4.2. Compounds
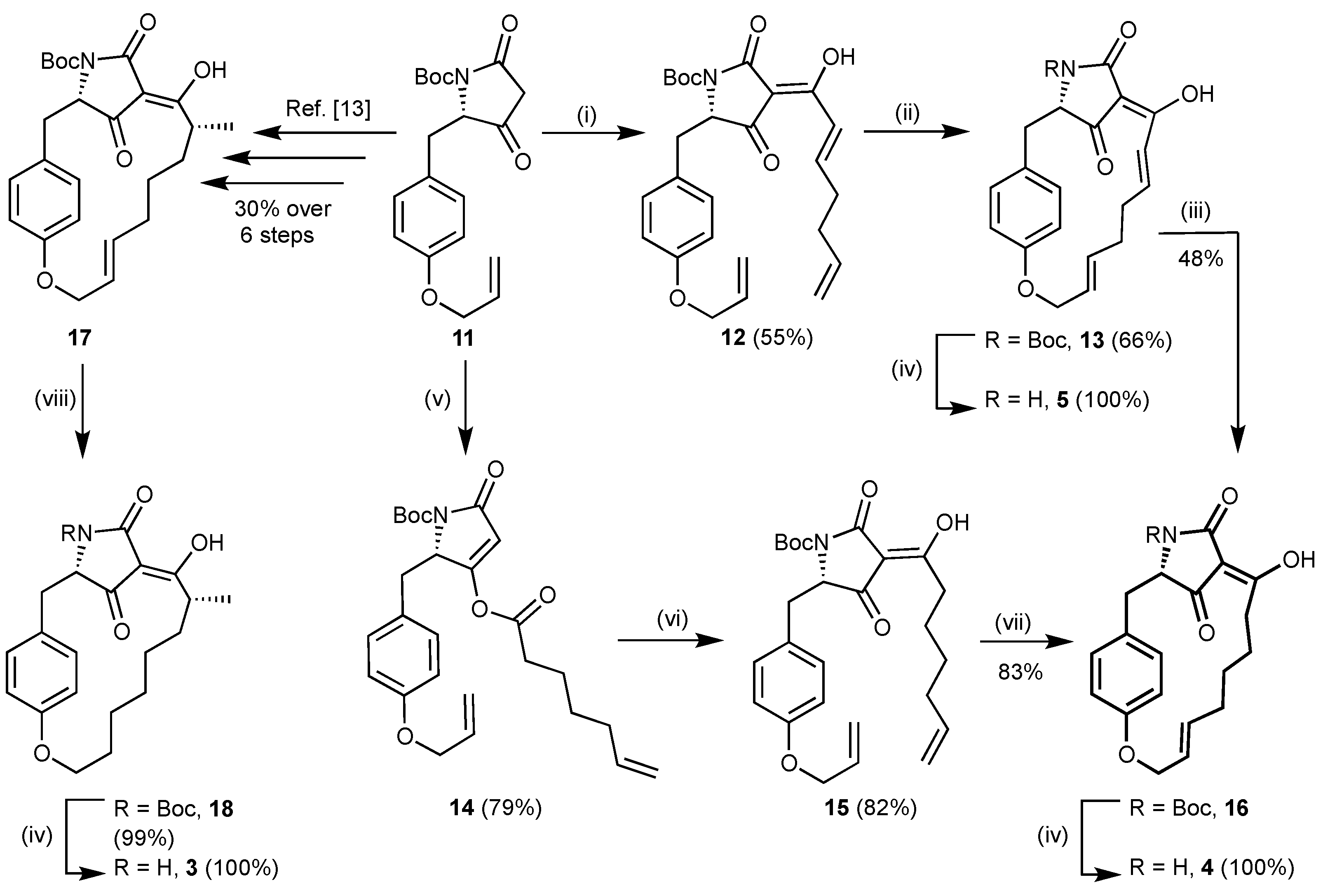
(S,Z)-tert-Butyl-2-(4-(allyloxy)benzyl)-4-((E)-1-hydroxyhepta-2,6-dien-1-ylidene)-3,5-dioxopyrrolidine-1-carboxylate (12). Tetramic acid
11 [
4] (1.90 g, 5.50 mmol, 1.10 eq) in dry THF (305 mL) was treated with ketenylidenetriphenylphosphorane (1.66 g, 5.50 mmol, 1.10 eq) in dry THF (140 mL) over 20 min while refluxing. After stirring for 2 h, KO
tBu (0.62 g, 5.50 mmol, 1.10 eq) was added. The solution was stirred for a further 20 min, before 4-pentenal (0.42 g, 5.00 mmol, 1.00 eq) in dry THF (65 mL) was added over a period of 15 min. Stirring at reflux was continued for 4 h and for 17 h at room temperature. The solvent was concentrated under reduced pressure and the crude product was dissolved in CH
2Cl
2 (300 mL). It was washed with sat. NH
4Cl solution (200 mL). The aqueous phase was extracted with CH
2Cl
2 (3 × 100 mL), the combined organic phases were washed with brine (300 mL) and dried over Na
2SO
4. Removal of the solvent and purification by column chromatography on reversed phase silica gel (RP18, 40% MeCN in H
2O + 0.1% HCOOH → 60% MeCN in H
2O + 0.1% HCOOH → 70% MeCN in H
2O + 0.1% HCOOH → 80% MeCN in H
2O + 0.1% HCOOH → 100% MeCN + 0.1% HCOOH) afforded 3-acyltetramic acid
12 (1.24 g, 2.74 mmol, 55%).
Rf = 0.88 (10% MeOH in CH
2Cl
2);
−95.0° (c 1.00, MeOH); Major tautomer:
1H NMR (500 MHz, CD
3OD)
δ 7.29 (dt,
J = 15.6, 6.9 Hz, 1H), 7.12 (d,
J = 15.6 Hz, 1H), 6.90 (m, 2H), 6.76 (m, 2H), 6.01 (ddt,
J = 17.3, 10.7, 5.3 Hz, 1H), 5.83 (ddt,
J = 17.0, 10.3, 6.7 Hz, 1H), 5.34 (dq,
J = 17.3, 1.6 Hz, 1H), 5.20 (dq,
J = 10.7, 1.6 Hz, 1H), 5.08 (dq,
J = 17.0, 1.4 Hz, 1H), 5.02 (dq,
J = 10.3, 1.6 Hz, 1H), 4.53 (m, 1H), 4.47 (dt,
J = 5.3, 1.6 Hz, 2H), 3.35 (dd,
J = 14.0, 5.5 Hz, 1H), 3.20 (dd,
J = 14.0, 2.6 Hz, 1H), 2.45 (q,
J = 6.9 Hz, 2H), 2.27 (q,
J = 6.9 Hz, 2H), 1.62 (s, 9H) ppm; Significant signals minor tautomer:
1H NMR (500 MHz, CD
3OD)
δ 5.00 (m, 2H), 4.60 (m, 1H), 4.43 (m, 2H), 3.39 (m, 1H), 3.07 (m, 1H), 2.10 (m, 2H) ppm;
13C NMR (125 MHz, CD
3OD)
δ 159.3, 153.9, 138.2, 134.9, 131.8, 127.6, 122.1, 117.4, 116.2, 115.6, 69.7, 35.9, 35.7, 33.7, 33.0, 28.4 ppm; Major tautomer:
1H NMR (500 MHz, CDCl
3)
δ 7.30–7.11 (m, 2H), 6.92 (m, 2H), 6.75 (m, 2H), 6.01 (ddt,
J = 17.2, 10.5, 5.3 Hz, 1H), 5.78 (ddt,
J = 17.0, 10.3, 6.5 Hz, 1H), 5.37 (dq,
J = 17.2, 1.6 Hz, 1H), 5.25 (dq,
J = 10.5, 1.6 Hz, 1H), 5.11–4.91 (m, 2H), 4.46 (dt,
J = 5.3, 1.6 Hz, 2H), 4.38 (m, 1H), 3.40–3.30 (m, 1H), 3.25 (m, 1H), 2.45 (q,
J = 6.7 Hz, 2H), 2.27 (q,
J = 6.7 Hz, 2H), 1.61 (s, 9H) ppm; Significant signals minor tautomer:
1H NMR (500 MHz, CDCl
3)
δ 4.60 (m, 1H), 3.38 (m, 1H), 3.20 (m, 1H), 2.40 (m, 2H), 2.24 (m, 2H) ppm; Major tautomer:
13C NMR (125 MHz, CDCl
3)
δ 192.5, 176.3, 173.8, 157.7, 152.3, 149.0, 136.8, 133.3, 130.84, 126.5, 121.7, 117.8, 116.0, 114.7, 100.7, 84.1, 68.8, 65.7, 35.1, 32.7, 32.1, 28.2 ppm; Significant signals minor tautomer:
13C NMR (125 MHz, CDCl
3)
δ 201.0, 178.4, 164.6, 157.8, 153.2, 150.2, 130.8, 126.4, 121.4, 114.8, 102.9, 83.4, 63.2, 34.9, 32.8, 32.0, 28.3 ppm; IR
νmax 2981 (w), 2937 (w), 2367 (w), 1769 (w), 1712 (m), 1642 (m), 1610 (m), 1578 (m), 1511 (m), 1414 (w), 1396 (w), 1370 (m), 1349 (m), 1304 (s), 1248 (m), 1150 (m), 1028 (w), 996 (w) cm
–1; HRMS (ESI)
m/
z [M + Na]
+ calcd. for C
26H
31NO
6Na
+ 476.20436, found 476.20380.
(3S,6Z,8E,12E)-4-Aza-N-(tert-butoxycarbonyl)-7-hydroxy-15-oxa-5,21-dioxo-tricyclo-[14.2.2.13,6]henicosa-1(18),6,8,12,16(17),19-hexaene (13). 3-Acyltetramic acid 12 (207 mg, 456 μmol, 1.00 eq) in dry, degassed CH2Cl2 (90 mL) was treated with 2nd generation Grubbs catalyst (39 mg, 46 μmol, 10 mol%). The solution was stirred at reflux for 18 h. The solvent was removed under reduced pressure and the crude product was purified by column chromatography on reversed phase silica gel (RP18, 40% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH → 70% MeCN in H2O + 0.1% HCOOH → 80% MeCN in H2O + 0.1% HCOOH → 100% MeCN + 0.1% HCOOH) to afford 13 as pale brown resin (128 mg, 301 µmol, 66%). Rf = 0.75 (5% MeOH in CH2Cl2); −19.6° (c 1.00, CHCl3); 1H NMR (500 MHz, CD3OD) δ 6.90 (dt, J = 15.6, 7.6 Hz, 1H), 6.80 (d, J = 8.5 Hz, 2H), 6.74–6.61 (m, 2H), 6.58 (d, J = 15.6 Hz, 1H), 5.54 (dt, J = 15.3, 7.6 Hz, 1H), 5.41 (dt, J = 15.3, 5.7 Hz, 1H), 4.63–4.48 (m, 3H), 3.29 (dd, J = 13.8, 3.2 Hz, 1H), 3.04 (dd, J = 13.8, 3.2 Hz, 1H), 2.52–2.26 (m, 4H), 1.63 (s, 9H) ppm; Major tautomer: 13C NMR (125 MHz, CD3OD) δ 174.7 (HMBC correlation), 158.4, 153.1, 134.1, 131.1, 128.3, 126.6, 122.9, 118.0, 117.6, 115.3, 84.9, 67.7, 66.0, 37.0, 33.6, 31.9, 28.4 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CD3OD) δ 159.9, 132.3, 117.2, 114.9 ppm; Major tautomer: 1H NMR (500 MHz, CDCl3) δ 6.95–6.48 (m, 6H), 5.50 (m, 1H), 5.35 (m, 1H), 4.62–4.39 (m, 3H), 3.27 (dd, J = 13.6, 3.6 Hz, 1H), 3.07 (m, 1H), 2.57–2.41 (m, 2H), 2.33–2.12 (m, 2H), 1.63 (s, 9H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CDCl3) δ 3.38 (dd, J = 13.6, 3.6 Hz, 1H) ppm; C2, C4 and C3 not observed; IR νmax 2973 (w), 2931 (w), 2934 (m), 1764 (m), 1713 (m), 1644 (s), 1608 (w), 1579 (s), 1508 (m), 1394 (w), 1369 (m), 1350 (s), 1306 (s), 1274 (m), 1254 (m), 1222 (m), 1159 (s), 1141 (m), 1109 (w), 978 cm–1; HRMS (ESI) m/z [M + Na]+ calcd. for C24H27NO6Na+ 448.17306, found 448.17270.
(3S,6Z,8E,12E)-4-Aza-7-hydroxy-15-oxa-5,21-dioxo-tri-cyclo [14.2.2.13,6]henicosa-1(18),6,8,12,16(17),19-hexaene (5). Tetramic acid 13 (245 mg, 576 μmol, 1.00 eq) in dry CH2Cl2 (11 mL) was treated with TFA (1.10 mL) and stirred for 15 min at room temperature. Toluene (75 mL) was added and the solvent was concentrated under reduced pressure. This was repeated once to yield 5 as a pale brown foam (187 mg, 576 µmol, quant.). Rf = 0.63 (10% MeOH in CH2Cl2 + 0.1% HCOOH); +92.9° (c 1.00, MeOH); 1H NMR (500 MHz, CD3OD): Diastereotopic H-atoms indicated as a, b: δ 7.03–6.41 (m, 6H, OCHC=CHCH2, CHAr), 5.57 (m, 1H, OCH2HC=CH), 5.35 (m, 1H, OCH2HC=CH), 4.60/4.48 (m, 2H, ArOCH2), 4.05 (brs, 1H, CHN), 3.01a (dd, J = 13.6, 3.9 Hz, 1H), 2.85b (dd, J = 13.6, 2.0 Hz, 1H, ArCH), 2.53–2.13 (m, 4H, OCH2HC=CH(CH2)2) ppm; 13C NMR (125 MHz, CD3OD) δ 172.8 (HNCO), 157.9 (OCq,Ar), 149.8 (OCHC=CHCH2), 132.8 (OCH2HC=CH), 131.2 (CH2CCHAr), 128.0 (OCH2HC=CH), 126.5 (CH2Cq,Ar), 123.2 (OCHC=CHCH2), 117.6 (OCCHAr), 67.4 (ArOCH2), 38.0 (ArCH2), 33.5 (OCHC=CHCH2), 32.5 (OCH2HC=CHCH2) ppm; Major tautomer: 1H NMR (500 MHz, CDCl3): δ 7.07–6.42 (m, 6H), 5.57–5.31 (m, 2H), 4.55 (m, 2H), 4.10 (m, 1H), 3.13 (dd, J = 13.8, 4.1 Hz, 1H), 2.84 (m, 1H), 2.55–2.11 (m, 4H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CDCl3) δ 4.20 (m, 1H), 2.90 (m, 1H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 195.6, 176.1, 171.8, 156.5, 148.6, 133.5, 132.2, 130.0, 126.8, 125.0, 122.3, 117.0, 111.6, 100.9, 66.5, 62.2, 37.6, 32.6, 31.7 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3) δ 203.7, 172.7, 170.2, 157.1, 149.7, 131.6, 130.8, 127.1, 125.5, 122.0, 115.7, 115.4, 104.0, 67.5, 60.2, 38.1, 32.1, 30.4 ppm; IR νmax 3303 (m), 2927 (w), 2934 (m), 2070 (w), 1643 (s), 1576 (s), 1507 (m), 1428 (m), 1369 (w), 1338 (w), 1254 (m), 1219 (m), 1177 (m), 1115 (m), 975 (s) cm–1; HRMS (ESI) m/z [M + H+] calcd. for C19H20NO4+ 326.13868, found 326.13785.
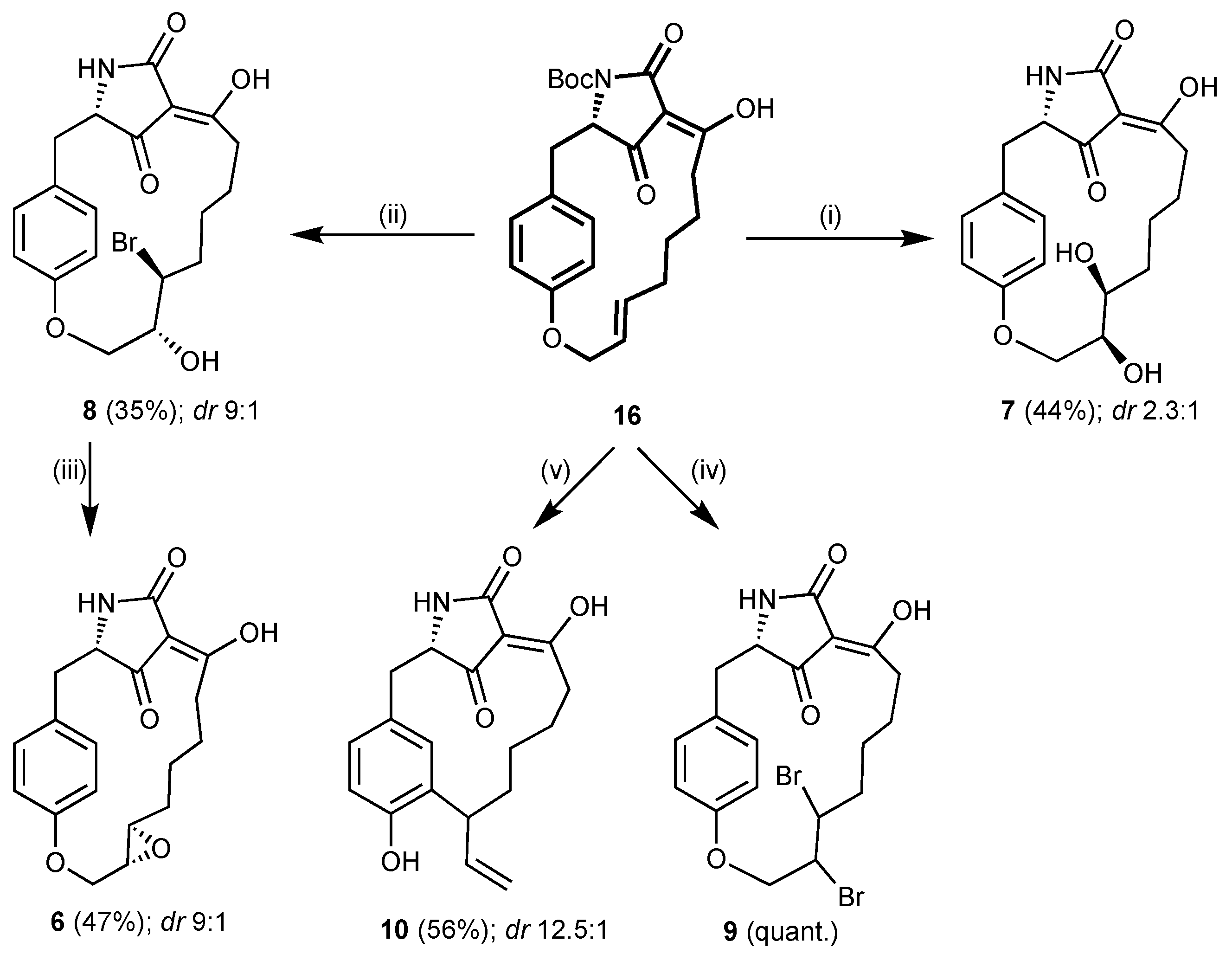
(3S,6Z,12E)-4-Aza-N-(tert-butoxycarbonyl)-7-hydroxy-15-oxa-5,21-dioxo-tricyclo-[14.2.2.13,6]henicosa-1(18),6,12, 16(17),19-pentaene (16). Tetramic acid 13 (77.0 mg, 181 μmol, 1.00 eq) and Wilkinson’s catalyst (17 mg, 18 μmol, 10 mol%) in dry CH2Cl2 (2.5 mL) were treated with Et3SiH (144 μL, 905 μmol, 5.00 eq). The solution was stirred for 19 h under reflux and the solvent was removed under reduced pressure. The crude product was dissolved in dry MeOH (2.6 mL) and KF (26.3 mg, 453 μmol, 2.50 eq) was added. After stirring for 20 h at −15 °C, more KF (26.3 mg, 453 μmol, 2.50 eq) was added and stirring was continued for a further 7 h at −15 °C. Chilled H2O (50 mL) and chilled brine (20 mL) were added. The aqueous phase was extracted with EtOAc (3 × 50 mL), and the combined organic phases were washed with 0.5M H2SO4 (40 mL) and dried over Na2SO4. Removal of the solvent and purification by column chromatography on reversed phase silica gel (RP18, 30% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH) gave 16 as a colourless foam (37 mg, 86.6 µmol, 48%). Rf = 0.83 (10% MeOH in CH2Cl2); +12.4° (c 1.00, MeOH); Major tautomer: 1H NMR (500 MHz, CD3OD) δ 6.90–6.60 (m, 4H), 5.53 (dt, J = 15.4, 7.9 Hz, 1H), 5.40 (dt, J = 15.4, 5.3 Hz, 1H), 4.64–4.49 (m, 3H), 3.41 (dd, J = 14.1, 4.5 Hz, 1H), 3.10 (dd, J = 14.1, 2.9 Hz, 1H), 2.36 (m, 2H), 2.09–1.90 (m, 2H), 1.64 (s, 9H), 1.30 (m, 2H), 1.08 (m, 2H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CDCl3) δ 3.29 (dd, J = 14.1, 4.5 Hz, 1H), 3.03 (dd, J = 14.1, 2.9 Hz, 1H), 2.91 (m, 2H) ppm; Major tautomer: 13C NMR (125 MHz, CD3OD) δ 158.0, 150.8, 135.8, 131.7, 127.1, 127.0, 117.9, 84.7, 68.0, 35.7, 33.6, 33.2, 29.6, 28.4, 28.0 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CD3OD) δ 158.3, 128.2, 118.5, 67.7, 37.0, 33.4, 29.8, 28.3, 27.7 ppm; 1H NMR (500 MHz, CDCl3) δ 7.02–6.42 (m, 4H), 5.56–5.26 (m, 2H), 4.69–4.37 (m, 3H), 3.49–3.34 (dd, J = 14.0, 4.9 Hz, 1H), 3.29 (m, 1H), 3.15–3.03 (m, 1H), 2.48–1.86 (m, 3H), 1.63 (s, 9H), 1.47–0.99 (m, 4H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 197.4, 196.8, 164.3, 156.6, 149.8, 134.5, 131.9, 125.8, 125.70, 118.4, 102.2, 83.5, 67.6, 62.1, 35.0, 32.7, 32.22, 28.7, 28.3, 26.9 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CD3OD) δ 192.0, 191.8, 156.1, 136.6, 130.0, 125.75, 114.0, 105.5, 84.4, 67.0, 65.8, 34.7, 34.5, 32.24, 28.4, 27.6, 26.7, 26.0 ppm; IR νmax 2978 (w), 2935 (w), 2863 (w), 1778 (m), 1744 (m), 1712 (s), 1662 (m), 1607 (s), 1509 (s), 1475 (w), 1456 (w), 1440 (m), 1423 (w), 1395 (w), 1366 (m), 1350 (s), 1305 (s), 1272 (m), 1258 (s), 1217 (s), 1150 (s), 1111 (m), 1081 (w), 1017 (w), 971 (m) cm–1; HRMS (ESI) m/z [M + Na+] calcd. for C24H29NO6Na+ 450.18871, found 450.18776.
(S)-tert-Butyl-2-(4-(allyloxy)benzyl)-3-(hept-6-enoyloxy)-5-oxo-2,5-dihydro-1H-pyrrole-1-carboxylate (14). 6-Heptenoic acid (2.11 mL, 15.6 mmol, 1.00 eq) in dry CH2Cl2 (78 mL) was treated with EDC∙HCl (3.59 g, 18.7 mmol, 1.20 eq) and DMAP (0.38 g, 3.12 mmol, 0.20 eq) at 0 °C. The solution was stirred for 20 min, before tetramic acid 11 (5.93 g, 17.2 mmol, 1.1 eq) was added at room temperature. After stirring for 4 h, the reaction was quenched with 0.5M H2SO4 (250 mL). The organic phase was separated and the aqueous phase was extracted with EtOAc (3 × 150 mL). The combined organic phases were washed with brine (200 mL) and dried over Na2SO4. After removal of the solvent under reduced pressure, the crude product was purified by column chromatography (silica gel 60, 10% EtOAc in hexanes → 15% EtOAc in hexanes → 20% EtOAc in hexanes → 25% EtOAc in hexanes) to obtain 14 as an orange resin (5.64 g, 12.4 mmol, 79%). Rf = 0.48 (30% EtOAc in hexanes); +107.5° (c 1.00, MeOH); 1H NMR (500 MHz, CDCl3) δ 6.93–6.83 (m, 2H), 6.81–6.71 (m, 2H), 6.03 (m, 1H), 5.88 (s, 1H), 5.79 (ddt, J = 17.0, 10.3, 6.7 Hz, 1H), 5.40 (m, 1H), 5.28 (m, 1H), 5.06–4.94 (m, 2H), 4.77 (dd, J = 6.0, 2.8 Hz, 1H), 4.48 (m, 2H), 3.29 (dd, J = 14.3, 6.2 Hz, 1H), 3.14 (dd, J = 14.3, 2.8 Hz, 1H), 2.48 (td, J = 7.4, 1.9 Hz, 2H), 2.09 (m, 2H), 1.74–1.65 (m, 2H), 1.60 (s, 9H), 1.46 (qn, J = 7.7 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 168.8, 168.2, 165.3, 157.9, 149.5, 138.0, 133.3, 130.5, 126.2, 117.9, 115.3, 114.8, 108.3, 83.3, 68.9, 60.7, 35.0, 34.3, 33.4, 28.4, 28.2, 23.9 ppm; IR νmax 3075 (w), 2975 (w), 2939 (w), 2863 (w), 1777 (s), 1744 (s), 1712 (s), 1633 (m), 1611 (w), 1582 (w), 1514 (s), 1478 (w), 1457 (w), 1424 (w), 1392 (w), 1370 (m), 1356 (m), 1320 (s), 1248 (s), 1226 (m), 1172 (s), 1158 (s), 1115 (m), 1064 (s), 1032 (m), 996 (m) cm–1; HRMS (ESI) m/z [M + Na+] calcd. for C26H33O6NNa+ 478.22001, found 478.21968.
(S,Z)-tert-Butyl-2-(4-(allyloxy)benzyl)-4-(1-hydroxyhept-6-en-1-ylidene)-3,5-dioxopyrrolidine-1-carboxylate (15). 4-O-acyltetramic acid 14 (5.54 g, 12.2 mmol, 1.0 eq) in dry CH2Cl2 (122 mL) was treated with dry NEt3 (2.04 mL, 14.6 mmol, 1.2 eq) at room temperature and stirred for 10 min. DMAP (743 mg, 6.1 mmol, 0.5 eq) was added and the solution was stirred for a further 24 h. NaHCO3 (200 mL) was added and the aqueous phase was extracted with EtOAc (2 × 150 mL). The combined organic phases were washed with brine (200 mL) and dried over Na2SO4. Removal of the solvent under reduced pressure and purification by column chromatography on reversed phase silica gel (RP18, 40% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH → 80% MeCN in H2O + 0.1% HCOOH → 100% MeCN + 0.1% HCOOH) afforded 15 as an orange resin (4.54 g, 9.97 mmol, 82%). Rf = 0.91 (10% MeOH in CH2Cl2); −31.8° (c 1.00, MeOH); 1H NMR (500 MHz, CD3OD) δ 6.90 (m, 2H), 6.77 (m, 2H), 6.01 (m, 1H), 5.78 (ddt, J = 17.1, 10.5, 7.2 Hz, 1H), 5.35 (dd, J = 17.1, 1.5 Hz, 1H), 5.21 (m, 1H), 5.01 (m, 1H), 4.94 (m, 1H), 4.58 (s, 1H), 4.46 (d, J = 5.2 Hz, 2H), 3.38 (dd, J = 14.2, 5.4 Hz, 1H), 3.18 (dd, J = 14.2, 2.3 Hz, 1H), 2.75 (t, J = 6.4 Hz, 2H), 2.04 (q, J = 7.2 Hz, 2H), 1.62 (s, 9H), 1.57–1.45 (m, 2H), 1.34 (m, 2H) ppm; 13C NMR (125 MHz, CD3OD) δ 195.2 (HMBC correlation), 159.3, 139.5, 134.9, 131.9, 127.4, 117.4, 115.5, 115.3, 84.8, 69.7, 64.6 (HMBC correlation), 35.8, 34.4, 29.4, 28.4, 26.2 ppm; Major tautomer: 1H NMR (500 MHz, CDCl3) δ 6.92 (m, 2H), 6.75 (m, 2H), 6.02 (m, 1H), 5.85–5.70 (m, 1H), 5.38 (m, 1H), 5.27 (m, 1H), 5.07–4.91 (m, 2H), 4.39 (dd, J = 5.4, 2.7 Hz, 1H), 4.46 (d, J = 5.3 Hz, 2H), 3.33 (dd, J = 14.1, 5.8 Hz, 1H), 3.24 (dd, J = 14.1, 2.6 Hz, 1H), 2.93–2.61 (m, 2H), 2.07 (q, J = 7.1 Hz, 2H), 1.61 (s, 9H), 1.60–1.29 (m, 4H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CDCl3) δ 7.10 (m, 2H), 6.87 (m, 2H), 4.64 (dd, J = 5.4, 2.7 Hz, 1H), 4.52 (d, J = 5.3 Hz, 2H), 3.41 (dd, J = 14.1, 5.8 Hz, 1H), 3.21 (dd, J = 14.1, 2.6 Hz, 1H), 2.01 (q, J = 7.1 Hz, 2H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 196.5, 192.4, 164.4, 157.8, 149.1, 138.3, 133.32, 130.9, 126.4, 117.8, 115.1, 114.7, 102.5, 84.2, 68.82, 65.8, 35.0, 33.4, 32.9, 28.3, 28.2, 25.5 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3) δ 157.9, 150.0, 138.4, 133.30, 130.8, 126.0, 117.9, 115.3, 114.9, 105.6, 83.5, 68.80, 61.9, 34.9, 34.8, 32.7, 28.5, 28.4, 24.7 ppm; IR νmax 3075 (w), 2978 (w), 2932 (w), 2860 (w), 1770 (w), 1744 (m), 1716 (s), 1667 (m), 1640 (m), 1604 (s), 1510 (s), 1457 (w), 1421 (m), 1395 (w), 1366 (m), 1349 (s), 1301 (s), 1241 (s), 1223 (s), 1172 (s), 1151 (s), 1025 (m), 996 (m), 967 (m), 913 (s) cm–1; HRMS (ESI) m/z [M + Na+] calcd. for C26H33O6NNa+ 478.22001, found 478.21944.
(3S,6Z,12E)-4-Aza-N-(tert-butoxycarbonyl)-7-hydroxy-15-oxa-5,21-dioxo-tricyclo[14.2.2.13,6]henicosa-1(18),6,12,16(17),19-pentaene (16) from 15. 3-Acyltetramic acid 15 (2.30 g, 5.04 mmol, 1.00 eq) in dry, degassed CH2Cl2 (1.00 L) was treated with 2nd generation Grubbs catalyst (428 mg, 504 μmol, 10 mol%). The solution was stirred at reflux for 24 h. The solvent was removed under reduced pressure and the crude product was purified by column chromatography on reversed phase silica gel (RP18, 40% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH → 80% MeCN in H2O + 0.1% HCOOH → 100% MeCN + 0.1% HCOOH) to yield 16 as a pale brown foam (1.78 g, 4.16 mmol, 83%). For analytical data see above.
(3S,6Z,12E)-4-Aza-7-hydroxy-15-oxa-5,21-dioxo-tricyclo[14.2.2.13,6]henicosa- 1(18),6,12,16(17),19-pentaene (4). Carbamate 16 (926 mg, 2.17 mmol, 1.00 eq) in dry CH2Cl2 (43 mL) was treated with TFA (4.3 mL) and stirred for 15 min at room temperature. Toluene (100 mL) was added and the solvent was concentrated under reduced pressure. This was repeated once to yield 4 as a pale brown foam (709 mg, 2.17 mmol, quant.). Rf = 0.64 (10% MeOH in CH2Cl2 + 0.01% HCOOH); +52.2° (c 1.00, MeOH); 1H NMR (500 MHz, CD3OD): Diastereotopic H-atoms indicated as a, b: δ 7.09–6.68 (m, 4H, CHAr), 5.58 (dt, J = 15.1, 7.8 Hz, 1H, OCH2HC=CH), 5.33 (m, 1H, OCH2HC=CH), 4.64a (dd, J = 13.9, 7.7 Hz, 1H, ArOCH), 4.53b (m, 1H, ArOCH), 4.15 (t, J = 3.3 Hz, 1H, CHN), 3.11–2.67a (brs, 1H, OCCH), 3.07a (dd, J = 14.1, 3.8 Hz, 1H, ArCH), 2.91b (dd, J = 14.1, 3.5 Hz, 1H, ArCH), 2.42–1.86b (brs, 1H, OCCH), 2.05a (m, 1H, HC=CHCH), 1.94b (m, 1H, HC=CHCH), 1.34–1.05 (m, 4H, HC=CHCH2(CH2)2) ppm; 13C NMR (125 MHz, CD3OD) δ 157.5 (OCq,Ar), 127.4 (OCH2CH=CH), 126.9 8 (CH2Cq,Ar), 118.3 (OCCHAr), 67.8 (ArOCH2), 36.6 (ArCH2), 33.6 (OCCH2), 33.4 (HC=CHCH2), 29.8 (HC=CHCH2(CH2)2), 28.5 (HC=CHCH2(CH2)2) ppm; Major tautomer: 1H NMR (500 MHz, CDCl3) δ 7.10–6.58 (m, 4H), 5.54 (m, 1H), 5.39 (m, 1H), 4.60 (m, 2H), 4.16 (m, 1H), 3.28–3.16 (m, 2H), 2.87 (dd, J = 14.1, 1.7 Hz, 1H), 2.12–1.84 (m, 3H), 1.40–1.04 (m, 4H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CDCl3) δ 5.49 (m, 1H), 5.32 (m, 1H), 4.29 (s, 1H), 2.87 (m, 1H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 194.5, 188.6, 175.8, 156.0, 136.8, 132.0, 130.3, 125.7, 125.6, 117.9, 114.1, 101.5, 67.0, 62.4, 36.2, 32.43, 32.41, 28.7, 27.6 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3) δ 201.2 (HMBC correlation), 192.4, 170.3, 156.5, 134.9, 131.2, 118.2, 104.7, 67.6, 59.8, 36.4, 33.1, 32.1, 28.5, 27.1 ppm; IR νmax 3255 (w), 2929 (w), 2857 (w), 1770 (w), 1646 (s), 1608 (s), 1508 (s), 1433 (m), 1367 (w), 1305 (w), 1259 (m), 1214 (s), 1176 (s), 1159 (s), 1113 (m), 1075 (m), 1062 (m), 1015 (m), 973 (s) cm–1. HRMS (ESI) m/z [M + H+] calcd. for C19H22NO4+ 328.15433, found 328.15343.
(3S,6Z,12S,13S)-4-Aza-7,12,13-trihydroxy-15-oxa-5,21-dioxo-tricyclo[14.2.2.13,6]henicosa-1(18),6,16(17),19-tetraene (7). AD-mix α (2.86 g, 1.4 g/mmol) in H2O (10.3 mL) was treated with alkene 16 (874 mg, 2.04 mmol, 1.00 eq) in tBuOH (10.3 mL) at 0 °C. The two-phase mixture was stirred at 7 °C for 5 d, before more AD-mix α (1.43 g, 0.7 g/mmol) was added. After stirring for a further 4 d, the mixture was treated with Na2SO3 (3.60 g, 28.6 mmol, 14.0 eq) and stirred for 2 h at room temperature. H2O was added to dissolve the precipitate. The aqueous phase was washed with EtOAc (10 mL) and the organic phase was extracted with H2O (10 mL). The solvent was removed under reduced pressure and the crude product was suspended in MeOH. The precipitate was filtered off and washed with MeOH. Concentration of the filtrate and purification of the residue by column chromatography on reversed phase silica gel (RP18, H2O + 0.1% HCOOH → 20% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH) gave a mixture of Boc-protected and deprotected tetramic acid 7. This was dissolved in dry CH2Cl2 (25 mL) and treated with TFA (1.5 mL). After stirring for 15 min at room temperature, toluene (100 mL) was added. The solvent was concentrated under reduced pressure and more toluene (100 mL) was added. Removal of the solvent under reduced pressure gave diol 7 as a yellowish foam and as a mixture of two diastereomers according to HPLC and NMR spectra. Yield: 323 mg (894 µmol, 44%, dr 2.3:1). Rf = 0.63 (10% MeOH in CH2Cl2 + 0.1% HCOOH); 1H NMR (500 MHz, CD3OD): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B; diastereotopic H-atoms indicated as a, b: δ 7.13–6.75 (m, 4H, CHAr), 4.26 (m, 1H, ArOCHa), 4.22 (m, 1H, CHN), 4.07 (dd, J = 11.7, 8.8 Hz, 1H, ArOCHb,B), 4.05 (m, 1H, J = 11.8, 8.9 Hz, 1H, ArOCHb,A), 3.80 (m, 1H, ArOCH2CHOHA), 3.60 (m, 2H, ArOCH2CHOHB, ArOCH2CHOHCHOHB), 3.46 (dt, J = 9.6, 2.7, 3.9 Hz, 1H, ArOCH2CHOHCHOHA), 3.10 (dt, J = 14.0, 3.8 Hz, 1H, ArCHa), 2.97 (dt, J = 14.0, 3.2 Hz, 1H, ArCHb), 1.67–1.17 (m, 4H, (CH2)2), 0.88–0.58 (m, 2H, OCCH2CH2) ppm; OCCH2 not observed; Major diastereomer: 13C NMR (125 MHz, CD3OD): δ 189.0 (CHO, HMBC correlation), 157.8 (OCq,Ar), 131.1 (CH2CCHAr, HMBC correlation), 127.2 (CH2Cq,Ar, HMBC correlation), 118.4 (OCCHAr), 113.7 (OCCHAr), 69.9 (ArOCH2CHOHCHOH), 67.5 (ArOCH2), 67.4 (ArOCH2CHOH, HSQC correlation), 36.6 (ArCH2), 34.5 (ArOCH2(HCOH)2CH2), 32.9 (OCCH2), 27.3 (ArOCH2(HCOH)2CH2CH2), 25.4 (OCCH2CH2) ppm; Minor diastereomer 13C NMR (125 MHz, CD3OD): δ 131.9 (HMBC correlation), 126.5 (HMBC correlation), 119.4, 115.2, 69.7, 68.5, 32.8, 25.9 ppm; IR νmax 3354 (m), 2933 (m), 1646 (s), 1607 (s), 1508 (s), 1433 (w), 1338 (w), 1255 (m), 1216 (m), 1177 (w), 1113 (w), 1016 (w) cm–1; HRMS (ESI) m/z [M + H+] calcd. for C19H24NO4+ 362.15981, found 362.15894.
(3S,6Z)-4-Aza-12-bromo-7,13-dihydroxy-15-oxa-5,21-dioxo-tricyclo[14.2.2.13,6]henicosa-1(18),6,16(17),19-tetraene (8). Alkene 16 (660 mg, 1.54 mmol, 1.00 eq) in DMSO (8 mL) was treated with H2O (41.7 μL, 2.32 mmol, 1.50 eq) and NBS (412 mg, 2.23 mmol, 1.50 eq) at 8 °C. After stirring the solution for 22 h at room temperature, sat. NaHCO3 solution (50 mL) was added. The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic phases were dried over Na2SO4. The solvent was removed under reduced pressure and the crude mixture of products was purified by column chromatography on reversed phase silica gel (RP18, 20% MeCN in H2O + 0.1% HCOOH → 30% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH → 80% MeCN in H2O + 0.1% HCOOH). Bromohydrins 8 and a side product with an additional bromo substituent were obtained separately and only partially deprotected. The mixture of bromohydrin 8 and its N-Boc-protected derivative was dissolved in dry CH2Cl2 (9 mL) and treated with TFA (900 μL). The solution was stirred for 15 min at room temperature and toluene (100 mL) was added. The mixture was concentrated under reduced pressure and toluene (50 mL) was added again. Removal of the solvent under reduced pressure gave 8 as a yellowish foam and as a mixture of two inseparable diastereomers of initially unknown dr according to 13C and 1H NMR spectra. Yield (8): 196 mg (462 µmol, 30%). Rf = 0.41 (10% MeOH in CH2Cl2); 1H NMR (500 MHz, CD3OD): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B; diastereotopic H-atoms indicated as a, b: δ 7.24–6.75 (m, 4H, CHAr), 4.67–4.19 (m, 4H, CHN, ArOCH2, CHBr), 3.73 (m, 1H, CHOH), 3.14 (m, 2H, ArCHa, OCCHa,A), 2.96 (dd, J = 14.0, 3.9 Hz, 1H, ArCHb), 1.60–0.91 (m, 5H, CBrCH2CHaCH2), 0.76–0.26 (m, 1H, CBrCH2CHbCH2) ppm; 1H NMR (500 MHz, CDCl3) δ 7.20–6.72 (m, 4H), 4.60–4.11 (m, 4H), 3.78 (m, 1H), 3.60–3.20 (m, 2H), 2.94 (m, 1H), 2.20–2.00 (m, 1H), 1.63–1.33 (m, 3H), 1.30–0.96 (m, 2H), 0.66–0.30 (m, 1H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B: δ 194.3B (CO), 194.2A (CO), 188.2 (COH), 175.6B (HNCO),175.5A (HNCO), 157.1 (OCq,Ar), 131.4B (CH2CCHAr), 130.1A (CH2CCHAr), 127.6 (CH2Cq,Ar), 117.0 (OCCHAr), 101.90B (NCOCCO), 101.88A (NCOCCO), 73.0 (CHOH), 69.6 (ArOCH2), 62.0B (HCNH), 61.9A (HCNH), 55.9 (CHBr), 36.2 (ArCH2), 34.2 (CHBrCH2), 33.1 (OCCH2), 27.4 (CHBrCH2), 26.0 (OCCH2CH2), 24.6 (CHBrCH2CH2) ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B: δ 202.6B, 202.5A, 189.03B, 189.00A, 169.1B, 169.0A, 156.3, 133.3A, 132.4B, 127.5, 116.2A, 115.3B, 106.0, 67.7, 59.6B, 59.5A, 53.7, 36.6, 33.4, 31.4, 24.6 ppm; IR νmax 3356 (m), 2936 (m), 1652 (s), 1609 (s), 1508 (s), 1462 (w), 1374 (w), 1254 (m), 1217 (m), 1177 (w), 1114 (w), 1043 (w) cm–1; HRMS (ESI) m/z [M + H+] calcd. for C19H23NO5Br+ 424.07451, found 424.07521.
(3S,6E)-4-Aza-13,16-dioxa-5,22-dioxo-tetracyclo[15.2.2.13,6.012,14]docosa-1(19),6,17(18),20-tetraene (6). Bromohydrin 8 (248 mg, 473 μmol, 1.00 eq) in dry THF (1.5 mL) was treated with KOtBu (86.7 mg, 709 μmol, 1.50 eq) at 0 °C. The suspension was stirred for 4 d at room temperature and then more KOtBu (58 mg, 473 μmol, 1.00 eq) was added. Stirring was continued for 1 d and H2O (5 mL) as well as EtOAc (5 mL) were added. The organic phase was separated and extracted with H2O (5 mL). The combined aqueous phases were concentrated under reduced pressure. The crude product was purified by column chromatography on reversed phase silica gel (RP18, 20% MeCN in H2O + 0.1% HCOOH → 30% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH → 80% MeCN in H2O + 0.1% HCOOH) to yield a virtually pure product. Another column chromatography on reversed phase silica gel (RP18, 0% MeCN in H2O + 0.1% HCOOH → 20% MeCN in H2O + 0.1% HCOOH → 30% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH→ 80% MeCN in H2O + 0.1% HCOOH) afforded epoxide 6 as a pale brown foam and as a 9:1 mixture of two diastereomers according to 13C and 1H NMR spectra. Yield: 76.0 mg (221 µmol, 47%). Rf = 0.28 (10% MeOH in CH2Cl2); 1H NMR (500 MHz, CD3OD): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B¸ diastereotopic H-atoms indicated as a, b: δ 7.05 (m, 2H, OC(CH)2), 6.77 (m, 2H, CH2C(CH)2), 4.55A,a (dd, J = 14.0, 3.1 Hz, 1H, ArOCH), 4.40B,a (m, 1H, ArOCH), 4.21 (t, J = 3.2 Hz, 1H, CHN), 4.03A,b (dd, J = 14.0, 3.1 Hz, 1H, ArOCH), 3.95B,b (m, 1H, ArOCH), 3.14a (dd, J = 14.1, 3.5 Hz, 1H, ArCH), 3.22–2.69a (brs, 1H, OCCH), 3.05 (m, 1H, ArOCH2CHO), 2.93b (dd, J = 14.1, 3.5 Hz, 1H, ArCH), 2.65 (m, 1H, ArOCH2CHOCH), 1.71a (m, 1H, ArOCH2CHOCHCH), 1.63–1.15 (m, 3H, OCCHb, ArOCH2CHOCHCH2CH2), 1.15–0.41 (m, 3H, ArOCH2CHOCHCHb, OCCH2CH2) ppm; 13C NMR (125 MHz, CD3OD): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B: δ 187.5 (HMBC correlation), 157.7 (OCq,Ar), 132.7A(CH2CCHAr), 132.5B (CH2CCHAr), 128.2 (CH2Cq,Ar), 116.5B (OCCHAr), 116.0A (OCCHAr), 113.2A (OCCHAr), 66.6B (ArOCH2), 66.4A (ArOCH2), 62.4 (HCNH, HSQC correlation), 58.5 (OCH2CHOCH), 58.4 (OCH2CHOCH), 36.3 (CH2Ar), 33.3 (ArOCH2CHOCHCH2), 31.9 (OCCH2), 27.3 (ArOCH2CHOCHCH2CH2), 24.8A (OCCH2CH2), 24.7B (OCCH2CH2) ppm; 1H NMR (500 MHz, CDCl3) δ 7.25–6.43 (m, 5H), 4.53 (t, J = 11.7 Hz, 1H), 4.44–3.83 (m, 2H), 3.40 (m, 1H), 3.24 (m, 1H), 3.12–2.80 (m, 2H), 2.71 (m, 1H), 2.21 (m, 1H), 1.92–1.42 (m, 3H), 1.19–0.35 (m, 3H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B: δ 194.6, 187.6, 175.3, 156.2, 131.5, 126.65, 115.1, 103.5, 65.5, 62.4, 57.2, 57.08, 35.7, 32.1, 30.6, 26.0, 23.5 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B: δ 202.2, 190.0, 169.2, 156.5, 131.8, 126.5, 115.2, 106.3, 66.2, 59.5, 58.0, 57.12, 36.1, 32.4, 31.4, 26.6, 23.4 ppm; IR νmax 3283 (w), 2931 (m), 1706 (s), 1654 (s), 1609 (s), 1509 (s), 1436 (m), 1373 (m), 1306 (w), 1239 (s), 1221 (s), 1180 (m), 1113 (w), 1072 (w), 1044 (m), 915 (m) cm–1; HRMS (ESI) m/z [M + H+] calcd. for C19H22NO5+ 344.14925, found 344.14904.
(3S,6Z)-4-Aza-12,13-dibromo-7-hydroxy-15-oxa-5,21-dioxo-tricyclo-[14.2.2.13,6] henicosa-1(18),6,16(17),19-tetraene (9). A solution of alkene 16 (500 mg, 1.17 mmol, 1.00 eq) in CCl4 (5.3 mL) was treated with bromine (90.3 μL, 1.75 mmol, 1.50 eq) and stirred in a sealed tube for 30 h at 80 °C. The solvent was removed under reduced pressure and the remainder was purified by column chromatography on reversed phase silica gel (RP18, 30% MeCN in H2O + 0.1% HCOOH → 35% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 45% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH → 100% MeCN in H2O + 0.1% HCOOH) to afford dibromide 9 as a yellow foam and as a mixture of two diastereomers of unknown dr. Yield: 228 mg (468 µmol, 40%). Rf = 0.62 (10% MeOH in CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.67/8.42/8.17 (s, 1H, NH), 7.24–6.72 (m, 4H, CHAr), 4.82–3.83 (m, 5H, CH2CHNH, ArOCH2, (CHBr)2), 3.39–2.94 (m, 2H, ArCH2), 2.41–0.75 (m, 8H, OCCH2, CHBr(CH2)3) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 198.3 (CO), 193.3 (COH), 168.5 (HNCO), 157.5 (OCq,Ar), 133.0 (CH2CCHAr), 130.4 (CH2CCHAr), 126.8 (CH2Cq,Ar), 115.6 (OCCHAr), 101.6 (NCOCCO, HMBC correlation), 70.0 (ArOCH2), 62.5 (HCNH), 59.6 ((CHBr)2), 54.3 ((CHBr)2), 38.2 (CH2CHBr), 34.9 (CH2Ar), 27.0, 21.9 (HOCCH2, CHBrCH2(CH2)2) ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3): δ 168.7, 132.7, 130.9, 62.4, 61.9, 55.0, 26.4/25.5 ppm; IR νmax 3241 (m), 2929 (m), 1693 (s), 1652 (s), 1605 (s), 1507 (s), 1460 (m), 1431 (m), 1350 (w), 1299 (w), 1250 (m), 1215 (s), 1177 (m), 1112 (w), 1112 (w), 1040 (w), 1016 (m), 925 (w), 906 (m), 858 (m), 811 (w), 769 (w), 733 (s) cm–1; HRMS (ESI) m/z [M + H+] calcd. for C19H22Br2NO4+ 487.98896, found 487.98884.
((3S,6Z)-4-Aza-7,14-dihydroxy-5,18-dioxo-12-vinyl-tricyclo[11.3.1.13,6]octadeca-1(17),6,13(14),15-tetraene (10). A solution of alkene 16 (500 mg, 1.17 mmol, 1.00 eq) in degassed diethylaniline (4.7 mL) was stirred in a sealed tube at 190 °C for 42 h. The solution was diluted with EtOAc (50 mL). The organic phase was washed with 2M HCl (2 × 75 mL) and the aqueous phase was extracted with EtOAc (30 mL). The combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. Purification of the crude product by column chromatography on reversed phase silica gel (RP18, 30% MeCN in H2O + 0.1% HCOOH → 40% MeCN in H2O + 0.1% HCOOH → 50% MeCN in H2O + 0.1% HCOOH → 60% MeCN in H2O + 0.1% HCOOH → 100% MeCN in H2O + 0.1% HCOOH) afforded 10 as a colourless foam and as an inseparable mixture of two diastereomers. Yield: 218 mg (666 µmol, 57%, dr 12.5:1). Rf = 0.28 (10% MeOH in CH2Cl2); 1H NMR (500 MHz, CD3OD): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B; diastereotopic H-atoms indicated as a, b: δ 6.88–6.72 (m, 2H, CHArCCH2), 6.60A (m, 1H, CHArCOH), 6.56B (m, 1H, CHArCOH), 6.36B (m, 1H, C=CH), 5.95A (ddd, J = 16.0, 10.4, 6.2 Hz, 1H, C=CH), 5.00–4.90 (m, 2H, CH2=C), 4.00A (t, J = 3.4 Hz, 1H, CH2CHNH), 3.96B (t, J = 3.4 Hz, 1H, CH2CHNH), 3.71 (m, 1H, CHC=C), 3.58a (m, 1H, HOCCH), 3.14A,a (dd, J = 14.0, 3.5 Hz, ArCH), 3.08B,a (dd, J = 14.0, 3.5 Hz, ArCH), 2.78b (dd, J = 13.7, 3.5 Hz, 1H, ArCH), 2.13b (dt, J = 14.0, 4.0 Hz, 1H, HOCCH), 1.99a (m, 1H, OCCH2CH), 1.67a (m, 1H, CHCH2CH), 1.55b (m, 1H, CHCH2CH), 1.45–1.26 (m, 2H, OC(CH2)2CH2), 0.89B,b (m, 1H, OCCH2CH), 0.44A,b (m, 1H, OCCH2CH) ppm; 13C NMR (125 MHz, CD3OD): Signals of major diastereomer marked as A, signals of minor diastereomer marked as B: δ 198.9 (CO), 188.8 (COH), 176.2 (HNCO), 155.4 (OCq,Ar), 144.1A (C=CH), 143.1B (C=CH), 131.1 (OCCHCHAr), 130.9 (CHCq,Ar), 130.7A (CCHArC), 129.9B (CCHArC), 126.2 (CH2Cq,Ar), 115.8B (OCCHAr), 115.2A (OCCHAr), 113.2B (C=CH2), 113.0A (C=CH2), 105.4 (NCOCCO), 63.9A (HCNH), 63.4B (HCNH), 40.7 (CHC=C), 37.4A (ArCH2), 37.3B (ArCH2), 35.4A (CHCH2CH2), 32.1B (CHCH2CH2), 29.3A (HOCCH2), 25.9B (HOCCH2), 24.9A, 24.8A (OC(CH2)2CH2, OCCH2CH2), 23.6B, 23.4B (OC(CH2)2CH2, OCCH2CH2) ppm; IR νmax 3294 (m), 2931 (m), 2863 (w), 1969 (w), 1656 (s), 1694 (s), 1511 (m), 1435 (m), 1336 (w), 1252 (m), 1169 (w), 1100 (w), 911 (m) cm–1; HRMS (ESI) m/z [M + H+] calcd. for C19H22NO4+ 328.15433, found 328.15411.
(3S,6Z,8R,12E)-4-Aza-N-(tert-butoxycarbonyl)-7-hydroxy-8-methyl-15-oxa-5,21-dioxo-tricyclo[14.2.2.13,6]henicosa-1(18),6,12,16(17),19-pentaene (18). A solution of alkene 17 (263 mg, 596 μmol, 1.00 eq) in EtOAc (6 mL) was treated with Pd on charcoal (26.3 mg, 10 wt%). The resulting suspension was stirred under a H2-atmosphere for 31 h at room temperature. The solid was filtered off over Celite® and washed with EtOAc. The combined filtrates were concentrated under reduced pressure to give 18 as an orange foam. Yield: 261 mg (588 µmol, 99%). Rf = 0.93 (10% MeOH in CH2Cl2); +38.8° (c 0.75, MeOH); Major tautomer: 1H NMR (500 MHz, CD3OD) δ 7.02–6.57 (m, 4H), 4.50 (m, 1H), 4.26–4.06 (m, 2H), 3.41 (dd, J = 14.4, 3.0 Hz, 1H), 3.37 (m, 1H), 3.08 (dd, J = 14.8, 3.0 Hz, 1H), 1.63 (s, 9H), 1.60–1.15 (m, 7H), 1.08 (d, J = 6.7 Hz, 3H), 1.03 (m, 1H), 0.73–0.42 (m, 2H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CD3OD) δ 4.68 (m, 1H), 3.54 (m,1H) ppm¸ Major tautomer: 13C NMR (125 MHz, CD3OD) δ 193.7, 174.9, 157.4, 150.6, 131.8, 127.9, 116.1, 104.2, 85.2, 67.8, 66.4, 38.3, 35.4, 34.9, 29.4, 28.4, 27.5, 26.1, 24.7, 17.7 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CD3OD) δ 63.3, 32.8, 30.4, 28.44, 23.7, 14.3 ppm; Major tautomer: 1H NMR (500 MHz, CDCl3) δ 7.06–6.65 (m, 4H), 4.44 (m, 1H), 4.26–4.06 (m, 2H), 3.42 (m, 1H), 3.41 (dd, J = 14.6, 3.0 Hz, 1H), 3.12 (dd, J = 14.6, 4.0 Hz, 1H), 1.64 (s, 9H), 1.50–1.13 (m, 7H), 1.06 (m, 3H), 1.04 (m, 1H), 0.73–0.42 (m, 2H) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CDCl3) δ 4.62 (m, 1H), 3.55 (m, 2H), 1.60 (s, 9H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 195.6, 191.6, 174.1, 155.8, 149.0, 132.6, 129.3, 126.6, 117.6, 117.0, 102.8, 84.4, 67.0, 65.5, 36.5, 34.6, 34.1, 28.36, 28.3, 26.1, 24.9, 23.5, 17.6 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3) δ 131.1, 130.7, 116.5, 115.4, 62.2, 36.6, 34.9, 33.8, 28.4, 28.0, 26.4, 25.8, 25.4, 23.8, 16.6 ppm; IR νmax 2971 (m), 2935 (m), 2233 (w), 2078 (m), 1740 (s), 1611 (s), 1509 (m), 1440 (m), 1354 (s), 1302 (m), 1256 (w), 1228 (s), 1218 (s), 1156 (m), 1116 (s), 972 (s) cm–1; HRMS (ESI) m/z [M + Na+] calcd. for C25H33NO6Na+ 466.22001, found 466.21899.
(3S,6Z,8R)-4-Aza-7-hydroxy-8-methyl-15-oxa-5,21-dioxo-tricyclo[14.2.2.13,6] henicosa-1(18),6,16(17),19-tetraene (3). To a solution of tetramic acid 18 (227 mg, 512 μmol, 1.00 eq) in CH2Cl2 (9.5 mL) was added TFA (950 μL). After stirring for 15 min at room temperature, toluene (100 mL) was added and the solvent was removed under reduced pressure. Toluene (50 mL) was added again and the solvent was removed, which afforded 3 as an orange foam. Yield: 176 mg (512 µmol, quant.). Rf = 0.51 (10% MeOH in CH2Cl2); –17.7° (c 0.74, MeOH); Major tautomer: 1H NMR (500 MHz, CD3OD): diastereotopic H-atoms indicated as a, b: δ 7.14 (m, 1H, CHArCCH2), 6.96 (m, 1H, CHArCCH2), 6.78 (m, 2H, CHArCO), 4.23–4.07 (m, 3H, CHN, ArOCH2), 3.33 (m, 1H, CHMe), 3.07a (dd, J = 14.3, 4.1 Hz, 1H, ArCH), 2.97b (dd, J = 14.4, 2.4 Hz, 1H, ArCHb), 1.52 (m, 2H, OCH2CH2), 1.44–1.16 (m, 5H, CHMeCHa, CMeCH2CH2, O(CH2)2CH2), 1.08b (m, 1H, CHMeCH), 1.05 (d, J = 6.9 Hz, 3H, CH3), 0.68–0.40 (m, 2H, CMe(CH2)2CH2) ppm; Significant signals minor tautomer: 1H NMR (500 MHz, CD3OD) δ 6.64 (m, 2H), 3.53 (m, 1H) ppm; Major tautomer: 13C NMR (125 MHz, CD3OD) δ 198.2 (CO, HMBC correlation), 190.2 (COH), 177.5 (HNCO, HMBC correlation), 157.0 (OCq,Ar), 133.3 (CHArCCH2), 130.8 (CHArCCH2), 128.1 (CH2Cq,Ar), 118.2 (OCCHAr), 117.4 (OCCHAr), 103.2 (NCOCCO), 67.6 (ArOCH2), 63.6 (CHN), 37.3 (CHMe), 36.4 (ArCH2), 35.2 (CMeCH2), 29.5 (CMeCH2CH2), 27.3 (CMe(CH2)2CH2), 25.9 (OCH2CH2), 24.7 (O(CH2)2CH2), 17.8 (CH3) ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CD3OD) δ 131.9, 116.0 ppm; Major tautomer: 1H NMR (500 MHz, CDCl3) δ 7.19–7.00 (m, 2H), 6.79 (m, 2H), 4.35–4.09 (m, 3H), 3.38 (m, 1H), 3.28–2.83 (m, 2H), 1.52 (m, 2H), 1.44–1.12 (m, 5H), 1.12 (m, 1H), 1.07 (d, J = 6.8 Hz, 3H), 0.67–0.41 (m, 2H) ppm; Significant signal minor tautomer: 1H NMR (500 MHz, CD3OD) δ 3.63 (m, 1H) ppm; Major tautomer: 13C NMR (125 MHz, CDCl3) δ 194.3, 192.1, 175.9, 155.7, 132.6, 129.6, 126.4, 117.3, 116.7, 102.0, 66.8, 62.2, 36.0, 34.4, 28.4, 26.0, 24.9, 23.6, 17.6 ppm; Significant signals minor tautomer: 13C NMR (125 MHz, CDCl3) δ 130.5, 116.3, 115.9, 66.9, 63.7, 36.4, 36.1, 29.4, 25.9, 25.1, 23.9, 17.1 ppm; IR νmax 2929 (m), 2853 (m), 1654 (s), 1608 (s), 1509 (s), 1456 (m), 1340 (m), 1259 (m), 1222 (m), 1174 (m) cm–1; HRMS (ESI) m/z [M + H]+ calcd. for C20H26NO4+ 344.18563, found 344.18481.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




