Helicobacter pylori Biofilm Confers Antibiotic Tolerance in Part via A Protein-Dependent Mechanism


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
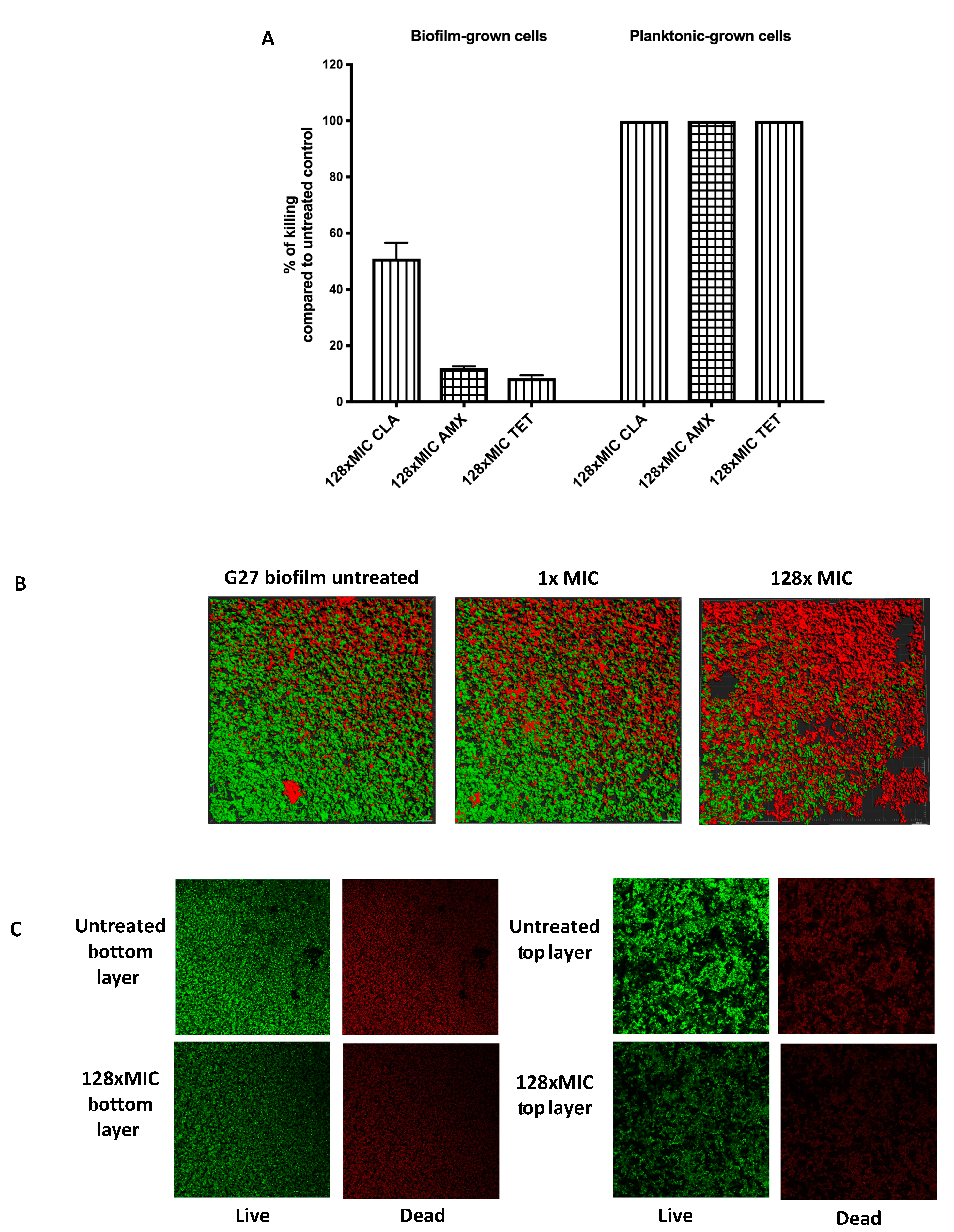
2.1. H. Pylori G27 Biofilm Cells are Antibiotic Tolerant
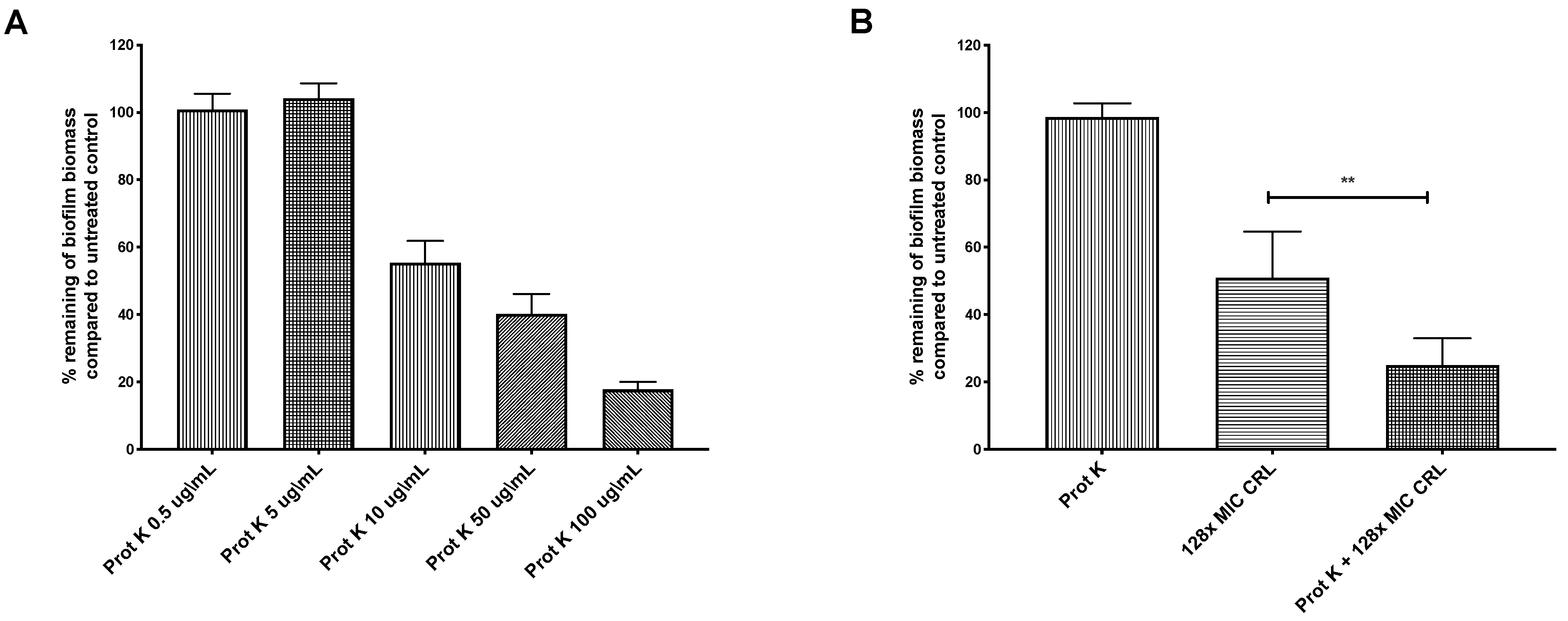
2.2. Biofilm-Associated Proteins Restrict Clarithromycin Effects
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Growth Conditions
4.2. Biofilm Formation and Crystal Violet Assays
4.3. Antibiotic Minimum Inhibitory Concentration (MIC) Determination
4.4. Planktonic and Biofilm Antimicrobial Susceptibility
4.5. Biofilm Dispersion Assay
4.6. Confocal Laser Scanning Microscopy
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Testerman, T.L.; Morris, J. Beyond the stomach: An updated view ofHelicobacter pyloripathogenesis, diagnosis, and treatment. World J. Gastroenterol. 2014, 20, 12781–12808. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Pisani, P.; Ferlay, J. Global cancer statistics. CA A Cancer J. Clin. 1999, 49, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.C.; Gurusamy, K.; Delaney, B.; Forman, D.; Moayyedi, P. Eradication therapy for peptic ulcer disease in Helicobacter pylori-positive people. Cochrane Database Syst. Rev. 2016, 2016, CD003840. [Google Scholar] [CrossRef] [PubMed]
- Vakil, N.; Vaira, B. Treatment for H. pylori Infection. J. Clin. Gastroenterol. 2013, 47, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chey, W.D.; Leontiadis, G.I.; Howden, C.W.; Moss, S.F. ACG Clinical Guideline: Treatment of Helicobacter pylori Infection. Am. J. Gastroenterol. 2017, 112, 212–239. [Google Scholar] [CrossRef]
- Fallone, C.A.; Chiba, N.; Van Zanten, S.V.; Fischbach, L.; Gisbert, J.P.; Hunt, R.H.; Jones, N.L.; Render, C.; Leontiadis, G.I.; Moayyedi, P.; et al. The Toronto Consensus for the Treatment of Helicobacter pylori Infection in Adults. Gastroenterology 2016, 151, 51–69.e14. [Google Scholar] [CrossRef] [Green Version]
- Crowe, S.E. Helicobacter pylori Infection. N. Engl. J. Med. 2019, 380, 1158–1165. [Google Scholar] [CrossRef]
- Mégraud, F.; Coenen, S.; Versporten, A.; Kist, M.; Lopez-Brea, M.; Hirschl, A.M.; Andersen, L.P.; Goossens, H.; Glupczynski, Y. Helicobacter pyloriresistance to antibiotics in Europe and its relationship to antibiotic consumption. Gut 2012, 62, 34–42. [Google Scholar] [CrossRef]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of Antibiotic Resistance in Helicobacter pylori: A Systematic Review and Meta-analysis in World Health Organization Regions. Gastroenterology 2018, 155, 1372–1382.e17. [Google Scholar] [CrossRef] [Green Version]
- Talarico, S.; Korson, A.S.; Leverich, C.K.; Park, S.; Jalikis, F.G.; Upton, M.P.; Broussard, E.; Salama, N.R. High prevalence of Helicobacter pylori clarithromycin resistance mutations among Seattle patients measured by droplet digital PCR. Helicobacter 2018, 23, e12472. [Google Scholar] [CrossRef]
- Marcus, E.A.; Inatomi, N.; Nagami, G.T.; Sachs, G.; Scott, D. The effects of varying acidity on Helicobacter pylori growth and the bactericidal efficacy of ampicillin. Aliment. Pharmacol. Ther. 2012, 36, 972–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.; Sachs, G.; Marcus, E.A. The role of acid inhibition in Helicobacter pylori eradication. F1000Research 2016, 5, 1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hathroubi, S.; Mekni, M.A.; Domenico, P.; Nguyen, D.; Jacques, M. Biofilms: Microbial Shelters against Antibiotics. Microb. Drug Resist. 2017, 23, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Genet. 2006, 5, 48–56. [Google Scholar] [CrossRef]
- Bernier, S.P.; Lebeaux, D.; DeFrancesco, A.S.; Valomon, A.; Soubigou, G.; Coppée, J.-Y.; Ghigo, J.-M.; Beloin, C. Starvation, Together with the SOS Response, Mediates High Biofilm-Specific Tolerance to the Fluoroquinolone Ofloxacin. PLoS Genet. 2013, 9, e1003144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents-How P. aeruginosa Can Escape Antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.S. Antimicrobial Tolerance in Biofilms. Microbiol. Spectr. 2015, 3, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Yonezawa, H.; Osaki, T.; Hanawa, T.; Kurata, S.; Ochiai, K.; Kamiya, S. Impact of Helicobacter pylori Biofilm Formation on Clarithromycin Susceptibility and Generation of Resistance Mutations. PLoS ONE 2013, 8, e73301. [Google Scholar] [CrossRef]
- Yonezawa, H.; Osaki, T.; Hojo, F.; Kamiya, S. Effect of Helicobacter pylori biofilm formation on susceptibility to amoxicillin, metronidazole and clarithromycin. Microb. Pathog. 2019, 132, 100–108. [Google Scholar] [CrossRef]
- Hathroubi, S.; Zerebinski, J.; Ottemann, K.M.; Hengge, R.; Cover, T. Helicobacter pylori Biofilm Involves a Multigene Stress-Biased Response, Including a Structural Role for Flagella. MBio 2018, 9, e01973-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windham, I.H.; Servetas, S.L.; Whitmire, J.M.; Pletzer, D.; Hancock, R.E.W.; Merrell, D.S. Helicobacter pylori Biofilm Formation Is Differentially Affected by Common Culture Conditions, and Proteins Play a Central Role in the Biofilm Matrix. Appl. Environ. Microbiol. 2018, 84, AEM.00139-18. [Google Scholar] [CrossRef] [Green Version]
- Grande, R.; Di Giulio, M.; Bessa, L.J.; Di Campli, E.; Baffoni, M.; Guarnieri, S.; Cellini, L. Extracellular DNA in Helicobacter pylori biofilm: A backstairs rumour. J. Appl. Microbiol. 2010, 110, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Grande, R.; Di Marcantonio, M.C.; Robuffo, I.; Pompilio, A.; Celia, C.; Di Marzio, L.; Paolino, D.; Codagnone, M.; Muraro, R.; Stoodley, P.; et al. Helicobacter pylori ATCC 43629/NCTC 11639 Outer Membrane Vesicles (OMVs) from Biofilm and Planktonic Phase Associated with Extracellular DNA (eDNA). Front. Microbiol. 2015, 6, 1114. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Cai, Y.; Chen, Z.; Gao, S.; Geng, X.; Li, Y.; Jia, J.; Sun, Y.; Li, Y. Bifunctional Enzyme SpoT Is Involved in Biofilm Formation ofHelicobacter pyloriwith Multidrug Resistance by Upregulating Efflux Pump Hp1174 (gluP). Antimicrob. Agents Chemother. 2018, 62, e00957-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.P.; Harwood, J.; Lee, R.; She, R.; Guiney, D.G. Characterization of Monospecies Biofilm Formation by Helicobacter pylori. J. Bacteriol. 2004, 186, 3124–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.K.; Huang, J.Y.; Wreden, C.; Sweeney, E.G.; Goers, J.; Remington, S.J.; Guillemin, K. Chemorepulsion from the Quorum Signal Autoinducer-2 Promotes Helicobacter pylori Biofilm Dispersal. mBio 2015, 6, e00379-15. [Google Scholar] [CrossRef] [Green Version]
- Yonezawa, H.; Osaki, T.; Kurata, S.; Fukuda, M.; Kawakami, H.; Ochiai, K.; Hanawa, T.; Kamiya, S. Outer Membrane Vesicles of Helicobacter pylori TK1402 are Involved in Biofilm Formation. BMC Microbiol. 2009, 9, 197. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.H.J.; Ng, C.G.; Goh, K.L.; Vadivelu, J.; Ho, B.; Loke, M.F. Metabolomic analysis of low and high biofilm-forming Helicobacter pylori strains. Sci. Rep. 2018, 8, 1409. [Google Scholar] [CrossRef] [Green Version]
- Sause, W.E.; Castillo, A.R.; Ottemann, K.M. The Helicobacter pylori Autotransporter ImaA (HP0289) Modulates the Immune Response and Contributes to Host Colonization. Infect. Immun. 2012, 80, 2286–2296. [Google Scholar] [CrossRef] [Green Version]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of Antibiotic Penetration, Oxygen Limitation, and Low Metabolic Activity to Tolerance of Pseudomonas aeruginosa Biofilms to Ciprofloxacin and Tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Sahore, S.; Kaur, P.; Rani, A.; Ray, P. Penetration barrier contributes to bacterial biofilm-associated resistance against only select antibiotics, and exhibits genus-, strain- and antibiotic-specific differences. Pathog. Dis. 2016, 74, ftw056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oubekka, S.D.; Briandet, R.; Fontaine-Aupart, M.-P.; Steenkeste, K. Correlative Time-Resolved Fluorescence Microscopy To Assess Antibiotic Diffusion-Reaction in Biofilms. Antimicrob. Agents Chemother. 2012, 56, 3349–3358. [Google Scholar] [CrossRef] [Green Version]
- Voss, B.J.; Gaddy, J.A.; McDonald, W.H.; Cover, T.L. Analysis of Surface-Exposed Outer Membrane Proteins in Helicobacter pylori. J. Bacteriol. 2014, 196, 2455–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, U.; Lee, C.-R. Distinct Roles of Outer Membrane Porins in Antibiotic Resistance and Membrane Integrity in Escherichia coli. Front. Microbiol. 2019, 10, 953. [Google Scholar] [CrossRef]
- Iyer, R.; Moussa, S.; Durand-Réville, T.F.; Tommasi, R.; Miller, A.A. Acinetobacter baumanniiOmpA Is a Selective Antibiotic Permeant Porin. ACS Infect. Dis. 2017, 4, 373–381. [Google Scholar] [CrossRef]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Mach, T.; Neves, P.; Spiga, E.; Weingart, H.; Winterhalter, M.; Ruggerone, P.; Ceccarelli, M.; Gameiro, P. Facilitated Permeation of Antibiotics across Membrane Channels—Interaction of the Quinolone Moxifloxacin with the OmpF Channel. J. Am. Chem. Soc. 2008, 130, 13301–13309. [Google Scholar] [CrossRef]
- Samsudin, F.; Ortiz-Suarez, M.L.; Piggot, T.J.; Bond, P.J.; Khalid, S. OmpA: A Flexible Clamp for Bacterial Cell Wall Attachment. Structure 2016, 24, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Odenbreit, S.; Swoboda, K.; Barwig, I.; Ruhl, S.; Borén, T.; Koletzko, S.; Haas, R. Outer Membrane Protein Expression Profile in Helicobacter pylori Clinical Isolates. Infect. Immun. 2009, 77, 3782–3790. [Google Scholar] [CrossRef] [Green Version]
- Taglialegna, A.; Navarro, S.; Ventura, S.; Garnett, J.A.; Matthews, S.J.; Penades, J.R.; Lasa, I.; Valle, J. Staphylococcal Bap Proteins Build Amyloid Scaffold Biofilm Matrices in Response to Environmental Signals. PLoS Pathog. 2016, 12, e1005711. [Google Scholar] [CrossRef] [Green Version]
- Rao, T.S.; Shukla, S.K. Staphylococcus aureus biofilm removal by targeting biofilm-associated extracellular proteins. Indian J. Med. Res. 2017, 146, S1–S8. [Google Scholar] [CrossRef]
- Latasa, C.; Solano, C.; Penadés, J.R.; Lasa, I. Biofilm-associated proteins. Comptes Rendus Boil. 2006, 329, 849–857. [Google Scholar] [CrossRef]
- Boyd, C.D.; Smith, T.J.; El-Kirat-Chatel, S.; Newell, P.D.; Dufrêne, Y.F.; O’Toole, G.A. Structural Features of the Pseudomonas fluorescens Biofilm Adhesin LapA Required for LapG-Dependent Cleavage, Biofilm Formation, and Cell Surface Localization. J. Bacteriol. 2014, 196, 2775–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.; Wurster, J.; Belenky, P. Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions. Pharmaceuticals 2018, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, D.N.; Shepherd, B.; Kraemer, P.; Hall, M.K.; Sycuro, L.K.; Pinto-Santini, D.M.; Salama, N.R. Identification of Helicobacter pylori Genes That Contribute to Stomach Colonization. Infect. Immun. 2006, 75, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydorn, A.; Nielsen, A.T.; Hentzer, M.; Sternberg, C.; Givskov, M.; Ersbøll, B.K.; Molin, S. Quantification of biofilm structures by the novel computer program comstat. Microbiology 2000, 146 Pt 10, 2395–2407. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hathroubi, S.; Zerebinski, J.; Clarke, A.; Ottemann, K.M. Helicobacter pylori Biofilm Confers Antibiotic Tolerance in Part via A Protein-Dependent Mechanism. Antibiotics 2020, 9, 355. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060355
Hathroubi S, Zerebinski J, Clarke A, Ottemann KM. Helicobacter pylori Biofilm Confers Antibiotic Tolerance in Part via A Protein-Dependent Mechanism. Antibiotics. 2020; 9(6):355. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060355
Chicago/Turabian StyleHathroubi, Skander, Julia Zerebinski, Aaron Clarke, and Karen M. Ottemann. 2020. "Helicobacter pylori Biofilm Confers Antibiotic Tolerance in Part via A Protein-Dependent Mechanism" Antibiotics 9, no. 6: 355. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060355