi vaccine strain in human volunteers. Vaccine 1996, 14, 19–24. [Google Scholar] [CrossRef]
- Matsui, H.; Isshiki, Y.; Eguchi, M.; Ogawa, Y.; Shimoji, Y. Evaluation of the live vaccine efficacy of virulence plasmid-cured, and phoP- or aroA-deficient Salmonella enterica serovar Typhimurium in mice. J. Vet. Med. Sci. 2015, 77, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Suzuki, M.; Isshiki, Y.; Kodama, C.; Eguchi, M.; Kikuchi, Y.; Motokawa, K.; Takaya, A.; Tomoyasu, T.; Yamamoto, T. Oral immunization with ATP-dependent protease-deficient mutants protects mice against subsequent oral challenge with virulent Salmonella enterica serovar typhimurium. Infect. Immun. 2003, 71, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Dilts, D.A.; Riesenfeld-Örn, I.; Fulginiti, J.P.; Ekwall, E.; Granert, C.; Nonenmacher, J.; Brey, R.N.; Cryz, S.J.; Karlsson, K.; Bergman, K.; et al. Phase I clinical trials of aroA aroD and aroA aroD htrA attenuated S. typhi vaccines; effect of formulation on safety and immunogenicity. Vaccine 2000, 18, 1473–1484. [Google Scholar] [CrossRef]
- Hone, D.M.; Tacket, C.O.; Harris, A.M.; Kay, B.; Losonsky, G.; Levine, M.M. Evaluation in volunteers of a candidate live oral attenuated Salmonella typhi vector vaccine. J. Clin. Invest. 1992, 90, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Tacket, C.O.; Hone, D.M.; Curtiss, R., 3rd; Kelly, S.M.; Losonsky, G.; Guers, L.; Harris, A.M.; Edelman, R.; Levine, M.M. Comparison of the safety and immunogenicity of delta aroC delta aroD and delta cya delta crp Salmonella typhi strains in adult volunteers. Infect. Immun. 1992, 60, 536–541. [Google Scholar] [PubMed]
- Casanova, J.L.; Jouanguy, E.; Lamhamedi, S.; Blanche, S.; Fischer, A. Immunological conditions of children with BCG disseminated infection. Lancet 1995, 346, 581. [Google Scholar] [CrossRef]
- Hesseling, A.C.; Rabie, H.; Marais, B.J.; Manders, M.; Lips, M.; Schaaf, H.S.; Gie, R.P.; Cotton, M.F.; van Helden, P.D.; Warren, R.M.; et al. Bacille Calmette-Guerin vaccine-induced disease in HIV-infected and HIV-uninfected children. Clin. Infect. Dis. 2006, 42, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Felmy, B.; Songhet, P.; Slack, E.M.C.; Müller, A.J.; Kremer, M.; Van Maele, L.; Cayet, D.; Heikenwalder, M.; Sirard, J.-C.; Hardt, W.D. NADPH oxidase deficient mice develop colitis and bacteremia upon infection with normally avirulent, TTSS-1- and TTSS-2-deficient Salmonella Typhimurium. PLoS ONE 2013, 8, e77204. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, V.; Pati, N.B.; Chandel, H.S.; Sahoo, S.S.; Saha, B.; Suar, M. Evaluation of Salmonella enterica serovar Typhimurium TTSS-2 deficient fur mutant as safe live-attenuated vaccine candidate for immunocompromised mice. PLoS ONE 2012, 7, e52043. [Google Scholar] [CrossRef] [PubMed]
- Periaswamy, B.; Maier, L.; Vishwakarma, V.; Slack, E.; Kremer, M.; Andrews-Polymenis, H.L.; McClelland, M.; Grant, A.J.; Suar, M.; Hardt, W.-D. Live attenuated S. Typhimurium vaccine with improved safety in immuno-compromised mice. PLoS ONE 2012, 7, e45433. [Google Scholar] [CrossRef] [PubMed]
- Pati, N.B.; Vishwakarma, V.; Selvaraj, S.K.; Dash, S.; Saha, B.; Singh, N.; Suar, M. Salmonella Typhimurium TTSS-2 deficient mig-14 mutant shows attenuation in immunocompromised mice and offers protection against wild-type Salmonella Typhimurium infection. BMC Microbiol. 2013, 13, 236. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, V.; Pati, N.B.; Ray, S.; Das, S.; Suar, M. TTSS2-deficient hha mutant of Salmonella Typhimurium exhibits significant systemic attenuation in immunocompromised hosts. Virulence 2014, 5, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Diard, M.; Stecher, B.; Hardt, W.-D. The streptomycin mouse model for Salmonella diarrhea: functional analysis of the microbiota, the pathogen's virulence factors, and the host's mucosal immune response. Immunol. Rev. 2012, 245, 56–83. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Stecher, B.; Freed, N.E.; Songhet, P.; Hardt, W.-D.; Doebeli, M. Self-destructive cooperation mediated by phenotypic noise. Nature 2008, 454, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Diard, M.; Garcia, V.; Maier, L.; Remus-Emsermann, M.N.P.; Regoes, R.R.; Ackermann, M.; Hardt, W.-D. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 2013, 494, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Hardt, W.D. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 2011, 14, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Germanier, R.; Fuer, E. Isolation and characterization of Gal E mutant Ty 21a of Salmonella typhi: a candidate strain for a live, oral typhoid vaccine. J. Infect. Dis. 1975, 131, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Cisarb, J.O.; Polyc, F.; Yangb, J.; Albanesea, J.; Dharmasenaa, M.; Waia, T.; Guerryc, P.; Kopeckoa, D.J. Genome Sequence of Salmonella enterica Serovar Typhi Oral Vaccine Strain Ty21a. Genome Announc. 2013, 1, e00650-13. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Regoes, R.R.; Dolowschiak, T.; Wotzka, S.Y.; Lengefeld, J.; Slack, E.; Grant, A.J.; Ackermann, M.; Hardt, W.-D. Cecum lymph node dendritic cells harbor slow-growing bacteria phenotypically tolerant to antibiotic treatment. PLoS Biol. 2014, 12, e1001793. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.J.; Morgan, F.J.E.; McKinley, T.J.; Foster, G.L.; Maskell, D.J.; Mastroeni, P. Attenuated Salmonella Typhimurium lacking the pathogenicity island-2 type 3 secretion system grow to high bacterial numbers inside phagocytes in mice. PLoS Pathog. 2012, 8, e1003070. [Google Scholar] [CrossRef] [PubMed]
- Endt, K.; Stecher, B.; Chaffron, S.; Slack, E.; Tchitchek, N.; Benecke, A.; Van Maele, L.; Sirard, J.-C.; Mueller, A.J.; Heikenwalder, M.; et al. The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog. 2010, 6, e1001097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtiss, R., 3rd; Wanda, S.-Y.; Gunn, B.M.; Zhang, X.; Tinge, S.A.; Ananthnarayan, V.; Mo, H.; Wang, S.; Kong, W. Salmonella enterica serovar typhimurium strains with regulated delayed attenuation in vivo. Infect. Immun. 2009, 77, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Wanda, S.-Y.; Gunn, B.M.; Zhang, X.; Tinge, S.A.; Ananthnarayan, V.; Mo, H.; Wang, S.; Kong, W. Regulated programmed lysis of recombinant Salmonella in host tissues to release protective antigens and confer biological containment. Proc. Natl. Acad. Sci. USA 2008, 105, 9361–9366. [Google Scholar] [CrossRef]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Curtiss, R., 3rd. Development of Vaccines Using Live Vectors. Vaccines (Basel) 2014, 2, 49–88. [Google Scholar] [CrossRef] [PubMed]
- Hapfelmeier, S.; Lawson, M.A.E.; Slack, E.; Kirundi, J.K.; Stoel, M.; Heikenwalder, M.; Cahenzli, J.; Velykoredko, Y.; Balmer, M.L.; Endt, K.; et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010, 328, 1705–1709. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Taguchi, K.; Tokoro, S.; Tatsuno, I.; Mori, H. Adhesion of enterohemorrhagic Escherichia coli O157:H7 to the intestinal epithelia is essential for inducing secretory IgA antibody production in the intestine of mice. Biol. Pharm. Bull. 2014, 37, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Garside, P.; Millington, O.; Smith, K.M. The anatomy of mucosal immune responses. Ann. NY Acad. Sci. 2004, 1029, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Langridge, W.; Denes, B.; Fodor, I. Cholera toxin B subunit modulation of mucosal vaccines for infectious and autoimmune diseases. Curr. Opin. Investig. Drugs 2010, 11, 919–928. [Google Scholar] [PubMed]
- Svennerholm, A.M.; Holmgren, J. Synergistic protective effect in rabbits of immunization with Vibrio cholerae lipopolysaccharide and toxin/toxoid. Infect. Immun. 1976, 13, 735–740. [Google Scholar] [PubMed]
- Black, R.E.; Levine, M.M.; Clements, M.L.; Young, C.R.; Svennerholm, A.M.; Holmgren, J. Protective efficacy in humans of killed whole-vibrio oral cholera vaccine with and without the B subunit of cholera toxin. Infect. Immun. 1987, 55, 1116–1120. [Google Scholar] [PubMed]
- Barthel, M.; Hapfelmeier, S.; Quintanilla-Martínez, L.; Kremer, M.; Rohde, M.; Hogardt, M.; Pfeffer, K.; Rüssmann, H.; Hardt, W.-D. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 2003, 71, 2839–2858. [Google Scholar] [CrossRef] [PubMed]
- Hapfelmeier, S.; Hardt, W.D. A mouse model for S. typhimurium-induced enterocolitis. Trends Microbiol. 2005, 13, 497–503. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Ehrbar, K.; Stecher, B.; Barthel, M.; Kremer, M.; Hardt, W.-D. Role of the Salmonella pathogenicity island 1 effector proteins SipA, SopB, SopE, and SopE2 in Salmonella enterica subspecies 1 serovar Typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 2004, 72, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Neutra, M.R.; Mantis, N.J.; Kraehenbuhl, J.P. Collaboration of epithelial cells with organized mucosal lymphoid tissues. Nat. Immunol. 2001, 2, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Donaldson, D.S.; Ohno, H.; Williams, I.R.; Mahajan, A. Microfold (M) cells: Important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013, 6, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.E.; Lickteig, D.J.; Plunkett, K.N.; Ryerse, J.S.; Konjufca, V. The Uptake of Soluble and Particulate Antigens by Epithelial Cells in the Mouse Small Intestine. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Strindelius, L.; Degling Wikingsson, L.; Sjoholm, I. Extracellular antigens from Salmonella enteritidis induce effective immune response in mice after oral vaccination. Infect. Immun. 2002, 70, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Strindelius, L.; Filler, M.; Sjoholm, I. Mucosal immunization with purified flagellin from Salmonella induces systemic and mucosal immune responses in C3H/HeJ mice. Vaccine 2004, 22, 3797–3808. [Google Scholar] [CrossRef] [PubMed]
- Strindelius, L.; Folkesson, A.; Normark, S.; Sjöholm, I. Immunogenic properties of the Salmonella atypical fimbriae in BALB/c mice. Vaccine 2004, 22, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L.; da Cunha, A.P.; Quintana, F.; Wu, H. Oral tolerance. Immunol. Rev. 2011, 241, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.E.; Konjufca, V.H. Per-oral immunization with antigen-conjugated nanoparticles followed by sub-cutaneous boosting immunization induces long-lasting mucosal and systemic antibody responses in mice. PLoS ONE 2015, 10, e0118067. [Google Scholar] [CrossRef] [PubMed]
- De Smet, R.; Demoor, T.; Verschuere, S.; Dullaers, M.; Ostroff, G.R.; Leclercq, G.; Allais, L.; Pilette, C.; Dierendonck, M.; De Geest, B.G.; et al. beta-Glucan microparticles are good candidates for mucosal antigen delivery in oral vaccination. J. Control. Release 2013, 172, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Rochereau, N.; Drocourt, D.; Perouzel, E.; Pavot, V.; Redelinghuys, P.; Brown, G.D.; Tiraby, G.; Roblin, X.; Verrier, B.; Genin, C.; et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013, 11, e1001658. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Gupta, P.N. Implication of nanoparticles/microparticles in mucosal vaccine delivery. Expert Rev. Vaccines 2007, 6, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, (Pt. 1). 1–13. [Google Scholar] [CrossRef] [PubMed]
- Glenn, G.M.; Francis, D.H.; Danielsen, E.M. Toxin-mediated effects on the innate mucosal defenses: implications for enteric vaccines. Infect. Immun. 2009, 77, 5206–5215. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; O’Neal, C.J.; Mitchell, D.D.; Robien, M.A.; Zhang, Z.; Pickens, J.C.; Tan, X.-J.; Korotkov, K.; Roach, C.; Krumm, B.; et al. Structural biology and structure-based inhibitor design of cholera toxin and heat-labile enterotoxin. Int. J. Med. Microbiol. 2004, 294, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lycke, N.; Bemark, M. Mucosal adjuvants and long-term memory development with special focus on CTA1-DD and other ADP-ribosylating toxins. Mucosal Immunol. 2010, 3, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.-Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Mosovsky, K.L.; Ross, A.C. Retinoic acid and alpha-galactosylceramide regulate the expression of costimulatory receptors and transcription factors responsible for B cell activation and differentiation. Immunobiology 2013, 218, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 2006, 314, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Pantazi, E.; Iwata, M.; Eksteen, B.; Song, S.-Y.; Junt, T.; Senman, B.; Otipoby, K.L.; Yokota, A.; Takeuchi, H.; Ricciardi-Castagnoli, P.; et al. Cutting Edge: Retinoic Acid Signaling in B Cells Is Essential for Oral Immunization and Microflora Composition. J. Immunol. 2015, 314, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.I.; Friedrichsen, M.; Boelter, J.; Lyszkiewicz, M.; Kremmer, E.; Pabst, O.; Förster, R. Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J. Clin. Invest. 2011, 121, 3051–3061. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Sande, J.L.; Pufnock, J.S.; Blattman, J.N.; Greenberg, P.D. Retinoic acid as a vaccine adjuvant enhances CD8+ T cell response and mucosal protection from viral challenge. J. Virol. 2011, 85, 8316–8327. [Google Scholar] [CrossRef] [PubMed]
- Milpied, P.J.; McHeyzer-Williams, M.G. High-affinity IgA needs TH17 cell functional plasticity. Nat. Immunol. 2013, 14, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Grassly, N.C.; Jafari, H.; Bahl, S.; Durrani, S.; Wenger, J.; Sutter, R.W.; Aylward, R.B. Mucosal immunity after vaccination with monovalent and trivalent oral poliovirus vaccine in India. J. Infect. Dis. 2009, 200, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Jiang, V.; Jafari, H.; Bahl, S.; Durrani, S.; Wenger, J.; Sutter, R.W.; Aylward, R.B. Performance of rotavirus vaccines in developed and developing countries. Hum. Vaccin 2010, 6, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Lopman, B.A.; Pitzer, V.E.; Sarkar, R.; Gladstone, B.; Patel, M.; Glasser, J.; Gambhir, M.; Atchison, C.; Grenfell, B.; Edmunds, W.J.; et al. Understanding reduced rotavirus vaccine efficacy in low socio-economic settings. PLoS ONE 2012, 7, e41720. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.M. Immunogenicity and efficacy of oral vaccines in developing countries: Lessons from a live cholera vaccine. BMC Biol. 2010, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Iwata, M.; von Andrian, U.H. Vitamin effects on the immune system: Vitamins A and D take centre stage. Nat. Rev. Immunol. 2008, 8, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.R.; De Calisto, J.; Simmons, N.L.; Cruz, A.N.; Villablanca, E.J.; Mora, J.R.; Barouch, D.H. Vitamin A deficiency impairs vaccine-elicited gastrointestinal immunity. J. Immunol. 2011, 187, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Ibs, K.H.; Rink, L. Zinc-altered immune function. J. Nutr. 2003, 133 (Suppl. 1), 1452S–1456S. [Google Scholar] [PubMed]
- Korpe, P.S.; Petri, W.A., Jr. Environmental enteropathy: Critical implications of a poorly understood condition. Trends Mol. Med. 2012, 18, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Keusch, G.T.; Denno, D.M.; Black, R.E.; Duggan, C.; Guerrant, R.L.; Lavery, J.V.; Nataro, J.P.; Rosenberg, I.H.; Ryan, E.T.; Tarr, P.I.; et al. Environmental enteric dysfunction: Pathogenesis, diagnosis, and clinical consequences. Clin. Infect. Dis. 2014, 59 (Suppl. 4), S207–S212. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.P.; Sarkar-Roy, N.; Staats, H.; Ramamurthy, T.; Maiti, S.; Chowdhury, G.; Whisnant, C.C.; Narayanasamy, K.; Wagener, D.K. Genomic correlates of variability in immune response to an oral cholera vaccine. Eur. J. Hum. Genet. 2013, 21, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Chaffron, S.; Käppeli, R.; Hapfelmeier, S.; Freedrich, S.; Weber, T.C.; Kirundi, J.; Suar, M.; McCoy, K.D.; von Mering, C.; et al. Like will to like: abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog. 2010, 6, e1000711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.; Baldridge, M.T.; Wallace, M.A.; Burnham, C.-A.D.; Virgin, H.W.; Stappenbeck, T.S. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 2015, 521, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Endt, K.; Maier, L.; Käppeli, R.; Barthel, M.; Misselwitz, B.; Kremer, M.; Hardt, W.-D. Peroral ciprofloxacin therapy impairs the generation of a protective immune response in a mouse model for Salmonella enterica serovar Typhimurium diarrhea, while parenteral ceftriaxone therapy does not. Antimicrob. Agents Chemother. 2012, 56, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Chassaing, B.; Zhang, B.; Gewirtz, A.T. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J. Infect. Dis. 2014, 210, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, I.; John, S.M.; Bandyopadhyay, R.; Kang, G. Probiotics, antibiotics and the immune responses to vaccines. Philos. T. R. Soc. B 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Lindner, C.; Wahl, B.; Föhse, L.; Suerbaum, S.; Macpherson, A.J.; Prinz, I.; Pabst, O. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J. Exp. Med. 2012, 209, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Kole, A.; Maloy, K.J. Control of intestinal inflammation by interleukin-10. Curr. Top. Microbiol. Immunol. 2014, 380, 19–38. [Google Scholar] [PubMed]
- Bollrath, J.; Powrie, F.M. Controlling the frontier: regulatory T-cells and intestinal homeostasis. Semin. Immunol. 2013, 25, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Geuking, M.B.; Cahenzli, J.; Lawson, M.A.E.; Ng, D.C.K.; Slack, E.; Hapfelmeier, S.; McCoy, K.D.; Macpherson, A.J. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011, 34, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Lindner, C.; Thomsen, I.; Wahl, B.; Ugur, M.; Sethi, M.K.; Friedrichsen, M.; Smoczek, A.; Ott, S.; Baumann, U.; Suerbaum, S.; et al. Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat. Immunol. 2015, 16, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.; Inman, R.D. Immunodominance: A pivotal principle in host response to viral infections. Clin. Immunol. 2012, 143, 99–115. [Google Scholar] [PubMed]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moor, K.; Slack, E. What Makes A Bacterial Oral Vaccine a Strong Inducer of High-Affinity IgA Responses? Antibodies 2015, 4, 295-313. https://0-doi-org.brum.beds.ac.uk/10.3390/antib4040295
Moor K, Slack E. What Makes A Bacterial Oral Vaccine a Strong Inducer of High-Affinity IgA Responses? Antibodies. 2015; 4(4):295-313. https://0-doi-org.brum.beds.ac.uk/10.3390/antib4040295
Chicago/Turabian StyleMoor, Kathrin, and Emma Slack. 2015. "What Makes A Bacterial Oral Vaccine a Strong Inducer of High-Affinity IgA Responses?" Antibodies 4, no. 4: 295-313. https://0-doi-org.brum.beds.ac.uk/10.3390/antib4040295