Thinking outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
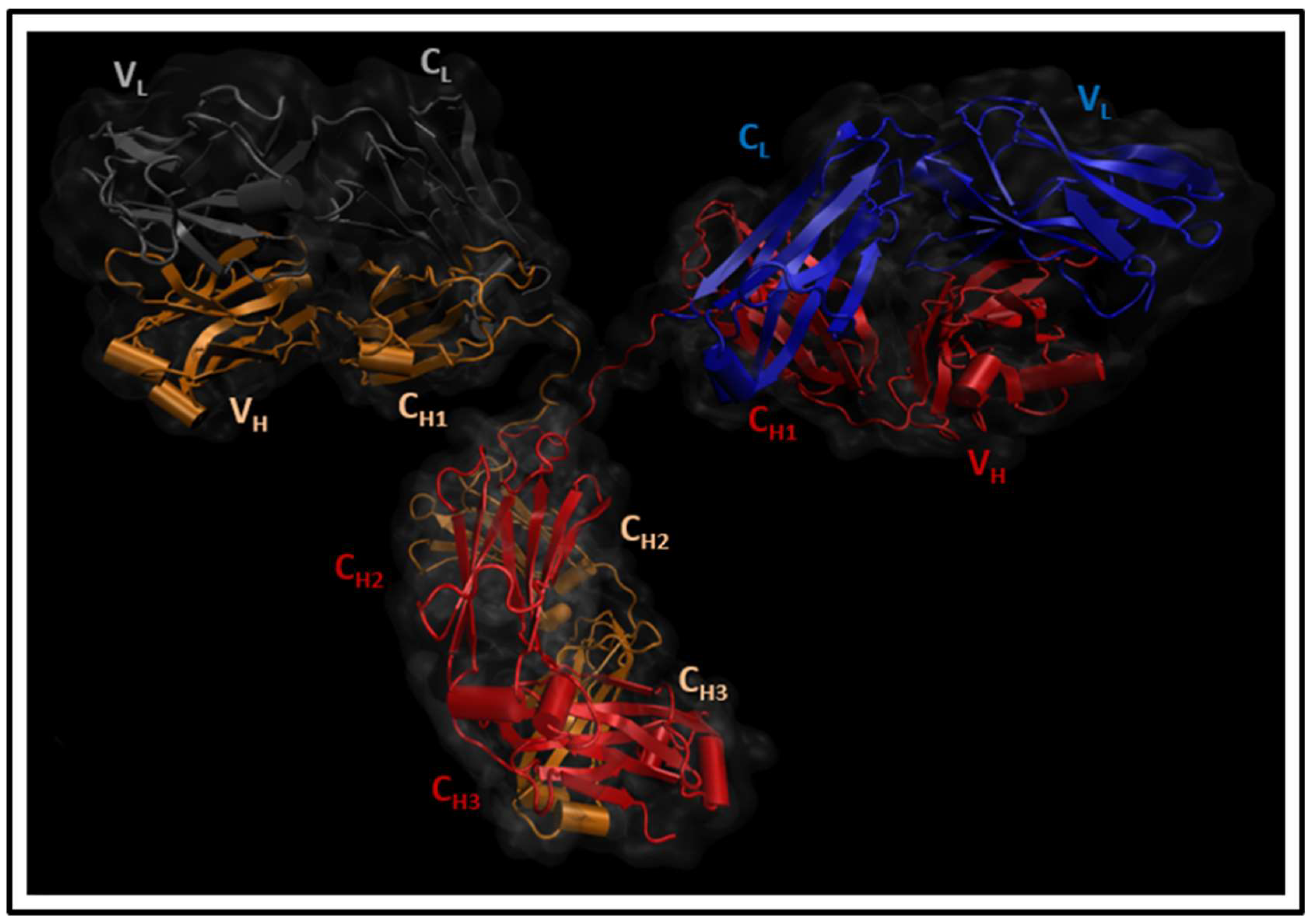
2. Materials and Methods
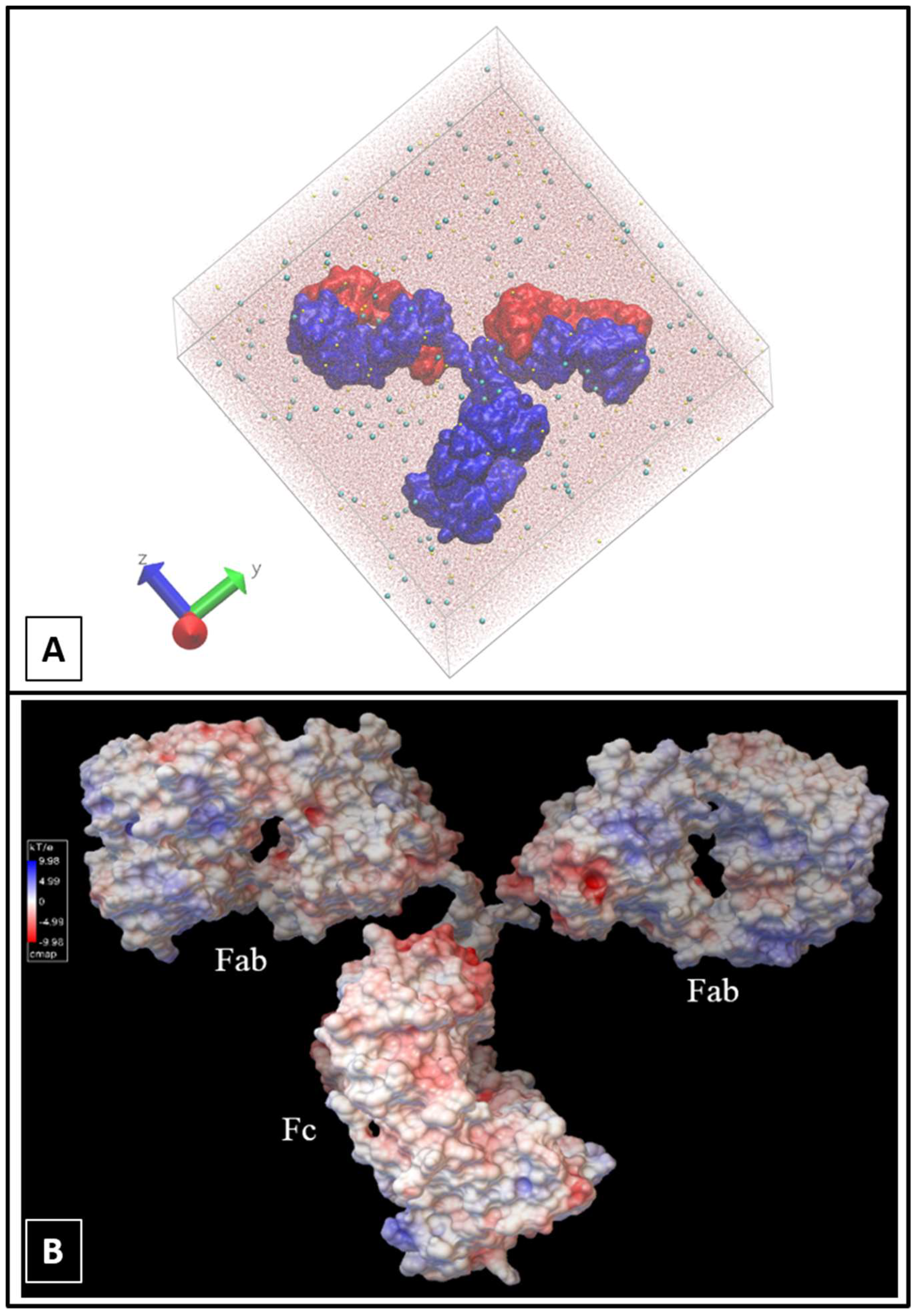
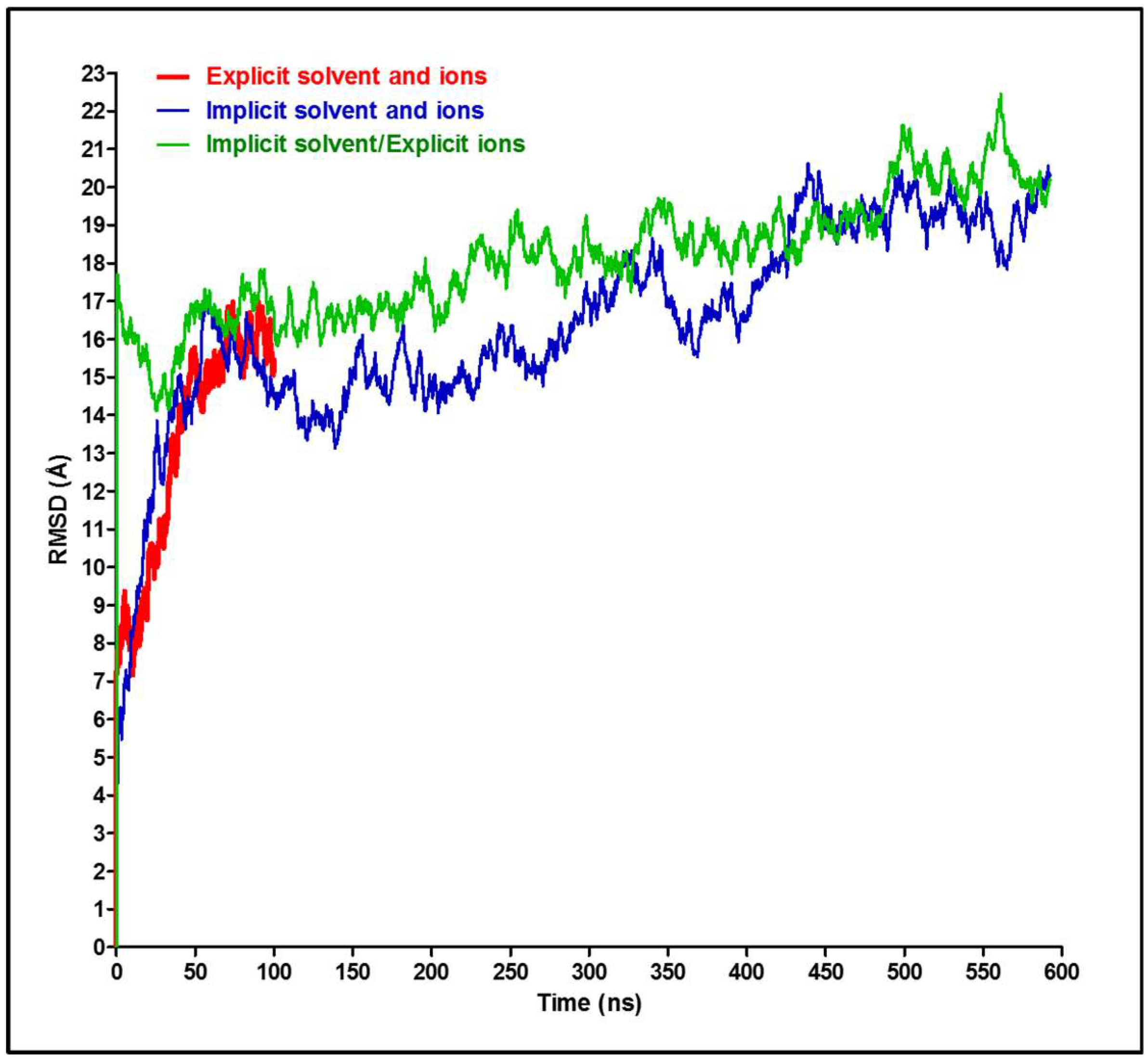
2.1. Molecular Dynamics Simulations
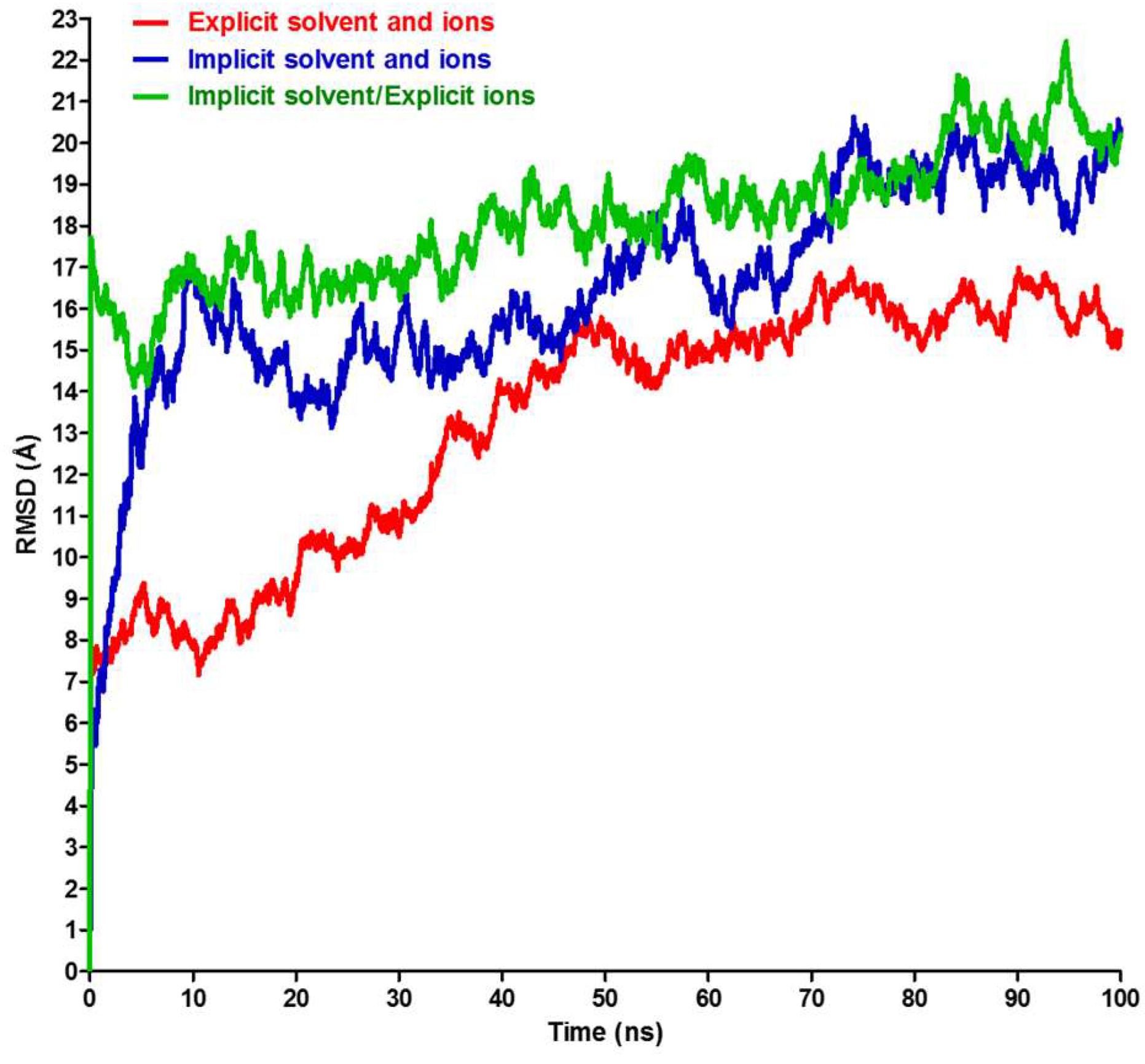
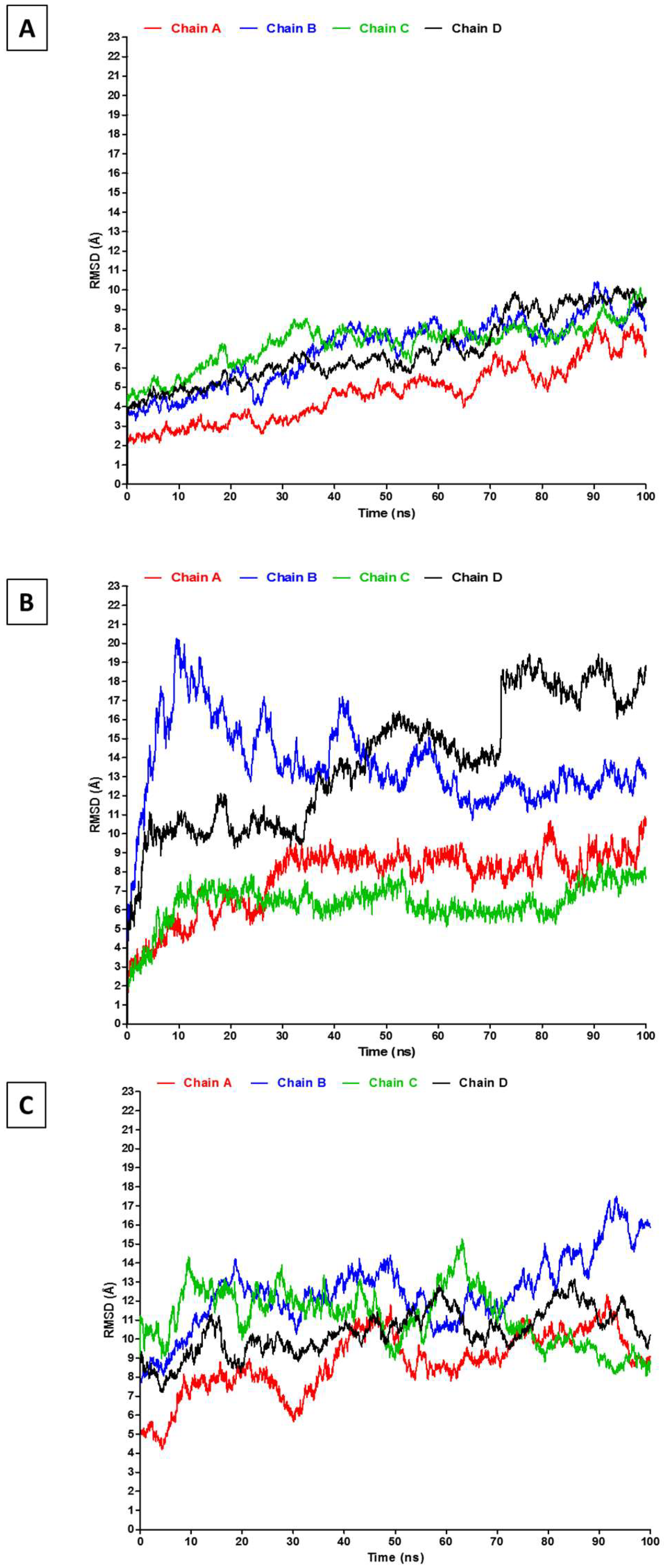
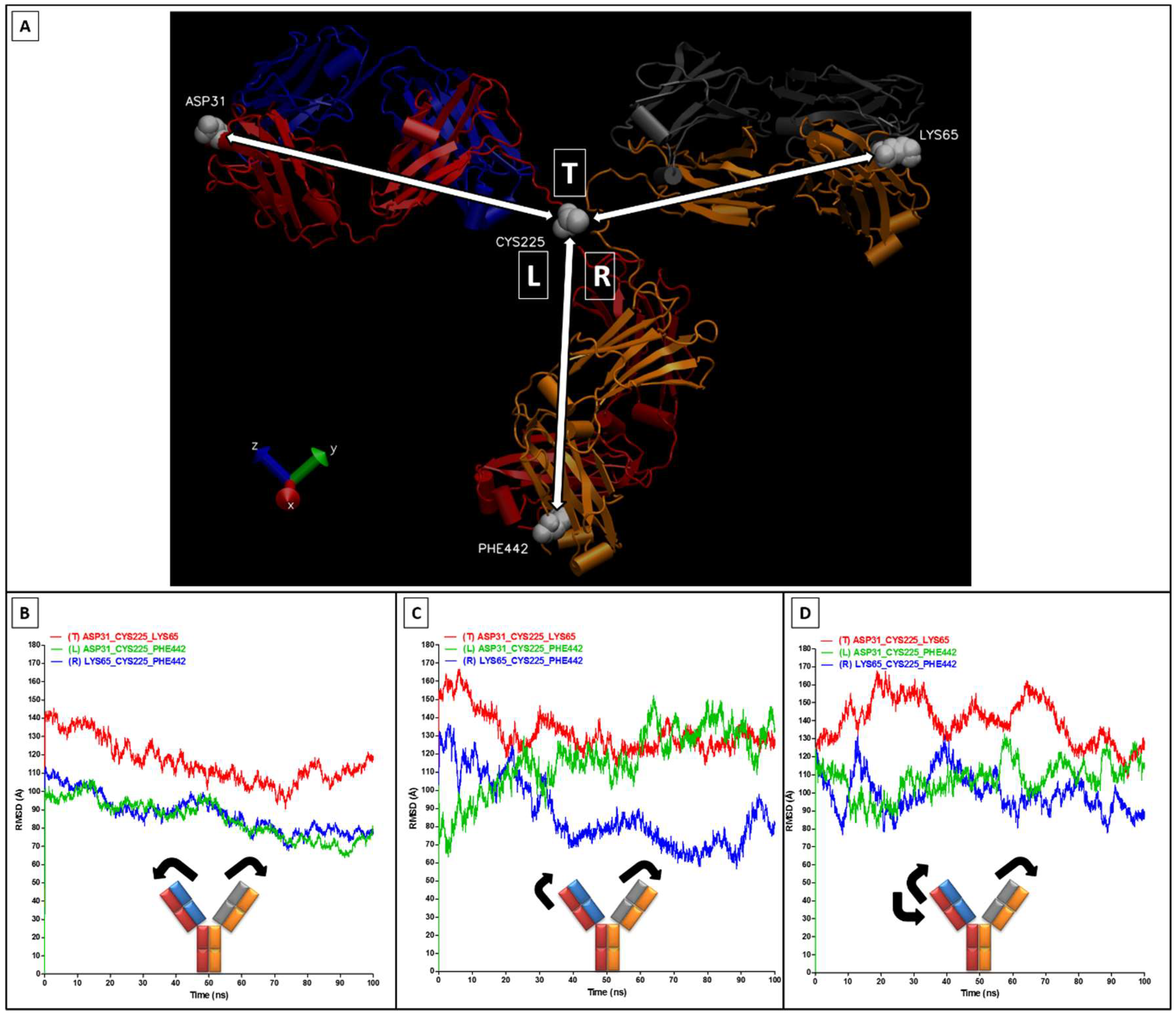
2.2. RMSD, RMSF, and Interchain Angles
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Reichert, J.M. Antibodies to watch in 2016. mAbs 2016, 8, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2018. mAbs 2018, 10, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Edelman, G.M. Dissociation of γ-globulin. J. Am. Chem. Soc. 1959, 81, 3155–3156. [Google Scholar] [CrossRef]
- Charles, A.; Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. The structure of a typical antibody molecule. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Maynard, J.; Georgiou, G. Antibody engineering. Annu. Rev. Biomed. Eng. 2000, 2, 339–376. [Google Scholar] [CrossRef] [PubMed]
- Irani, V.; Guy, A.J.; Andrew, D.; Beeson, J.G.; Ramsland, P.A.; Richards, J.S. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol. Immunol. 2015, 67, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Marcon, E.; Jain, H.; Bhattacharya, A.; Guo, H.; Phanse, S.; Pu, S.; Byram, G.; Collins, B.C.; Dowdell, E.; Fenner, M.; et al. Assessment of a method to characterize antibody selectivity and specificity for use in immunoprecipitation. Nat. Methods 2015, 12, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, G.; Semple, A.; James, J.K.; Cheung, J.K.; Shameem, M. A comparison of biophysical characterization techniques in predicting monoclonal antibody stability. mAbs 2016, 8, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Karim, M.R.; Ahsan, M.Q.; Khalipha, A.B.R.; Chowdhury, M.R.; Saifuzzaman, M. Use of computer in drug design and drug discovery: A review. Int. J. Pharm. Life Sci. 2012, 1. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. New technologies in computer-aided drug design: Toward target identification and new chemical entity discovery. Drug Discov. Today Technol. 2006, 3, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, L.; Wang, Y.; Chen, S. Molecular dynamics simulations of the adsorption of DNA segments onto graphene oxide. J. Phys. Appl. Phys. 2014, 47, 505401. [Google Scholar] [CrossRef]
- Margreitter, C.; Mayrhofer, P.; Kunert, R.; Oostenbrink, C. Antibody humanization by molecular dynamics simulations—In-silico guided selection of critical backmutations. J. Mol. Recognit. 2016, 29, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.; Chen, R.; Poblete, H.; Vergara-Jaque, A.; Riviere, J.E. Predicting Adsorption Affinities of Small Molecules on Carbon Nanotubes Using Molecular Dynamics Simulation. ACS Nano 2015, 9, 11761–11774. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Zhang, Q.; Chen, L.Y. Insights into scFv:drug binding using the molecular dynamics simulation and free energy calculation. J. Mol. Model. 2011, 17, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; Hasenhindl, C.; Obinger, C.; Oostenbrink, C. Molecular Dynamics Simulation of the Crystallizable Fragment of IgG1—Insights for the Design of Fcabs. Int. J. Mol. Sci. 2014, 15, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kumar, S.; Buck, P.M.; Singh, S.K. Impact of deglycosylation and thermal stress on conformational stability of a full length murine IgG2a monoclonal antibody: Observations from molecular dynamics simulations. Proteins 2013, 81, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Kortkhonjia, E.; Brandman, R.; Zhou, J.Z.; Voelz, V.A.; Chorny, I.; Kabakoff, B.; Patapoff, T.W.; Dill, K.A.; Swartz, T.E. Probing antibody internal dynamics with fluorescence anisotropy and molecular dynamics simulations. mAbs 2013, 5, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.P.; Huang, H.; Roy, A.; Post, C.B. Evaluating the dynamics and electrostatic interactions of folded proteins in implicit solvents. Protein Sci. Publ. Protein Soc. 2016, 25, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, M.; Brooks, C.L. Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr. Opin. Struct. Biol. 2004, 14, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Drozdetski, A.; Walker, R.C.; Onufriev, A.V. Speed of Conformational Change: Comparing Explicit and Implicit Solvent Molecular Dynamics Simulations. Biophys. J. 2015, 108, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Feig, M. Kinetics from Implicit Solvent Simulations of Biomolecules as a Function of Viscosity. J. Chem. Theory Comput. 2007, 3, 1734–1748. [Google Scholar] [CrossRef] [PubMed]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Implicit Solvent Model for Million-Atom Atomistic Simulations: Insights into the Organization of 30-nm Chromatin Fiber. J. Chem. Theory Comput. 2016, 12, 5946–5959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, K.A.; Lin, Y.-S.; Das, R.; Pande, V.S. Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. J. Chem. Theory Comput. 2012, 8, 1409–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins 2003, 53, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Kubiak-Ossowska, K.; Tokarczyk, K.; Jachimska, B.; Mulheran, P.A. Bovine Serum Albumin Adsorption at a Silica Surface Explored by Simulation and Experiment. J. Phys. Chem. B 2017, 121, 3975–3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, B.A.; Jachimska, B.; Komorek, P.; Mulheran, P.A.; Chen, Y. Lysozyme encapsulated gold nanoclusters: Effects of cluster synthesis on natural protein characteristics. Phys. Chem. Chem. Phys. PCCP 2017, 19, 7228–7235. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry (Mosc.) 1997, 36, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Carugo, O.; Pongor, S. A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci. Publ. Protein Soc. 2001, 10, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O. Statistical validation of the root-mean-square-distance, a measure of protein structural proximity. Protein Eng. Des. Sel. PEDS 2007, 20, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, J.P.; Patapoff, T.W.; Aragon, S.R. Construction, MD Simulation, and Hydrodynamic Validation of an All-Atom Model of a Monoclonal IgG Antibody. Biophys. J. 2010, 99, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Hanaki, H.; Inoue, M.; Akama, H.; Nakae, T.; Sunakawa, K.; Omura, S. Development of an Immunochromatographic Strip for Simple Detection of Penicillin-Binding Protein 2′. Clin. Vaccine Immunol. 2011, 18, 248–253. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Qaraghuli, M.M.; Kubiak-Ossowska, K.; Mulheran, P.A. Thinking outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations. Antibodies 2018, 7, 21. https://0-doi-org.brum.beds.ac.uk/10.3390/antib7030021
Al Qaraghuli MM, Kubiak-Ossowska K, Mulheran PA. Thinking outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations. Antibodies. 2018; 7(3):21. https://0-doi-org.brum.beds.ac.uk/10.3390/antib7030021
Chicago/Turabian StyleAl Qaraghuli, Mohammed M., Karina Kubiak-Ossowska, and Paul A. Mulheran. 2018. "Thinking outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations" Antibodies 7, no. 3: 21. https://0-doi-org.brum.beds.ac.uk/10.3390/antib7030021