Catalytic Antioxidants in the Kidney


1
Department of Internal Medicine, College of Medicine, The Catholic University of Korea, Seoul 06591, Korea
2
Institute for Aging and Metabolic Diseases, College of Medicine, The Catholic University of Korea, Seoul 06591, Korea
*
Author to whom correspondence should be addressed.
Antioxidants 2021, 10(1), 130; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010130
Submission received: 28 December 2020
/
Revised: 9 January 2021
/
Accepted: 12 January 2021
/
Published: 18 January 2021
(This article belongs to the Special Issue Catalytic Antioxidants)
Abstract
:Reactive oxygen species and reactive nitrogen species are highly implicated in kidney injuries that include acute kidney injury, chronic kidney disease, hypertensive nephropathy, and diabetic nephropathy. Therefore, antioxidant agents are promising therapeutic strategies for kidney diseases. Catalytic antioxidants are defined as small molecular mimics of antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase, and some of them function as potent detoxifiers of lipid peroxides and peroxynitrite. Several catalytic antioxidants have been demonstrated to be effective in a variety of in vitro and in vivo disease models that are associated with oxidative stress, including kidney diseases. This review summarizes the evidence for the role of antioxidant enzymes in kidney diseases, the classifications of catalytic antioxidants, and their current applications to kidney diseases.

1. Introduction
Oxidative stress describes an imbalance between the formation of reactive species and the defense of antioxidants that occurs to a disturbance in redox signaling or molecular damage [1]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generated as toxic byproducts of the oxygen metabolism that is essential for living organisms. These free radicals consist of superoxide (O2•−), hydrogen peroxide (H2O2), nitric oxide (NO•), hydroxyl radicals (OH•), peroxynitrite (ONOO−), and lipid peroxyl radicals (LOO•) [1]. During respiration, cellular O2•− is produced endogenously in the mitochondria, and ROS are generated by complexes in the electron transport chain and partially reduced metabolites of molecular oxygen formed in biological systems [2]. Excessive ROS production develops through the activation of specific oxidases, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), xanthine oxidase, uncoupled nitric oxide synthase (NOS), and arachidonic acid-metabolizing enzymes [3]. ROS can induce the damage of cellular proteins, lipids, carbohydrates, and DNA, finally leading to cellular dysfunction. Therefore, they are being explored since early times as important modulating agents in numerous cellular signaling pathways (Figure 1) [4]. Antioxidant defense mechanisms are complicated and compartmentalized, enabling the independent regulation of cytoplasmic, mitochondrial, and nuclear levels of ROS [5]. ROS levels are regulated in living systems by numerous antioxidant enzymes, including superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), peroxiredoxin (Prx), thioredoxin (Trx), and cytochrome c oxidase [6,7].
Oxidative stress is implicated in the pathogenesis of various kidney diseases, including acute kidney injury (AKI), chronic kidney disease (CKD), hypertensive nephropathy, and diabetic nephropathy [8,9,10,11]. Therefore, antioxidant agents are promising therapeutic strategies for kidney disease. Catalytic antioxidants are small, molecular mimics of antioxidant enzymes such as SOD, CAT, and GPx, and some of them act as detoxifiers of lipid peroxides and ONOO− [12]. Since these compounds are catalytic and not simply free-radical scavengers, they display more potent antioxidant activity than other dietary supplements [12]. This article summarizes the evidence for the role of antioxidant enzymes in kidney disease, the classifications of catalytic antioxidants, and their current applications to kidney diseases.
2. Antioxidant Enzymes and Kidney Disease
Cells have crucial antioxidant defense mechanisms to protect themselves against toxic injury by free-radicals. Antioxidants can have endogenous or exogenous origins, with endogenous synthesis producing enzymes and small molecules or diet providing important exogenous defenses. Based on their activity, antioxidants can be divided into enzymatic and non-enzymatic. The primary enzymatic antioxidants are SOD, CAT, and GPx. Endogenous non-enzymatic antioxidants include L-arginine, lipoic acid, coenzyme Q10, melatonin, albumin, and uric acid [13]. Exogenous non-enzymatic antioxidants include nutrients such as ascorbic acid (vitamin C), α-tocopherol (vitamin E), phenolic antioxidants, lecithin oil, and drugs such as acetylcysteine [14]. There are also several antioxidant systems in the kidney to protect renal tissue and related cells from oxidative stress.
2.1. Superoxide Dismutase and Kidney Disease
Superoxide radical anions are a potentially harmful species produced from the one-electron reduction of molecular oxygen during respiration. SODs are key antioxidant enzyme systems, and most organisms that live in the presence of oxygen express at least one SOD. The coordinated metals at the active site can be used to classify SODs: copper-zinc SOD (Cu/Zn-SOD), manganese SOD (Mn-SOD), iron SOD (Fe-SOD), and nickel SOD (Ni-SOD) [4]. As a group of metalloenzymes that catalyze dismutation reactions to detoxify ROS [12], SODs catalyze the dismutation of two O2•− to yield H2O2 and molecular O2, which is decomposed into water and oxygen through CAT [15].
2O2•− + 2H3O+ → O2 + H2O2 + 2H2O
SODs are also classified into three major isoforms based on their localization in subcellular compartments: SOD1 (Cu/Zn-SOD), SOD2 (Mn-SOD), and SOD3 (Extracellular SOD, EC-SOD), all of which are normally presented in the kidney. SOD1 constitutively exists in the cytosol and intermembrane space on mitochondria, and SOD2 is found in the mitochondria of eukaryotic cells. SOD3 is a Cu/Zn-SOD that is secreted into the extracellular space [16]. Of those three SODs, SOD1 is abundant in most tissues and accounts for 60–80% of SOD activity and about 30% of SOD activity in the renal vasculature in the kidneys [17]. SOD2 is also expressed in most tissue cells, e.g., stomach, lung, skeletal muscle, spleen, heart, liver, kidney, and brain [18]. SOD3 is highly expressed in the blood vessels, kidneys, lungs, and heart [4]. Although SOD1 accounts for the highest proportion of SOD activity in the kidney, SOD2 deficiency has been associated with more severe pathological changes than SOD1 deficiency [19], because ROS and RNS are mainly formed in the mitochondria [11].
All three of the SOD isoforms play a crucial role in the deterioration and alleviation of various kidney diseases. Several experimental studies have provided evidence that deleting or overexpressing SODs through genetic manipulation or medication changes oxidative stress and the disease severity of AKI or CKD. The depletion of SOD1 causes a significant increase in nuclear factor κ light chain enhancer of activated B cells (NF-κB)-mediated signal transduction and oxidative DNA damage in the kidneys [20,21]. Indeed, SOD1 knockout mice had severely decreased renal function after renal ischemia/reperfusion (I/R) injury [22], and treatment with recombinant human SOD1 significantly decreased ROS and improved renal function by decreasing tissue levels of tumor necrosis factor (TNF)-α and interleukin (IL)-1 in renal I/R injury [23]. SOD1 deficiency augmented salt sensitive–hypertension and tubulointerstitial fibrosis in unilateral ureteral obstruction (UUO) mice, whereas SOD1 overexpression using transgenic mice or chronic tempol treatment abolished those findings in UUO mice [24]. SOD1 also regulates renal microvascular remodeling, arteriolar responsiveness, and sensitivity to angiotensin II (Ang II). SOD1 knockout mice displayed increased blood pressure and decreased afferent arteriole diameter during Ang II infusion, and those changes were mitigated in SOD1-transgenic mice [25]. In diabetic nephropathy, advanced glycation end products (AGEs) augmented oxidative stress via the ROS production by NOX in the mitochondria, and the interaction between AGEs and the receptor of AGEs (RAGE) enhanced the initiation of related signal transduction [26]. Antioxidant enzymes, such as SOD and CAT inhibit AGE-mediated ROS production. SOD1-transgenic db/db mice and streptozotocin (STZ)-treated SOD1-transgenic mice exhibited reduced albuminuria, transforming growth factor (TGF)-β1, and collagen IV expression, along with mesangial matrix expansion and decreased oxidative stress markers compared with control diabetic mice [27,28].
SOD2 dysfunction has been reported to aggravate renal dysfunction, tubulointerstitial fibrosis, inflammation, and apoptosis in the kidney [29]. Parajuli et al. found that kidney-specific SOD2-deficient mice had lighter and smaller kidneys than wild type mice and enhanced oxidative stress and tubular injury, including the dilation of distal tubules, protein cast formation, and epithelial cell swelling in distal tubules [30]. In renal I/R injury, SOD2 knockout mice exhibited lower expression of SOD2 in the distal nephrons and exacerbated renal function compared with control mice [31]. Pretreatment with recombinant SOD2 significantly increased SOD activity and ameliorated renal function declines and tubular necrosis in a rat model of radiocontrast-induced AKI [32]. Furthermore, a high salt diet in SOD2-deficient mice caused a significant increase in arterial pressure and urinary albumin excretion through the upregulation of NOX and the activation of NF-κB [33]. Another study also showed that SOD2 deficiency aggravated renal interstitial inflammation and accelerated glomerulosclerosis, tubulointerstitial damage, and salt-sensitive hypertension, especially in aged mice [34]. The mechanism of impaired microvascular function proposed by those authors was that SOD2 deficiency increased O2•− levels and impaired the flow and agonist-induced vasodilation of isolated mesenteric arteries [35].
Excess mitochondrial O2•− production and related mitochondrial dysfunction have been associated with the pathogenesis of diabetic nephropathy [36,37]. Several experiments have reported reductions in SOD2 activity in animal models of type 1 and type 2 diabetic nephropathy [38,39,40]. In contrast, other studies have reported no significant differences in SOD2 expression between diabetic and control mice [41,42]. Dugan et al. showed that SOD2-deficient mice with diabetes had increased renal ROS, but they found no evidence of an increase in albuminuria or mesangial matrix expansion [43]. Therefore, the role of SOD2 in diabetic nephropathy is controversial, and additional research is needed to determine the mechanisms of SOD2 activity in diabetic nephropathy.
As with SOD1 and SOD2, several studies have used SOD3 knockout animal models to demonstrate the role of SOD3 in protecting against or accelerating kidney damage in response to oxidative stress. Ang II treatment after renal artery clipping in SOD3 knockout mice developed higher blood pressure and induced endothelial dysfunction, and recombinant SOD3 treatment selectively decreased blood pressure in hypertensive SOD3 knockout mice [44]. Another study reported that SOD3 localizes predominantly in the proximal tubules and colocalizes with erythropoietin (EPO). Compared with the control animals, hypoxia-exposed SOD3 knockout mice showed smaller increases in their EPO levels and lesser accumulations of nuclear translocated hypoxia-inducible factor (HIF)-1α by the activation of NOX in the kidneys [45]. In line with that finding, the deletion of SOD3 blunted renal blood flow recovery after renal ischemia and significantly increased tubular necrosis and tubular cast formation after reperfusion [46]. SOD3 knockout mice also had increased proteinuria and renal fibrosis and podocyte injury after adriamycin treatment, an experimental model of focal segmental glomerulosclerosis (FSGS), and that finding was associated with an upregulation of NOX2 and β-catenin signaling [47]. Therefore, SOD3 plays a crucial role in renal protection against diverse kidney diseases.
To assess the role of SOD isoforms in diabetic nephropathy, Fijuta et al. evaluated SOD activity and SOD isoform expression in the kidneys of diabetic mouse models and found the downregulation of SOD1 and SOD3, but not SOD2, in diabetic kidneys [42]. The same group reported using SOD1- and SOD3-knockout diabetic mice to confirm the distinct role of SOD isoforms in diabetic nephropathy [48]. They suggested that SOD1 deficiency, but not SOD3 deficiency, increases renal O2•− and causes overt renal injury in C57BL/6-Akita diabetic mice and that SOD1 plays a more prominent role than SOD3 in the pathogenesis of diabetic nephropathy. However, recent studies have reported that SOD3 has an independent role in protection against diabetic nephropathy [49,50]. Our study demonstrated that the expression of SOD3 was significantly increased in the glomerulus and tubular area of db/db mice after recombinant human SOD3 supplements [50]. Recombinant human SOD3 supplements ameliorated diabetic nephropathy by inhibiting ROS and the phosphorylation of extracellular signal-regulated kinase (ERK)1/2 or the activation of intrarenal 5′-AMP-activated protein kinase–peroxisome proliferator-activated receptor γ coactivator (PGC)-1α–nuclear factor erythroid-2-related factor (Nrf)2 signaling in animal models of type 1 and type 2 diabetic nephropathy [49,50]. Therefore, further experiments are needed to clarify the independent role of SOD3 in protecting against diabetic nephropathy.
2.2. Catalase and Kidney Disease
CAT is a 240-kDa homotetrameric heme-containing protein located predominantly in the peroxisome and abundantly present in the liver, lungs, and kidneys [51]. In the kidney, CAT is largely distributed in the cytoplasm of proximal tubules of the juxtamedullary cortex but is less expressed in the proximal tubules of the superficial cortex. On the other hand, CAT is not present in the glomeruli, distal tubules, loop of Henle, or collecting ducts [52]. CAT deficiency results in the overexpression of mitochondrial ROS and functional mitochondrial impairment [53]. CAT reduces the H2O2 generated by SOD into oxygen and water. Although CAT is highly efficient at reducing H2O2, its role in modulating H2O2 might not be central because it is mainly localized in the peroxisome.
2H2O2 → 2H2O + O2
CAT deficiency has been reported to increase tubulointerstitial fibrosis and the lipid peroxidation products of tubulointerstitial lesions in UUO mice [54]. Kobayashi et al. confirmed that CAT decreased renal function and accelerated progressive renal fibrosis through the upregulation of the epithelial to mesenchymal transition in the remnant kidneys of acatalasemic mice subjected to 5/6 nephrectomy [55]. In addition, adriamycin treatment in acatalasemic mice produced severe albuminuria, accelerated glomerulosclerosis and tubulointerstitial fibrosis, and enhanced lipid peroxide accumulation compared with wild-type mice [56].
In diabetic nephropathy, proximal tubule-specific overexpression of CAT inhibited renal ROS production and tubulointerstitial fibrosis and attenuated angiotensinogen, p53, and proapoptotic Bcl-2 associated X-protein (BAX) gene expression in STZ-treated diabetic mice and db/db mice [57,58]. Consistent with those studies, CAT overexpression in Akita mice significantly decreased systolic blood pressure by regulating the intrarenal renin-angiotensin system (RAS), which enhanced angiotensin-converting enzyme (ACE)2 and suppressed ACE and angiotensinogen expression [59], or by activating the nuclear factor erythroid 2–related factor 2 (Nrf2)-heme oxygenase (HO)-1 signaling pathway [60]. Godin et al. confirmed the association between CAT and intrarenal RAS actions in the development of hypertension and renal injury using proximal tubule–specific CAT and/or angiotensinogen transgenic mice [61]. Another researcher also reported that CAT deficiency accelerated diabetic nephropathy by impairing peroxisomal/mitochondrial biogenesis and fatty acid oxidation [53]. Therefore, endogenous CAT plays an important role in protecting against diabetic nephropathy by decreasing oxidative stress through the regulation of intrarenal RAS and peroxisomal metabolism.
2.3. Glutathione Peroxidase and Kidney Diseases
GPx, another H2O2 scavenger, converts peroxides and OH• into nontoxic substances by oxidizing reduced glutathione (GSH) into glutathione disulfide (GSSG), which is then reduced back to GSH by glutathione reductase using NADPH [62,63]. GPx cooperates with CAT to decompose H2O2 to H2O and oxidized glutathione, which is then reduced by glutathione reductase. GPx requires GSH as a hydrogen donor to decompose H2O2 into water and oxygen and selenium (Se) as a cofactor to participate in the reaction with peroxides [64].
GPxs are tetrameric proteins in which each monomer includes one atom of Se at the catalytic site. Each monomer contains a selenocysteine, which the sulfur in the cysteine has been replaced by selenium (R-SeH). Throughout the catalytic cycle, a selenol (protein-Se−) reacts with peroxide (H2O2 or lipid hydroperoxide, LOOH) to produce selenenic acid (protein-SeOH). Selenenic acid regenerates selenol by two GSH, and GSH are finally oxidized into a GSSG and LOOH. LOOH is reduced to its corresponding lipid alcohol (LOH) [65].
H2O2 + 2GSH → GSSG + 2H2O
LOOH + 2GSH → GSSG + H2O + LOH
LOOH + 2GSH → GSSG + H2O + LOH
To date, eight different GPxs have been found in mammals; however, only five isoforms contain the selenocysteine needed to catalyze the reduction of H2O2 and LOOHs using GSH as a reducing cofactor (GPx 1–4 and 6) [66]. In the kidney, substantial amounts of GPx have been found in the proximal and distal tubules and smooth muscle cells of the renal arteries [67]. Among the GPx isoforms, GPx1 and GPx4 expression is mainly detected in podocytes and mesangial cells [68]; GPx3 is produced in the basement membranes of the renal cortical proximal and distal convoluted tubules [69]; and GPx2 and GPx5 have not been detected in the kidney. As the first to be identified, GPx1 is highly expressed throughout the human body, and its role in the reduction of oxidative stress has been widely demonstrated [67]. GPx1 predominantly exists in normal kidneys, accounting for 96% of kidney GPx activity [70,71]. Esposito et al. demonstrated that GPx1 is substantially expressed in the mitochondria of the kidney cortex, and GPx1 deficiency reduced body weight and exacerbated an endogenous, age-dependent decline in overall cellular function [72]. Therefore, regulation of kidney GPx1 was postulated to play a principal role in protecting kidneys from oxidative stress [71].
Several previous studies have been evaluated the renoprotective role of GPx1 against kidney diseases. Genetic inhibition of GPx1 aggravated cocaine-induced AKI by activating the angiotensin II type-1 receptor (AT1R) through the inhibition of phosphoinositide 3-kinase (PI3K)-Akt signaling [73]. In addition, GPx1 overexpression improved glomerulosclerosis by attenuating oxidative stress and mitochondrial ROS in aged mice [74]. In diabetic nephropathy, Chiu et al. reported that plasma and urine GPx levels were substantially lower in patients with diabetic glomerulosclerosis than in those without glomerulosclerosis and that glomerular GPx expression was lower in diabetic rats than in the normal control rats [75]. However, GPx1-deficient diabetic mice showed levels of oxidative injury, glomerular damage, and renal fibrosis similar to those found in the control diabetic mice, and GPx1 deficiency was not endogenously compensated by the increases of CAT or other GPx isoforms in the early stage of diabetic kidney disease [71]. Enhanced GPx activity and GPx carboxylation did not accompany a concomitant increase in GPx expression in the kidneys of young diabetic mice, and GPx1 and GPx4 expression and activity in the kidney did not differ between aged diabetic and non-diabetic mice [68]. In contrast, Chew et al. demonstrated that in diabetic ApoE/GPx1 double knockout mice, GPx1 deficiency increased albuminuria, which is associated with increased mesangial matrix expansion and the upregulation of inflammatory and fibrotic mediators [76]. Therefore, the renoprotective role of GPx1 against diabetic kidney disease remains uncertain.
GPx3 is an extracellular antioxidant selenoprotein that is also called plasma GPx [77]. GPx3 is mainly synthesized in the basolateral compartment of the kidney and binds to the basement membrane of renal cortical epithelial cells [69]. GPx3 also binds to basement membranes of extra-renal epithelial cells in the gastrointestinal tract, lung, and epididymis though the bloodstream [78]. These findings suggest that GPx3 deficiency due to kidney injury may affect the remote organ. Indeed, GPx3 deficiency significantly decreased survival rates and promoted left ventricular dysfunction due to a ROS accumulation that exacerbated inflammatory signaling and platelet activation in a surgery-induced CKD model [79]. Therefore, GPx3 may play an important role in the crosstalk between kidney and other organs.
Recently, ferroptosis, an iron-dependent programmed cell death characterized by accumulating lipid hydroperoxides to lethal levels, has been reported to be involved in the pathophysiology of various renal diseases [80,81,82]. GPx4 is the primary enzyme that prevents ferroptosis, and a GPx4 inhibitor induced ferroptotic cell death by binding and inactivating GPx4 [83]. GPx4 deficiency also aggravated AKI through an increase of intracellular LOOH and the promotion of ferroptotic cell death; liproxstatin-1 prevented kidney injury associated with GPx4 depletion [84]. A recent study showed that diabetic mice had significantly increased levels of acyl-CoA synthetase long-chain family member 4 (ACSL4) and decreased GPx4, and those findings suggest that ferroptosis was involved in the pathogenesis of diabetic nephropathy [85]. To date, no association has been elucidated between GPx2 and GPx5 and renal disease.
3. Catalytic Antioxidants
Excessive ROS produces oxidative damage to cellular structures through an imbalance in the oxidant–antioxidant status, and therefore, antioxidants can be used therapeutically to recover the balance between ROS generation and removal [86]. Several exogenous, native antioxidants have proved unsuccessful as therapeutic strategies because of their short half-life, low cell permeability due to their large size, antigenicity, and high-manufacturing costs [12,87]. Catalytic antioxidants have caught the attention of experts for the treatment of diseases associated with ROS. Several catalytic antioxidants have been designed and developed based on the structures of the active sites of native antioxidative enzymes. Therefore, they have been documented to exhibit SOD activity, ONOO−-reduction activity, CAT activity, and GPx activity.
Catalytic antioxidants can be classified as independent catalytic antioxidants (ICAs) and cofactor-dependent catalytic antioxidants (DCAs) by how they perform their catalytic action [88]. ICAs decompose ROS/RNS without the need for any additional compounds. The representative ICAs are SOD and CAT mimics. The low-valence metal ions in those enzymes reduce O2•−, and the high-valent metal ion formed in that way oxidizes a second molecule of the toxin. DCAs require the help of other cofactors to complete their full catalytic cycle. GPx and Prx mimics are representative DCAs that require GSH and Trx, respectively, to reduce H2O2 to H2O. Catalytic antioxidants, specialized classes of organometallic complexes, are mimics of SOD, CAT, or GPx that can detoxify a broad range of ROS [12,89].
3.1. Catalytic Antioxidants as SOD and CAT Mimics
SODs are ubiquitous metalloproteins that act as the first line of defense enzymes against ROS via the dismutation of O2•− to H2O2 and molecular oxygen. Since heme is a naturally discovered native metalloporphyrin, Fe porphyrins, FeTM-4-PyP5+, were the first compounds proposed as SOD mimics in the late 1970s [90]. However, manganese (Mn) complexes remain the most stable and potential SOD mimics [91,92]. Currently, the main types of Mn-SOD mimetics are Mn cyclic polyamines, Mn and Fe porphyrin, Mn salen, and non-metal compounds such as nitrones and nitroxides [93]. Of the known synthetic compounds, nitrones and nitroxides cannot catalytically scavenge O2•−, but they can react with ONOO−.
3.1.1. Macrocyclics
Macrocyclics contain an Mn atom coordinated to five nitrogen ligands. Riley et al. designed Mn(II) cyclic polyamines, which is an optimized SOD mimetic (M40403, M series by Metaphore Pharmaceuticals) [94]. M40403 is a stable low molecular, Mn-containing, non-peptidic molecule that has the similar function and efficacy of native SOD enzymes [94]. The pentavalent coordination allows the Mn to participate in only single-electron transfers, which makes the compound specific for O2•− scavenging because H2O2 or ONOO− scavenging requires two-electron transfers [12,95,96]. In the kidney, only one study has demonstrated that M40403 protected gentamicin-induced AKI by suppressing nitrotyrosine formation and poly(ADP-ribose) synthetase activation [97].
3.1.2. Mn porphyrins
Metalloporphyrins are cell-permeable, redox-active, catalytic antioxidants that act as SOD mimetics, and have been demonstrated as potent catalysts of numerous redox reactions. Most metalloporphyrins have either a Fe or Mn moiety coordinated in four nitrogen axial ligands. Mn porphyrins (MnPs), the most potent Mn-SOD mimics, have been optimized to accumulate in the mitochondria, where they similarly act at the Mn-SOD catalytic site [89,98]. Meso-substituted metalloporphyrin analogs have varying degrees of SOD activity, net charge, and pharmacodynamic characteristics [99,100]. Generally, metalloporphyrins with stronger SOD activity possess greater CAT activity, but the CAT activity of SOD mimetics is less than 1% that of native CAT [89].
The Mn moiety of the SOD mimetics functions in the dismutation reaction with O2•− by alternately reducing and oxidizing, which changes its valence from Mn(III) to Mn(II), much like native SODs. The O2•− dismutation process of MnPs consists of two steps in which the Mn center cycles between Mn(III) and Mn(II). In the first step, Mn(III) reduced by O2•− to yield Mn(II) and O2, and this step is considered as the rate-limiting step. The second step is the oxidation of Mn(II) by O2•− to yield H2O2 and regenerate the Mn(III) porphyrins. This catalytic cycle is obviously modulated by the redox potential of the metal site [92]. The antioxidant efficiency of MnPs in vivo depends on their bioavailability, i.e., tissue, cellular, and subcellular distribution, and the nature of N-pyridyl substituents, which can alter their charge, size, shape, and lipophilicity [98,101,102,103].
MnP-based SOD mimics have been designed to imitate the kinetics and thermodynamics of SOD enzymes during the catalysis of O2•− dismutation [104]. Since Irwin Fridovich first developed MnPs as potential SOD mimics in 1994 [105], diverse MnPs have been synthesized as cellular redox modulators. The first MnP-based lead compound was the cationic porphyrin Mn(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin (MnTM-2-PyP5+, AEOL10112), along with Mn(III) meso-tetrakis(N-methylpyridinium-4-yl)porphyrin (MnTM-4-PyP5+) and the anionic porphyrin Mn(III) tetrakis(4-benzoic acid)porphyrin (MnTBAP3−, AEOL10201) (AEOL series by Aeolus Pharmaceuticals). In a next step of drug development, the ethyl analogue, Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+, AEOL10113, BMX-010) was synthesized [106,107]. MnTE-2-PyP5+ has increased bulkiness relative to MnTM-2-PyP5+, which reduces its interactions with nucleic acids and thereby its toxicity. Therefore, MnTE-2-PyP5+ emerged as one of the potent synthetic SOD mimetics and an effective ONOO− scavenger. Mn(III) meso-tetrakis(1,3-diethylimidazolium-2-yl)porphyrin (MnTDE-2-ImP5+, AEOL 10150) is structurally different from MnTE-2-PyP5+, which possesses imidazole side chain substitutions. MnTDE-2-ImP5+ has kinetics and thermodynamics similar to MnTE-2-PyP5+, but it is bulkier, so it has different bioavailability.
Later investigators demonstrated the relationship between MnP lipophilicity and its biological activity. More lipophilic SOD mimics are effective at lower concentrations, whereas less lipophilic compounds are highly effective only at concentrations above 10 μM [108]. Therefore, the next stages in the drug development of MnP were improvements in lipophilicity that produced Mn (III) meso-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin (MnTnHex-2-PyP5+) and decreases in toxicity while maintaining lipophilicity, which produced Mn(III) meso-tetrakis(N-n-butoxyethyl-pyridinium-2yl)porphyrin (MnTnBuOE-2-PyP5+, BMX-001) [103,109]. MnTnHex-2-PyP5+ has received much attention because it is significantly more lipophilic than MnTE-2-PyP5+ while having the same catalytic activity to eliminate O2•− and ONOO− [110]. Due to potent lipophilicity, MnTnHex-2-PyP5+ is distributed at the highest levels to all organs and accumulates in mitochondria better than most of the other analogs; it also shows decreased toxicity because of its micellar character [111]. MnTnHex-PyP5+ has a better therapeutic option than MnTE-2-PyP5+ due to its high bioavailability. To date, several MnPs, including MnTE-2-PyP5+ and MnTnBuOE-2-PyP5+, are currently being tested in clinical trials for cancerous and non-cancerous conditions [112].
Several cationic MnPs have been investigated in various models of kidney injury. Previous research reported that MnTM-4-PyP5+ administration attenuated tubulointerstitial damage in I/R injury by inhibiting apoptosis and proinflammatory cytokines [113,114]. Park et al. demonstrated that long-term administration of MnTM-4-PyP5+ ameliorated renal fibrosis after ischemic AKI by decreasing the deposition of collagen and accelerating the normalization of primary cilia length [115,116]. The same researchers later demonstrated the renoprotective mechanism of MnTM-4-PyP5+ using UUO mice. They found that it decreased ROS and prevented the elongation of primary cilia by inhibiting phosphorylated ERK, p21, and exocyst complex members Sec8 and Sec10 [117]. Another MnP, MnTnHex-2-PyP5+, also protected against renal I/R injury by inducing the production of ATP synthase-β subunit [110]. Similarly, the administration of MnTE-2-PyP5+, MnTM-4-PyP5+, or MnTM-2-PyP5+ conferred protection against the harmful effects associated with sepsis-induced AKI [118,119] and against diabetic nephropathy [120,121].
MnTBAP3− compounds were initially developed as stable and efficient anionic SOD mimics [122], but later neither SOD-like activity nor CAT-like activity of MnTBAP3− has been elucidated [123]. MnTBAP3− was not efficacious due to its poor kinetics and thermodynamics; negative charges repelled this compound from the negatively charged deprotonated protein cysteines. Therefore, Rebouças and colleagues suggested that pure MnTBAP3− could not interact with protein cysteines and catalyze H2O2 dismutation in aqueous media [124]. They suggested that MnTBAP3− has often been inappropriately described as a SOD- and CAT-mimic and that its therapeutic effects have been erroneously assigned to SOD-like activity [103,124]. In addition, pure MnTBAP3− can partially reduce ONOO−, but only if it is administered at high concentrations [125].
Despite the controversy, several experimental studies have demonstrated the renoprotective effects of MnTBAP3− in various models of kidney disease. Zahmatkesh et al. showed that administering MnTBAP3− prior to ischemia prevented renal I/R injury without changing plasma NOx levels [126,127]. Therefore, they suggested that MnTBAP3− is not a NO scavenger and that its action could be mediated by the inhibition of ONOO− production. Similarly, MnTBAP3− attenuated cisplatin-induced nephrotoxicity by enhancing HO-1 and reducing nitrative stress [128]. Other researchers reported that MnTBAP3− reduced ROS production and mitochondrial dysfunction by inhibiting the NLR family pyrin domain containing 3 (NLRP3) inflammasome and subsequently releasing proinflammatory cytokines in animal models of albumin- and aldosterone-induced renal tubular injury [129,130]. MnTBAP3− also prevented tubulointerstitial fibrosis and mitochondrial dysfunction by reducing the deposition of extracellular matrix components, including fibronectin, collagen I, and collagen III, in mice with 5/6 nephrectomy [131].
3.1.3. Manganosalens
The Mn(III)-containing salen compounds, i.e., EUK series (EUK series by Eukarion), are Mn complexes with a semi-cyclic ligand salen [12,92]. They have the catalytic activity of SOD, CAT, and peroxidase, and their mechanism of action is similar to those of the metalloporphyrins [132]. The EUK compounds have been shown to scavenge O2•− and H2O2, react with ONOO−, and maybe react with lipid peroxides [12,65,133]. Mn(III) salens have modest SOD-like activity, whereas Mn(II) cyclic polyamines and Mn(III) porphyrins possess high SOD-like activity [98]. The prototype salen Mn complex (EUK-8) and the improved CAT mimetics (EUK-134 and EUK-189) are effective in a wide range of disease models, including kidney disease [15,133].
In the kidney, several experiments using both EUK-8 and EUK-134 have been performed. EUK-134 prevented renal dysfunction and tubulointerstitial injury by reducing oxidative and nitrosative stress in renal I/R injury [134,135]. In renal proximal tubular cells, EUK-134 significantly improved cell viability and reduced paraquat-induced cell death by reducing the production of O2•− and OH• [136]. EUK-8 attenuated lipopolysaccharide (LPS)-induced renal injury and delayed hypotension caused by endotoxins [137], and EUK-134 also prevented the LPS-induced fall in renal blood flow, which was associated with a decrease in protein nitrotyrosinylation in the kidney [138]. In an in vitro model of CKD, endothelial cells exposed to serum from uremic patients decreased their expression of intercellular adhesion molecule (ICAM)-1 and increased the phosphorylation of p38 mitogen-activated protein kinase (p38MAPK)-NF-κB signaling, and EUK-118 and EUK-134 treatment significantly decreased both intracellular ROS and phosphorylated p38MAPK-NF-κB expression [139].
3.1.4. Nitroxides
Nitroxides, including tempol and Mito-TEMPO, are another class of non-metal-based SOD mimetics. Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl) is a redox-cycling, water-soluble nitroxide that exhibits SOD-like activity and O2•− scavenging activity [140]. Tempol is among the most potent of the nitroxides in protecting cells and tissues against ROS, but it does not sustain significant metabolism for more than a few hours due to a rapid exchange between the nitroxide, hydroxylamine, and oxammonium cation species [141]. To date, the renoprotective effects of tempol have been demonstrated in numerous experimental studies of diverse kidney diseases, especially hypertensive and diabetic kidney disease. Pretreatment with tempol attenuated renal dysfunction and decreased ROS in renal I/R injury and LPS-induced AKI models [142,143,144]. The decrease of SOD activity in diabetic nephropathy was widely known through previous experiments, and tempol treatment in diabetic nephropathy restored renal function and the activity of antioxidant enzymes, including SOD and GPx [42,145,146,147]. These effects were attributed to improved endothelial function [145], reduced renal vascular resistance associated with HO-1 expression [146], and the upregulation of transient receptor potential cation channel subfamily C member 6 (TRPC6) expression [147]. Consistent with the results from diabetic nephropathy, obese, diabetic, hypertensive ZSF1 rats treated with tempol showed increased SOD activity and significantly reduced lipid peroxidation and peroxidase activity in the kidney [148].
Since hypertension and renal vasoconstriction depend on O2•−, the biological effects of tempol on endothelial function have been studied extensively in various animal models of hypertension. Tempol treatment decreased the mean arterial pressure by decreasing the renal sympathetic nerve response [149], increasing plasma renin activity [150], and increasing medullary blood flow and sodium excretion [151] in hypertensive animal models, such as spontaneously hypertensive rats and fructose-hypertensive rats. Nishiyama et al. also demonstrated that tempol protects against glomerular injury by inhibiting MAPK and NOX signaling in a salt-dependent model of hypertension [152]. Another chronic renal hypoxia model using the two-kidney, one-clip hypertension technique decreased SOD1 expression, especially in the tubulointerstitial area, which was associated with increased TNF-α. Tempol treatment ameliorated tubulointerstitial injury and reduced macrophage infiltration in the renovascular hypertensive model [153]. Chronic Ang II infusion was also accompanied by extensive renal fibrosis, represented as upregulated NOX and suppressed SOD. Cotreatment with an NADPH inhibitor and tempol inhibited TGF-β1 expression and the related fibrogenic responses in the chronic Ang II infusion model of hypertensive kidney disease [154]. Consistent with chronic Ang II infusion, mice who underwent 5/6 nephrectomy downregulated SOD1 and SOD2, upregulated NOX, and increased atrial pressure and nitrotyrosine, and tempol treatment ameliorated the hypertension and increased the level of urinary NO metabolites [155].
Mito-TEMPO, a mitochondrial-targeted SOD mimetic, is a nitroxide linked to the triphenyl phosphonium cation, which promotes 1,000-fold accumulation into the mitochondrial matrix [156,157]. Mito-TEMPO restored renal mitochondrial function and attenuated sepsis-induced AKI by decreasing mitochondrial oxidative stress and increasing Mn-SOD activity [158]. Mito-TEMPO also prevented aldosterone-induced renal tubular injury by restoring mitochondrial function and suppressing the activation of the NLRP3 inflammasome and apoptosis [159]. In addition, mitochondrial dysfunction, inflammatory cytokine levels, oxidative stress, and endoplasmic reticulum (ER) stress were involved in 5/6 nephrectomy-induced renal fibrosis, and Mito-TEMPO attenuated tubulointerstitial fibrosis by ameliorating renal inflammation, mitochondrial dysfunction, and ER stress [160]. Furthermore, indoxyl sulfate treatment in the 5/6 nephrectomy model augmented renal fibrosis and decreased renal function by activating the NOX and RhoA/Rho-associated kinase (ROCK) pathway; Mito-TEMPO or tempol decreased NOX and increased SOD1 and SOD2 in the thoracic aorta of indoxyl sulfate-treated 5/6 nephrectomy model [161].
3.2. Catalytic Antioxidants as GPx Mimics
3.2.1. Ebselen
Organoselenium compounds exhibit potent antioxidant activity mediated by GPx mimetic properties. Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one or PZ51), the first Se-based GPx mimic, is one of the best studied GPx mimics [65]. Ebselen can metabolize peroxides using GSH or directly reduce thioredoxin reductase [11]. Ebselen also reduces H2O2 and lipid peroxides and scavenges ONOO− without affecting endogenous NO• [162]. Ebselen was less toxic than other treatments tested because of its stable isoselenazole moiety, and it proved to be an effective treatment for experimental models of diverse kidney diseases. The beneficial effects of ebselen were mainly investigated in cisplatin-induced AKI models [163,164,165,166]. Baldew et al. first demonstrated that pretreatment with ebselen prevented cisplatin-induced renal injury and that the protective effect of ebselen was dose-dependent [163]. Other researchers reported that ebselen treatment enhanced the activities of antioxidant enzymes such as SOD, CAT, and GPx without changing the cisplatin concentrations in cisplatin-induced AKI [165,166]. Ebselen treatment also prevented AKI from other causes, including gentamycin, I/R injury, and radiocontrast [167,168,169].
Ebselen also proved to be an effective treatment for reducing oxidative stress of the kidney in various models of diabetic nephropathy, including Zucker diabetic fat rats [170], diabetic ApoE−/− GPx−/− mice [76,171], and STZ-induced diabetic mice [172]. Ebselen treatment prevented decreases in capillary density and angiogenic competence related to vascular endothelial growth factor (VEGF) expression and restored acetylcholine-induced vasorelaxation in Zucker diabetic fat rats [170]. A recent study demonstrated that ebselen improved endothelial dysfunction by increasing endothelial GSH levels and reducing p38MAPK and NF-κB activation in uremic sera-exposed endothelial cells [139]. Ebselen was also shown to improve diabetes-associated atherosclerosis and renal injury by reducing oxidative stress and proatherogenic markers such as VEGF, connective tissue growth factor (CTGF), vascular cell adhesion molecule-1 (VCAM-1), and monocyte chemoattractant protein-1 (MCP-1) in diabetic ApoE−/− GPx−/− mice [76]. However, ebselen attenuated albuminuria and renal dysfunction in diabetic mice only with early intervention; it did not alter albuminuria and glomerulosclerosis with late intervention [172].
3.2.2. Diphenyl Diselenide
Diphenyl diselenide (PhSe2) is another organoselenium compound that has been reported to catalytically scavenge peroxides, with higher GPx-like activity than ebselen [173]. However, the diphenyl diselenides are electrophilic agents with cytotoxic, genotoxic, and mutagenic effects [174]. Diphenyl diselenide prevented the inhibition of δ-aminolevulinate dehydratase (δ-ALA-D), CAT and GPx activity, and enhanced ascorbic acid levels in glycerol-induced AKI [175]. In contrast, diphenyl diselenide in mercuric chloride nephropathy potentiated renal damage and oxidative stress, compared with mercuric chloride alone [176]. Diphenyl diselenide also attenuated STZ-induced toxicity by increasing platelet nucleoside triphosphate diphosphohydrolases (NTPDases) and 5′-nucleotidase, inhibitors of platelet aggregation, without increasing δ-ALA-D and Na+K+-ATPase [177]. A recent study reported that diphenyl diselenide was as effective as ebselen in treating cisplatin-induced AKI, and its mechanism was reducing oxidative stress by activating δ-ALA-D and Na+/K+-ATPase and upregulating the Nrf2/Keap-1/HO-1 pathway [178].
4. Conclusions
In this review, we discussed the role of antioxidant enzymes and the emerging evidence for the renoprotective effects of catalytic antioxidants in kidney disease (Table 1). Catalytic antioxidants, especially mimics of specific redox enzymes such as SOD, CAT, and GPx, have been demonstrated to have therapeutic advantages in various experimental models of kidney disease. Despite the protection against ROS shown by these compounds in in vitro and in vivo oxidative stress models, their practical application in kidney disease remains highly challenging. Therefore, further clinical trials are needed to assess the efficacy and toxicity of catalytic antioxidants in the human body and confirm their clinical applications in kidney disease. We expect this review to be helpful to researchers developing catalytic antioxidants applicable to various kidney diseases.
Author Contributions
Y.A.H. and C.W.P. designed and conceptualized the experiments. Y.A.H. and C.W.P. wrote the manuscript. Both the authors critically analyzed the manuscript and approved its final version for publication. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grants from the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (CWP; 2020R1F1A1071640). This research work was also supported by the National Research Foundation of Korea grants funded by the Korea government (MSIT; Ministry of Science and ICT) (YAH; 2018R1C1B5045006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sies, H. Oxidative Stress: A Concept in Redox Biology and Medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Jiang, S.; Persson, P.B.; Persson, E.A.G.; Lai, E.Y.; Patzak, A. Reactive Oxygen Species in Renal Vascular Function. Acta Physiol. 2020, 229, e13477. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Redox Compartmentalization in Eukaryotic Cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matés, J.M.; Pérez-Gómez, C.; Núñez de Castro, I. Antioxidant Enzymes and Human Diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant Responses and Cellular Adjustments to Oxidative Stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K. Obesity and Diabetic Kidney Disease: Role of Oxidant Stress and Redox Balance. Antioxid. Redox Signal. 2016, 25, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.M.; Witting, P.K. Protective Role for Antioxidants in Acute Kidney Disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, B.J. Catalytic Antioxidants: A Radical Approach to New Therapeutics. Drug Discov. Today 2004, 9, 557–566. [Google Scholar] [CrossRef]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous Non-Enzymatic Antioxidants in the Human Body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Rouco, L.; González-Noya, A.M.; Pedrido, R.; Maneiro, M. Pursuing the Elixir of Life: In Vivo Antioxidative Effects of Manganosalen Complexes. Antioxidants 2020, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Marklund, S.L. Extracellular Superoxide Dismutase and Other Superoxide Dismutase Isoenzymes in Tissues from Nine Mammalian Species. Biochem. J. 1984, 222, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Van Remmen, H.; Salvador, C.; Yang, H.; Huang, T.T.; Epstein, C.J.; Richardson, A. Characterization of the Antioxidant Status of the Heterozygous Manganese Superoxide Dismutase Knockout Mouse. Arch. Biochem. Biophys. 1999, 363, 91–97. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Brzoska, K.; Sochanowicz, B.; Siomek, A.; Olinski, R.; Kruszewski, M. Alterations in the Expression of Genes Related to NF-kappaB Signaling in Liver and Kidney of CuZnSOD-Deficient Mice. Mol. Cell. Biochem. 2011, 353, 151–157. [Google Scholar] [CrossRef]
- Siomek, A.; Brzoska, K.; Sochanowicz, B.; Gackowski, D.; Rozalski, R.; Foksinski, M.; Zarakowska, E.; Szpila, A.; Guz, J.; Bartlomiejczyk, T.; et al. Cu,Zn-superoxide Dismutase Deficiency in Mice Leads to Organ-Specific Increase in Oxidatively Damaged DNA and NF-kappaB1 Protein Activity. Acta Biochim. Pol. 2010, 57, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanobe, T.; Okada, F.; Iuchi, Y.; Onuma, K.; Tomita, Y.; Fujii, J. Deterioration of Ischemia/Reperfusion-Induced Acute Renal Failure in SOD1-Deficient Mice. Free Radic. Res. 2007, 41, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wheeler, M.D.; Connor, H.D.; Zhong, Z.; Bunzendahl, H.; Dikalova, A.; Samulski, R.J.; Schoonhoven, R.; Mason, R.P.; Swenberg, J.A.; et al. Cu/Zn-Superoxide Dismutase Gene Attenuates Ischemia-Reperfusion Injury in the Rat Kidney. J. Am. Soc. Nephrol. 2001, 12, 2691–2700. [Google Scholar] [PubMed]
- Carlström, M.; Brown, R.D.; Sällström, J.; Larsson, E.; Zilmer, M.; Zabihi, S.; Eriksson, U.J.; Persson, A.E. SOD1 Deficiency Causes Salt Sensitivity and Aggravates Hypertension in Hydronephrosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R82–R92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlström, M.; Lai, E.Y.; Ma, Z.; Steege, A.; Patzak, A.; Eriksson, U.J.; Lundberg, J.O.; Wilcox, C.S.; Persson, A.E. Superoxide Dismutase 1 Limits Renal Microvascular Remodeling and Attenuates Arteriole and Blood Pressure Responses to Angiotensin II via Modulation of Nitric Oxide Bioavailability. Hypertension 2010, 56, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- DeRubertis, F.R.; Craven, P.A.; Melhem, M.F.; Salah, E.M. Attenuation of Renal Injury in db/db Mice Overexpressing Superoxide Dismutase: Evidence for Reduced Superoxide-Nitric Oxide Interaction. Diabetes 2004, 53, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Craven, P.A.; Melhem, M.F.; Phillips, S.L.; DeRubertis, F.R. Overexpression of Cu2+/Zn2+ Superoxide Dismutase Protects against Early Diabetic Glomerular Injury in Transgenic Mice. Diabetes 2001, 50, 2114–2125. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Manganese Superoxide Dismutase Dysfunction and the Pathogenesis of Kidney Disease. Front. Physiol. 2020, 11, 755. [Google Scholar] [CrossRef]
- Parajuli, N.; Marine, A.; Simmons, S.; Saba, H.; Mitchell, T.; Shimizu, T.; Shirasawa, T.; Macmillan-Crow, L.A. Generation and Characterization of a Novel Kidney-Specific Manganese Superoxide Dismutase Knockout Mouse. Free Radic. Biol. Med. 2011, 51, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, N.; MacMillan-Crow, L.A. Role of Reduced Manganese Superoxide Dismutase in Ischemia-Reperfusion Injury: A Possible Trigger for Autophagy and Mitochondrial Biogenesis? Am. J. Physiol. Renal Physiol. 2013, 304, F257–F267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisani, A.; Sabbatini, M.; Riccio, E.; Rossano, R.; Andreucci, M.; Capasso, C.; De Luca, V.; Carginale, V.; Bizzarri, M.; Borrelli, A.; et al. Effect of a Recombinant Manganese Superoxide Dismutase on Prevention of Contrast-Induced Acute Kidney Injury. Clin. Exp. Nephrol. 2014, 18, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Vaziri, N.D. Salt-Sensitive Hypertension in Mitochondrial Superoxide Dismutase Deficiency Is Associated with Intra-Renal Oxidative Stress and Inflammation. Clin. Exp. Nephrol. 2014, 18, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Iturbe, B.; Sepassi, L.; Quiroz, Y.; Ni, Z.; Wallace, D.C.; Vaziri, N.D. Association of Mitochondrial SOD Deficiency with Salt-Sensitive Hypertension and Accelerated Renal Senescence. J. Appl. Physiol. 2007, 102, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Huang, A.; Wu, Z.; Kaminski, P.M.; Wolin, M.S.; Hintze, T.H.; Kaley, G.; Sun, D. Increased Superoxide Leads to Decreased Flow-Induced Dilation in Resistance Arteries of Mn-SOD-Deficient Mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2225–H2231. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Sharma, K. Mitochondrial Dysfunction in the Diabetic Kidney. Adv. Exp. Med. Biol. 2017, 982, 553–562. [Google Scholar]
- Li, C.; Matavelli, L.C.; Akhtar, S.; Siragy, H.M. (Pro)renin Receptor Contributes to Renal Mitochondria Dysfunction, Apoptosis and Fibrosis in Diabetic Mice. Sci. Rep. 2019, 9, 11667. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Ko, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol Prevents Renal Lipotoxicity and Inhibits Mesangial Cell Glucotoxicity in a Manner Dependent on the AMPK-SIRT1-PGC1alpha Axis in db/db Mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef] [Green Version]
- De Cavanagh, E.M.; Ferder, L.; Toblli, J.E.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Renal Mitochondrial Impairment Is Attenuated by AT1 Blockade in Experimental Type I Diabetes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H456–H465. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, T.W.; Kim, Y.; Yang, K.S.; Park, H.S.; Choi, S.R.; Chung, S.; Kim, H.W.; et al. Fenofibrate Improves Renal Lipotoxicity Through Activation of AMPK-PGC-1alpha in db/db Mice. PLoS ONE 2014, 9, e96147. [Google Scholar]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of Renal Superoxide Dismutase in Progressive Diabetic Nephropathy. J. Am. Soc. Nephrol. 2009, 20, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK Dysregulation Promotes Diabetes-Related Reduction of Superoxide and Mitochondrial Function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, O.; Marklund, S.L.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. Extracellular Superoxide Dismutase Is a Major Determinant of Nitric Oxide Bioavailability: In Vivo and Ex Vivo Evidence from ecSOD-Deficient Mice. Circ. Res. 2003, 93, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suliman, H.B.; Ali, M.; Piantadosi, C.A. Superoxide Dismutase-3 Promotes Full Expression of the EPO Response to Hypoxia. Blood 2004, 104, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.P.; Sullivan, J.C.; Wach, P.F.; Boesen, E.I.; Yamamoto, T.; Fukai, T.; Harrison, D.G.; Pollock, D.M.; Pollock, J.S. Protective Role of Extracellular Superoxide Dismutase in Renal Ischemia/Reperfusion Injury. Kidney Int. 2010, 78, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.J.; Zhou, D.; Xiao, L.; Zhou, L.; Li, Y.; Bastacky, S.I.; Oury, T.D.; Liu, Y. Extracellular Superoxide Dismutase Protects against Proteinuric Kidney Disease. J. Am. Soc. Nephrol. 2015, 26, 2447–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Fujishima, H.; Takahashi, K.; Sato, T.; Shimizu, T.; Morii, T.; Shimizu, T.; Shirasawa, T.; Qi, Z.; Breyer, M.D.; et al. SOD1, but Not SOD3, Deficiency Accelerates Diabetic Renal Injury in C57BL/6-Ins2(Akita) Diabetic Mice. Metabolism 2012, 61, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.W.; Shen, C.J.; Tung, Y.T.; Chen, H.L.; Chen, Y.H.; Chang, W.H.; Cheng, K.C.; Yang, S.H.; Chen, C.M. Extracellular Superoxide Dismutase Ameliorates Streptozotocin-Induced Rat Diabetic Nephropathy via Inhibiting the ROS/ERK1/2 Signaling. Life Sci. 2015, 135, 77–86. [Google Scholar] [CrossRef]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, H.W.; Kim, T.Y.; et al. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 28, 1543–1561. [Google Scholar] [CrossRef]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice Lacking Catalase Develop Normally but Show Differential Sensitivity to Oxidant Tissue Injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Kang, Y.J. Cellular and Subcellular Localization of Catalase in the Heart of Transgenic Mice. J. Histochem. Cytochem. 2000, 48, 585–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase Deficiency Accelerates Diabetic Renal Injury Through Peroxisomal Dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunami, R.; Sugiyama, H.; Wang, D.H.; Kobayashi, M.; Maeshima, Y.; Yamasaki, Y.; Masuoka, N.; Ogawa, N.; Kira, S.; Makino, H. Acatalasemia Sensitizes Renal Tubular Epithelial Cells to Apoptosis and Exacerbates Renal Fibrosis after Unilateral Ureteral Obstruction. Am. J. Physiol. Renal Physiol. 2004, 286, F1030–F1038. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sugiyama, H.; Wang, D.H.; Toda, N.; Maeshima, Y.; Yamasaki, Y.; Masuoka, N.; Yamada, M.; Kira, S.; Makino, H. Catalase Deficiency Renders Remnant Kidneys More Susceptible to Oxidant Tissue Injury and Renal Fibrosis in Mice. Kidney Int. 2005, 68, 1018–1031. [Google Scholar] [CrossRef] [Green Version]
- Takiue, K.; Sugiyama, H.; Inoue, T.; Morinaga, H.; Kikumoto, Y.; Kitagawa, M.; Kitamura, S.; Maeshima, Y.; Wang, D.H.; Masuoka, N.; et al. Acatalasemic Mice Are Mildly Susceptible to Adriamycin Nephropathy and Exhibit Increased Albuminuria and Glomerulosclerosis. BMC Nephrol. 2012, 13, 14. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Tran, S.; Sachetelli, S.; Zhang, S.L.; Guo, D.F.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Catalase Overexpression Attenuates Angiotensinogen Expression and Apoptosis in Diabetic Mice. Kidney Int. 2007, 71, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Chénier, I.; Godin, N.; Zhang, S.L.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Attenuation of Interstitial Fibrosis and Tubular Apoptosis in db/db Transgenic Mice Overexpressing Catalase in Renal Proximal Tubular Cells. Diabetes 2008, 57, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lo, C.S.; Chenier, I.; Maachi, H.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Overexpression of Catalase Prevents Hypertension and Tubulointerstitial Fibrosis and Normalization of Renal Angiotensin-Converting Enzyme-2 Expression in Akita Mice. Am. J. Physiol. Renal Physiol. 2013, 304, F1335–F1346. [Google Scholar] [CrossRef] [Green Version]
- Abdo, S.; Shi, Y.; Otoukesh, A.; Ghosh, A.; Lo, C.S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase Overexpression Prevents Nuclear Factor Erythroid 2-Related Factor 2 Stimulation of Renal Angiotensinogen Gene Expression, Hypertension, and Kidney Injury in Diabetic Mice. Diabetes 2014, 63, 3483–3496. [Google Scholar] [CrossRef] [Green Version]
- Godin, N.; Liu, F.; Lau, G.J.; Brezniceanu, M.L.; Chénier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase Overexpression Prevents Hypertension and Tubular Apoptosis in Angiotensinogen Transgenic Mice. Kidney Int. 2010, 77, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flohe, L.; Günzler, W.A.; Schock, H.H. Glutathione Peroxidase: A Selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox Environment of the Cell as Viewed Through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Lei, X.G.; Cheng, W.H. New Roles for an Old Selenoenzyme: Evidence from Glutathione Peroxidase-1 Null and Overexpressing Mice. J. Nutr. 2005, 135, 2295–2298. [Google Scholar] [CrossRef] [Green Version]
- Day, B.J. Catalase and Glutathione Peroxidase Mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Behne, D.; Kyriakopoulos, A. Mammalian Selenium-Containing Proteins. Annu. Rev. Nutr. 2001, 21, 453–473. [Google Scholar] [CrossRef]
- Muse, K.E.; Oberley, T.D.; Sempf, J.M.; Oberley, L.W. Immunolocalization of Antioxidant Enzymes in Adult Hamster Kidney. Histochem. J. 1994, 26, 734–753. [Google Scholar] [CrossRef]
- Wiedenmann, T.; Dietrich, N.; Fleming, T.; Altamura, S.; Deelman, L.E.; Henning, R.H.; Muckenthaler, M.U.; Nawroth, P.P.; Hammes, H.P.; Wagner, A.H.; et al. Modulation of Glutathione Peroxidase Activity by Age-Dependent Carbonylation in Glomeruli of Diabetic Mice. J. Diabetes Complicat. 2018, 32, 130–138. [Google Scholar] [CrossRef]
- Olson, G.E.; Whitin, J.C.; Hill, K.E.; Winfrey, V.P.; Motley, A.K.; Austin, L.M.; Deal, J.; Cohen, H.J.; Burk, R.F. Extracellular Glutathione Peroxidase (Gpx3) Binds Specifically to Basement Membranes of Mouse Renal Cortex Tubule Cells. Am. J. Physiol. Renal Physiol. 2010, 298, F1244–F1253. [Google Scholar] [CrossRef] [Green Version]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a Homozygous Null Mutation for the Most Abundant Glutathione Peroxidase, Gpx1, Show Increased Susceptibility to the Oxidative Stress-Inducing Agents Paraquat and Hydrogen Peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef] [Green Version]
- De Haan, J.B.; Stefanovic, N.; Nikolic-Paterson, D.; Scurr, L.L.; Croft, K.D.; Mori, T.A.; Hertzog, P.; Kola, I.; Atkins, R.C.; Tesch, G.H. Kidney Expression of Glutathione Peroxidase-1 Is Not Protective against Streptozotocin-Induced Diabetic Nephropathy. Am. J. Physiol. Renal Physiol. 2005, 289, F544–F551. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.A.; Kokoszka, J.E.; Waymire, K.G.; Cottrell, B.; MacGregor, G.R.; Wallace, D.C. Mitochondrial Oxidative Stress in Mice Lacking the Glutathione Peroxidase-1 Gene. Free Radic. Biol. Med. 2000, 28, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.N.; Chung, Y.H.; Shin, E.J.; Kim, D.J.; Jeong, J.H.; Nguyen, T.T.; Nam, Y.; Lee, Y.J.; Nah, S.Y.; Yu, D.Y.; et al. Genetic Depletion of Glutathione Peroxidase-1 Potentiates Nephrotoxicity Induced by Multiple Doses of Cocaine via Activation of Angiotensin II AT1 Receptor. Free Radic. Res. 2016, 50, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Lan, R.S.; Huang, R.; Feng, H.; Kumar, R.; Dayal, S.; Chan, K.S.; Dai, D.F. Glutathione Peroxidase-1 Overexpression Reduces Oxidative Stress, and Improves Pathology and Proteome Remodeling in the Kidneys of Old Mice. Aging Cell 2020, 19, e13154. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.W.; Kuo, M.C.; Kuo, H.T.; Chang, J.M.; Guh, J.Y.; Lai, Y.H.; Chen, H.C. Alterations of Glomerular and Extracellular Levels of Glutathione Peroxidase in Patients and Experimental Rats with Diabetic Nephropathy. J. Lab. Clin. Med. 2005, 145, 181–186. [Google Scholar] [CrossRef]
- Chew, P.; Yuen, D.Y.; Stefanovic, N.; Pete, J.; Coughlan, M.T.; Jandeleit-Dahm, K.A.; Thomas, M.C.; Rosenfeldt, F.; Cooper, M.E.; de Haan, J.B. Antiatherosclerotic and Renoprotective Effects of Ebselen in the Diabetic Apolipoprotein E/GPx1-Double Knockout Mouse. Diabetes 2010, 59, 3198–3207. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, F.G.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Regulation of the Extracellular Antioxidant Selenoprotein Plasma Glutathione Peroxidase (GPx-3) in Mammalian Cells. Mol. Cell. Biochem. 2009, 327, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Yin, D. Glutathione Peroxidase-3 Produced by the Kidney Binds to a Population of Basement Membranes in the Gastrointestinal Tract and in Other Tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G32–G38. [Google Scholar] [CrossRef] [Green Version]
- Pang, P.; Abbott, M.; Abdi, M.; Fucci, Q.A.; Chauhan, N.; Mistri, M.; Proctor, B.; Chin, M.; Wang, B.; Yin, W.; et al. Pre-Clinical Model of Severe Glutathione Peroxidase-3 Deficiency and Chronic Kidney Disease Results in Coronary Artery Thrombosis and Depressed Left Ventricular Function. Nephrol. Dial. Transplant. 2018, 33, 923–934. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Martinez-Moreno, J.M.; Ramos, A.M.; Sanchez-Niño, M.D.; Guerrero-Hue, M.; Moreno, J.A.; Ortiz, A.; Sanz, A.B. Ferroptosis and Kidney Disease. Nefrologia 2020, 40, 384–394. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yang, S.K.; Wu, X.; He, D.; Cao, K.; Zhang, W. Emerging Role of Ferroptosis in Acute Kidney Injury. Oxid. Med. Cell. Longev. 2019, 2019, 8010614. [Google Scholar] [CrossRef] [PubMed]
- Belavgeni, A.; Meyer, C.; Stumpf, J.; Hugo, C.; Linkermann, A. Ferroptosis and Necroptosis in the Kidney. Cell Chem. Biol. 2020, 27, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bi, R.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Cui, X.; Yang, H.; Gao, X.; Zhang, D. Ferroptosis Involves in Renal Tubular Cell Death in Diabetic Nephropathy. Eur. J. Pharmacol. 2020, 888, 173574. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. Oxidative Stress and Human Diseases: Origin, Link, Measurement, Mechanisms, and Biomarkers. Crit. Rev. Clin. Lab. Sci. 2009, 46, 241–281. [Google Scholar] [CrossRef]
- Kurutas, E.B. The Importance of Antioxidants Which Play the Role in Cellular Response against Oxidative/Nitrosative Stress: Current State. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Haber, A.; Gross, Z. Catalytic Antioxidant Therapy by Metallodrugs: Lessons from Metallocorroles. Chem. Commun. 2015, 51, 5812–5827. [Google Scholar] [CrossRef]
- Patel, M.; Day, B.J. Metalloporphyrin Class of Therapeutic Catalytic Antioxidants. Trends Pharmacol. Sci. 1999, 20, 359–364. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Halliwell, B. Superoxide Dismutase Activities of an Iron Porphyrin and Other Iron Complexes. J. Am. Chem. Soc. 1979, 101, 1026–1031. [Google Scholar] [CrossRef]
- Batinić-Haberle, I.; Spasojević, I.; Hambright, P.; Benov, L.; Crumbliss, A.L.; Fridovich, I. Relationship among Redox Potentials, Proton Dissociation Constants of Pyrrolic Nitrogens, and In Vivo and In Vitro Superoxide Dismutating Activities of Manganese(III) and Iron(III) Water-Soluble Porphyrins. Inorg. Chem. 1999, 38, 4011–4022. [Google Scholar] [CrossRef]
- Batinić-Haberle, I.; Rebouças, J.S.; Spasojević, I. Superoxide Dismutase Mimics: Chemistry, Pharmacology, and Therapeutic Potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetta, R. Potential Therapeutic Applications of MnSODs and SOD-Mimetics. Chemistry 2018, 24, 5032–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aston, K.; Rath, N.; Naik, A.; Slomczynska, U.; Schall, O.F.; Riley, D.P. Computer-Aided Design (CAD) of Mn(II) Complexes: Superoxide Dismutase Mimetics with Catalytic Activity Exceeding the Native Enzyme. Inorg. Chem. 2001, 40, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.P.; Lennon, P.J.; Neumann, W.L.; Weiss, R.H. Toward the Rational Design of Superoxide Dismutase Mimics: Mechanistic Studies for the Elucidation of Substituent Effects on the Catalytic Activity of Macrocyclic Manganese(II) Complexes. J. Am. Chem. Soc. 1997, 119, 6522–6528. [Google Scholar] [CrossRef]
- Golden, T.R.; Patel, M. Catalytic Antioxidants and Neurodegeneration. Antioxid. Redox Signal. 2009, 11, 555–570. [Google Scholar] [CrossRef] [Green Version]
- Cuzzocrea, S.; Mazzon, E.; Dugo, L.; Serraino, I.; Di Paola, R.; Britti, D.; De Sarro, A.; Pierpaoli, S.; Caputi, A.; Masini, E.; et al. A Role for Superoxide in Gentamicin-Mediated Nephropathy in Rats. Eur. J. Pharmacol. 2002, 450, 67–76. [Google Scholar] [CrossRef]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese Superoxide Dismutase, MnSOD and Its Mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar] [CrossRef] [Green Version]
- Azadmanesh, J.; Borgstahl, G.E.O. A Review of the Catalytic Mechanism of Human Manganese Superoxide Dismutase. Antioxidants 2018, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Soldevila-Barreda, J.J.; Sadler, P.J. Approaches to the Design of Catalytic Metallodrugs. Curr. Opin. Chem. Biol. 2015, 25, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Rajic, Z.; Tovmasyan, A.; Reboucas, J.S.; Ye, X.; Leong, K.W.; Dewhirst, M.W.; Vujaskovic, Z.; Benov, L.; Spasojevic, I. Diverse Functions of Cationic Mn(III) N-Substituted Pyridylporphyrins, Recognized as SOD Mimics. Free Radic. Biol. Med. 2011, 51, 1035–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovmasyan, A.; Sheng, H.; Weitner, T.; Arulpragasam, A.; Lu, M.; Warner, D.S.; Vujaskovic, Z.; Spasojevic, I.; Batinic-Haberle, I. Design, Mechanism of Action, Bioavailability and Therapeutic Effects of mn Porphyrin-Based Redox Modulators. Med. Princ. Pract. 2013, 22, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. An Educational Overview of the Chemistry, Biochemistry and Therapeutic Aspects of Mn Porphyrins—From Superoxide Dismutation to H2O2-Driven Pathways. Redox Biol. 2015, 5, 43–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, D.; Spasojevic, I.; Nguyen, H.; Deng, B.; Tovmasyan, A.; Weitner, T.; Sampaio, R.S.; Batinic-Haberle, I.; Huang, T.T. CNS Bioavailability and Radiation Protection of Normal Hippocampal Neurogenesis by a Lipophilic Mn Porphyrin-Based Superoxide Dismutase Mimic, MnTnBuOE-2-PyP(5). Redox Biol. 2017, 12, 864–871. [Google Scholar] [CrossRef]
- Faulkner, K.M.; Liochev, S.I.; Fridovich, I. Stable Mn(III) Porphyrins Mimic Superoxide Dismutase In Vitro and Substitute for It In Vivo. J. Biol. Chem. 1994, 269, 23471–23476. [Google Scholar]
- Spasojević, I.; Chen, Y.; Noel, T.J.; Fan, P.; Zhang, L.; Rebouças, J.S.; St Clair, D.K.; Batinić-Haberle, I. Pharmacokinetics of the Potent Redox-Modulating Manganese Porphyrin, MnTE-2-PyP(5+), in Plasma and Major Organs of B6C3F1 Mice. Free Radic. Biol. Med. 2008, 45, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Batinić-Haberle, I. Manganese Porphyrins and Related Compounds as Mimics of Superoxide Dismutase. Methods Enzymol. 2002, 349, 223–233. [Google Scholar]
- Tovmasyan, A.; Reboucas, J.S.; Benov, L. Simple Biological Systems for Assessing the Activity of Superoxide Dismutase Mimics. Antioxid. Redox Signal. 2014, 20, 2416–2436. [Google Scholar] [CrossRef] [Green Version]
- Gad, S.C.; Sullivan, D.W., Jr.; Spasojevic, I.; Mujer, C.V.; Spainhour, C.B.; Crapo, J.D. Nonclinical Safety and Toxicokinetics of MnTnBuOE-2-PyP5+ (BMX-001). Int. J. Toxicol. 2016, 35, 438–453. [Google Scholar] [CrossRef]
- Saba, H.; Batinic-Haberle, I.; Munusamy, S.; Mitchell, T.; Lichti, C.; Megyesi, J.; MacMillan-Crow, L.A. Manganese Porphyrin Reduces Renal Injury and Mitochondrial Damage during Ischemia/Reperfusion. Free Radic. Biol. Med. 2007, 42, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Tome, M.E. Thiol Regulation by Mn Porphyrins, Commonly Known as SOD Mimics. Redox Biol. 2019, 25, 101139. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue against Oxidative Injury. Antioxid. Redox Signal. 2018, 29, 1691–1724. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.L.; Hilton, G.; Mortensen, J.; Regner, K.; Johnson, C.P.; Nilakantan, V. MnTMPyP, a Cell-Permeant SOD Mimetic, Reduces Oxidative Stress and Apoptosis Following Renal Ischemia-Reperfusion. Am. J. Physiol. Renal Physiol. 2009, 296, F266–F276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, J.; Shames, B.; Johnson, C.P.; Nilakantan, V. MnTMPyP, a Superoxide Dismutase/Catalase Mimetic, Decreases Inflammatory Indices in Ischemic Acute Kidney Injury. Inflamm. Res. 2011, 60, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Seok, Y.M.; Jung, K.J.; Park, K.M. Reactive Oxygen Species/Oxidative Stress Contributes to Progression of Kidney Fibrosis Following Transient Ischemic Injury in Mice. Am. J. Physiol. Renal Physiol. 2009, 297, F461–F470. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.I.; Kim, J.; Jang, H.S.; Noh, M.R.; Lipschutz, J.H.; Park, K.M. Reduction of Oxidative Stress during Recovery Accelerates Normalization of Primary Cilia Length that Is Altered after Ischemic Injury in Murine Kidneys. Am. J. Physiol. Renal Physiol. 2013, 304, F1283–F1294. [Google Scholar] [CrossRef]
- Han, S.J.; Jang, H.S.; Kim, J.I.; Lipschutz, J.H.; Park, K.M. Unilateral Nephrectomy Elongates Primary Cilia in the Remaining Kidney via Reactive Oxygen Species. Sci. Rep. 2016, 6, 22281. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Holthoff, J.H.; Seely, K.A.; Pathak, E.; Spencer, H.J., III; Gokden, N.; Mayeux, P.R. Development of Oxidative Stress in the Peritubular Capillary Microenvironment Mediates Sepsis-Induced Renal Microcirculatory Failure and Acute Kidney Injury. Am. J. Pathol. 2012, 180, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jittikanont, S.; Falk, S.A.; Li, P.; Feng, L.; Gengaro, P.E.; Poole, B.D.; Bowler, R.P.; Day, B.J.; Crapo, J.D.; et al. Interaction among Nitric Oxide, Reactive Oxygen Species, and Antioxidants during Endotoxemia-Related Acute Renal Failure. Am. J. Physiol. Renal Physiol. 2003, 284, F532–F537. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Batinic-Haberle, I.; Benov, L.T. Effect of Potent Redox-Modulating Manganese Porphyrin, MnTM-2-PyP, on the Na(+)/H(+) Exchangers NHE-1 and NHE-3 in the Diabetic Rat. Redox Rep. 2009, 14, 236–242. [Google Scholar] [CrossRef]
- Ali, D.K.; Oriowo, M.; Tovmasyan, A.; Batinic-Haberle, I.; Benov, L. Late Administration of Mn Porphyrin-Based SOD Mimic Enhances Diabetic Complications. Redox Biol. 2013, 1, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, B.J.; Shawen, S.; Liochev, S.I.; Crapo, J.D. A Metalloporphyrin Superoxide Dismutase Mimetic Protects against Paraquat-Induced Endothelial Cell Injury, In Vitro. J. Pharmacol. Exp. Ther. 1995, 275, 1227–1232. [Google Scholar] [PubMed]
- Tovmasyan, A.; Maia, C.G.; Weitner, T.; Carballal, S.; Sampaio, R.S.; Lieb, D.; Ghazaryan, R.; Ivanovic-Burmazovic, I.; Ferrer-Sueta, G.; Radi, R.; et al. A Comprehensive Evaluation of Catalase-Like Activity of Different Classes of Redox-Active Therapeutics. Free Radic. Biol. Med. 2015, 86, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebouças, J.S.; Spasojević, I.; Batinić-Haberle, I. Pure Manganese(III) 5,10,15,20-tetrakis(4-benzoic acid)porphyrin (MnTBAP) Is Not a Superoxide Dismutase Mimic in Aqueous Systems: A Case of Structure-Activity Relationship as a Watchdog Mechanism in Experimental Therapeutics and Biology. J. Biol. Inorg. Chem. 2008, 13, 289–302. [Google Scholar] [CrossRef]
- Batinić-Haberle, I.; Cuzzocrea, S.; Rebouças, J.S.; Ferrer-Sueta, G.; Mazzon, E.; Di Paola, R.; Radi, R.; Spasojević, I.; Benov, L.; Salvemini, D. Pure MnTBAP Selectively Scavenges Peroxynitrite over Superoxide: Comparison of Pure and Commercial MnTBAP Samples to MnTE-2-PyP in Two Models of Oxidative Stress Injury, an SOD-Specific Escherichia coli Model and Carrageenan-Induced Pleurisy. Free Radic. Biol. Med. 2009, 46, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Zahmatkesh, M.; Kadkhodaee, M.; Moosavi, S.M.; Jorjani, M.; Kajbafzadeh, A.; Golestani, A.; Ghaznavi, R. Beneficial Effects of MnTBAP, a Broad-Spectrum Reactive Species Scavenger, in Rat Renal Ischemia/Reperfusion Injury. Clin. Exp. Nephrol. 2005, 9, 212–218. [Google Scholar] [CrossRef]
- Zahmatkesh, M.; Kadkhodaee, M.; Arab, H.A.; Shams, S. Effects of Co-Administration of an iNOS Inhibitor with a Broad-Spectrum Reactive Species Scavenger in Rat Renal Ischemia/Reperfusion Injury. Nephron. Exp. Nephrol. 2006, 103, e119–e125. [Google Scholar] [CrossRef]
- Pan, H.; Shen, K.; Wang, X.; Meng, H.; Wang, C.; Jin, B. Protective Effect of Metalloporphyrins against Cisplatin-Induced Kidney Injury in Mice. PLoS ONE 2014, 9, e86057. [Google Scholar] [CrossRef]
- Zhuang, Y.; Yasinta, M.; Hu, C.; Zhao, M.; Ding, G.; Bai, M.; Yang, L.; Ni, J.; Wang, R.; Jia, Z.; et al. Mitochondrial Dysfunction Confers Albumin-Induced NLRP3 Inflammasome Activation and Renal Tubular Injury. Am. J. Physiol. Renal Physiol. 2015, 308, F857–F866. [Google Scholar] [CrossRef]
- Bi, X.; Wang, J.; Liu, Y.; Wang, Y.; Ding, W. MnTBAP Treatment Ameliorates Aldosterone-Induced Renal Injury by Regulating Mitochondrial Dysfunction and NLRP3 Inflammasome Signalling. Am. J. Transl. Res. 2018, 10, 3504–3513. [Google Scholar]
- Yu, J.; Mao, S.; Zhang, Y.; Gong, W.; Jia, Z.; Huang, S.; Zhang, A. MnTBAP Therapy Attenuates Renal Fibrosis in Mice with 5/6 Nephrectomy. Oxid. Med. Cell. Longev. 2016, 2016, 7496930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudry, M.; Etienne, S.; Bruce, A.; Palucki, M.; Jacobsen, E.; Malfroy, B. Salen-Manganese Complexes Are Superoxide Dismutase-Mimics. Biochem. Biophys. Res. Commun. 1993, 192, 964–968. [Google Scholar] [CrossRef]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Musleh, W.; Bruce, A.; Baudry, M.; Malfroy, B. Salen-Manganese Complexes: Combined Superoxide Dismutase/Catalase Mimics with Broad Pharmacological Efficacy. Adv. Pharmacol. 1997, 38, 247–269. [Google Scholar]
- Gianello, P.; Saliez, A.; Bufkens, X.; Pettinger, R.; Misseleyn, D.; Hori, S.; Malfroy, B. EUK-134, a Synthetic Superoxide Dismutase and Catalase Mimetic, Protects Rat Kidneys from Ischemia-Reperfusion-Induced Damage. Transplantation 1996, 62, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.K.; Patel, N.S.; Kvale, E.O.; Brown, P.A.; Stewart, K.N.; Mota-Filipe, H.; Sharpe, M.A.; Di Paola, R.; Cuzzocrea, S.; Thiemermann, C. EUK-134 Reduces Renal Dysfunction and Injury Caused by Oxidative and Nitrosative Stress of the Kidney. Am. J. Nephrol. 2004, 24, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Samai, M.; Sharpe, M.A.; Gard, P.R.; Chatterjee, P.K. Comparison of the Effects of the Superoxide Dismutase Mimetics EUK-134 and Tempol on Paraquat-Induced Nephrotoxicity. Free Radic. Biol. Med. 2007, 43, 528–534. [Google Scholar] [CrossRef]
- McDonald, M.C.; d’Emmanuele di Villa Bianca, R.; Wayman, N.S.; Pinto, A.; Sharpe, M.A.; Cuzzocrea, S.; Chatterjee, P.K.; Thiemermann, C. A Superoxide Dismutase Mimetic with Catalase Activity (EUK-8) Reduces the Organ Injury in Endotoxic Shock. Eur. J. Pharmacol. 2003, 466, 181–189. [Google Scholar] [CrossRef]
- Magder, S.; Parthenis, D.G.; Ghouleh, I.A. Preservation of Renal Blood Flow by the Antioxidant EUK-134 in LPS-Treated Pigs. Int. J. Mol. Sci. 2015, 16, 6801–6817. [Google Scholar] [CrossRef] [Green Version]
- Vera, M.; Torramade-Moix, S.; Martin-Rodriguez, S.; Cases, A.; Cruzado, J.M.; Rivera, J.; Escolar, G.; Palomo, M.; Diaz-Ricart, M. Antioxidant and Anti-Inflammatory Strategies Based on the Potentiation of Glutathione Peroxidase Activity Prevent Endothelial Dysfunction in Chronic Kidney Disease. Cell. Physiol. Biochem. 2018, 51, 1287–1300. [Google Scholar] [CrossRef]
- Krishna, M.C.; Grahame, D.A.; Samuni, A.; Mitchell, J.B.; Russo, A. Oxoammonium Cation Intermediate in the Nitroxide-Catalyzed Dismutation of Superoxide. Proc. Natl. Acad. Sci. USA. 1992, 89, 5537–5541. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S.; Pearlman, A. Chemistry and Antihypertensive Effects of Tempol and Other Nitroxides. Pharmacol. Rev. 2008, 60, 418–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.K.; Cuzzocrea, S.; Brown, P.A.; Zacharowski, K.; Stewart, K.N.; Mota-Filipe, H.; Thiemermann, C. Tempol, a Membrane-Permeable Radical Scavenger, Reduces Oxidant Stress-Mediated Renal Dysfunction and Injury in the Rat. Kidney Int. 2000, 58, 658–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksu, U.; Ergin, B.; Bezemer, R.; Kandil, A.; Milstein, D.M.; Demirci-Tansel, C.; Ince, C. Scavenging Reactive Oxygen Species Using Tempol in the Acute Phase of Renal Ischemia/Reperfusion and Its Effects on Kidney Oxygenation and Nitric Oxide Levels. Intensive Care Med. Exp. 2015, 3, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, M.; Frank, S.; Olbrich, A.; Pfeilschifter, J.; Thiemermann, C. Decline in the Expression of Copper/Zinc Superoxide Dismutase in the Kidney of Rats with Endotoxic Shock: Effects of the Superoxide Anion Radical Scavenger, Tempol, on Organ Injury. Br. J. Pharmacol. 1998, 125, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, T.; Kadery, B.; Lotan, C.; Da’as, N.; Kleinman, Y.; Haj-Yehia, A. Effects of the Superoxide Dismutase-Mimetic Compound Tempol on Endothelial Dysfunction in Streptozotocin-Induced Diabetic Rats. Eur. J. Pharmacol. 2002, 436, 111–118. [Google Scholar] [CrossRef]
- Rodriguez, F.; Lopez, B.; Perez, C.; Fenoy, F.J.; Hernandez, I.; Stec, D.E.; Li Volti, G.; Salom, M.G. Chronic Tempol Treatment Attenuates the Renal Hemodynamic Effects Induced by a Heme Oxygenase Inhibitor in Streptozotocin Diabetic Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1540–R1548. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Li, W.; Han, J.; Zhang, W.; Gong, H.; Ma, R. Renal Protection of In Vivo Administration of Tempol in Streptozotocin-Induced Diabetic Rats. J. Pharmacol. Sci. 2012, 119, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Rafikova, O.; Salah, E.M.; Tofovic, S.P. Renal and Metabolic Effects of Tempol in Obese ZSF1 Rats—Distinct Role for Superoxide and Hydrogen Peroxide in Diabetic Renal Injury. Metabolism 2008, 57, 1434–1444. [Google Scholar] [CrossRef]
- Shokoji, T.; Nishiyama, A.; Fujisawa, Y.; Hitomi, H.; Kiyomoto, H.; Takahashi, N.; Kimura, S.; Kohno, M.; Abe, Y. Renal Sympathetic Nerve Responses to Tempol in Spontaneously Hypertensive Rats. Hypertension 2003, 41, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Welch, W.J.; Mendonca, M.; Blau, J.; Karber, A.; Dennehy, K.; Patel, K.; Lao, Y.S.; José, P.A.; Wilcox, C.S. Antihypertensive Response to Prolonged Tempol in the Spontaneously Hypertensive Rat. Kidney Int. 2005, 68, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Onuma, S.; Nakanishi, K. Superoxide Dismustase Mimetic Tempol Decreases Blood Pressure by Increasing Renal Medullary Blood Flow in Hyperinsulinemic-Hypertensive Rats. Metabolism 2004, 53, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Yoshizumi, M.; Hitomi, H.; Kagami, S.; Kondo, S.; Miyatake, A.; Fukunaga, M.; Tamaki, T.; Kiyomoto, H.; Kohno, M.; et al. The SOD Mimetic Tempol Ameliorates Glomerular Injury and Reduces Mitogen-Activated Protein Kinase Activity in Dahl Salt-Sensitive Rats. J. Am. Soc. Nephrol. 2004, 15, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, D.; Kojima, I.; Inagi, R.; Matsumoto, M.; Fujita, T.; Nangaku, M. Chronic Hypoxia Aggravates Renal Injury via Suppression of Cu/Zn-SOD: A Proteomic Analysis. Am. J. Physiol. Renal Physiol. 2008, 294, F62–F72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney Fibrosis in Hypertensive Rats: Role of Oxidative Stress. Am. J. Nephrol. 2008, 28, 548–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, N.D.; Dicus, M.; Ho, N.D.; Boroujerdi-Rad, L.; Sindhu, R.K. Oxidative Stress and Dysregulation of Superoxide Dismutase and NADPH Oxidase in Renal Insufficiency. Kidney Int. 2003, 63, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A Mitochondria-Targeted Nitroxide Is Reduced to Its Hydroxylamine by Ubiquinol in Mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross Talk between Mitochondria and NADPH Oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of Renal Mitochondrial Respiratory Complexes and Manganese Superoxide Dismutase during Sepsis: Mitochondria-Targeted Antioxidant Mitigates Injury. Am. J. Physiol. Renal Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Liu, T.; Bi, X.; Zhang, Z. Mitochondria-Targeted Antioxidant Mito-Tempo Protects against Aldosterone-Induced Renal Injury In Vivo. Cell. Physiol. Biochem. 2017, 44, 741–750. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Ding, W.; Wang, Y. Mito-TEMPO Alleviates Renal Fibrosis by Reducing Inflammation, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2018, 2018, 5828120. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl Sulfate Potentiates Endothelial Dysfunction via Reciprocal Role for Reactive Oxygen Species and RhoA/ROCK Signaling in 5/6 Nephrectomized Rats. Free Radic. Res. 2017, 51, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Feng, Q.; Kumagai, T.; Torikai, K.; Ohigashi, H.; Osawa, T.; Noguchi, N.; Niki, E.; Uchida, K. Ebselen, a Glutathione Peroxidase Mimetic Seleno-Organic Compound, as a Multifunctional Antioxidant. Implication for Inflammation-Associated Carcinogenesis. J. Biol. Chem. 2002, 277, 2687–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldew, G.S.; McVie, J.G.; van der Valk, M.A.; Los, G.; de Goeij, J.J.; Vermeulen, N.P. Selective Reduction of Cis-diamminedichloroplatinum(II) Nephrotoxicity by Ebselen. Cancer Res. 1990, 50, 7031–7036. [Google Scholar] [PubMed]
- Baldew, G.S.; Boymans, A.P.; Mol, J.G.; Vermeulen, N.P. The Influence of Ebselen on the Toxicity of Cisplatin in LLC-PK1 Cells. Biochem. Pharmacol. 1992, 44, 382–387. [Google Scholar] [CrossRef]
- Husain, K.; Morris, C.; Whitworth, C.; Trammell, G.L.; Rybak, L.P.; Somani, S.M. Protection by Ebselen against Cisplatin-Induced Nephrotoxicity: Antioxidant System. Mol. Cell. Biochem. 1998, 178, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Iizuka, K.; Terada, A.; Hara, M.; Nishijima, H.; Shimada, A.; Nakada, K.; Satoh, Y.; Akama, Y. Prevention of Nephrotoxicity of Cisplatin by Repeated Oral Administration of Ebselen in Rats. Tohoku J. Exp. Med. 2000, 191, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanarajan, R.; Abraham, P.; Isaac, B. Protective Effect of Ebselen, a Selenoorganic Drug, against Gentamicin-Induced Renal Damage in Rats. Basic Clin. Pharmacol. Toxicol. 2006, 99, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Kizilgun, M.; Poyrazoglu, Y.; Oztas, Y.; Yaman, H.; Cakir, E.; Cayci, T.; Akgul, O.E.; Kurt, Y.G.; Yaren, H.; Kunak, Z.I.; et al. Beneficial Effects of N-acetylcysteine and Ebselen on Renal Ischemia/Reperfusion Injury. Ren. Fail. 2011, 33, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Ozgur, T.; Tutanc, M.; Zararsiz, I.; Motor, S.; Ozturk, O.H.; Yaldiz, M.; Kurtgoz, O.Y. The Protective Effect of Ebselen on Radiocontrast-Induced Nephrotoxicity. Ren. Fail. 2012, 34, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Gealekman, O.; Brodsky, S.V.; Zhang, F.; Chander, P.N.; Friedli, C.; Nasjletti, A.; Goligorsky, M.S. Endothelial Dysfunction as a Modifier of Angiogenic Response in Zucker Diabetic Fat Rat: Amelioration with Ebselen. Kidney Int. 2004, 66, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.M.; Sharma, A.; Yuen, D.Y.; Stefanovic, N.; Krippner, G.; Mugesh, G.; Chai, Z.; de Haan, J.B. The Modified Selenenyl Amide, M-hydroxy Ebselen, Attenuates Diabetic Nephropathy and Diabetes-Associated Atherosclerosis in ApoE/GPx1 Double Knockout Mice. PLoS ONE 2013, 8, e69193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.M.; Sharma, A.; Stefanovic, N.; de Haan, J.B. Late-Intervention Study with Ebselen in an Experimental Model of Type 1 Diabetic Nephropathy. Free Radic. Res. 2015, 49, 219–227. [Google Scholar] [CrossRef]
- Huang, X.; Dong, Z.; Liu, J.; Mao, S.; Xu, J.; Luo, G.; Shen, J. Selenium-Mediated Micellar Catalyst: An Efficient Enzyme Model for Glutathione Peroxidase-Like Catalysis. Langmuir 2007, 23, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Rosa, R.M.; Sulzbacher, K.; Picada, J.N.; Roesler, R.; Saffi, J.; Brendel, M.; Henriques, J.A. Genotoxicity of Diphenyl Diselenide in Bacteria and Yeast. Mutat. Res. 2004, 563, 107–115. [Google Scholar] [CrossRef]
- Brandão, R.; Acker, C.I.; Leite, M.R.; Barbosa, N.B.; Nogueira, C.W. Diphenyl Diselenide Protects against Glycerol-Induced Renal Damage in Rats. J. Appl. Toxicol. 2009, 29, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.; Moresco, R.N.; Bellé, L.P.; Leite, M.R.; de Freitas, M.L.; Bianchini, A.; Nogueira, C.W. Diphenyl Diselenide Potentiates Nephrotoxicity Induced by Mercuric Chloride in Mice. J. Appl. Toxicol. 2011, 31, 773–782. [Google Scholar] [CrossRef]
- Barbosa, N.B.; Rocha, J.B.; Soares, J.C.; Wondracek, D.C.; Gonçalves, J.F.; Schetinger, M.R.; Nogueira, C.W. Dietary Diphenyl Diselenide Reduces the STZ-Induced Toxicity. Food Chem. Toxicol. 2008, 46, 186–194. [Google Scholar] [CrossRef]
- Fulco, B.C.W.; Jung, J.T.K.; Chagas, P.M.; Rosa, S.G.; Prado, V.C.; Nogueira, C.W. Diphenyl Diselenide Is as Effective as Ebselen in a Juvenile Rat Model of Cisplatin-Induced Nephrotoxicity. J. Trace Elem. Med. Biol. 2020, 60, 126482. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of endogenous sources of oxidative stress and antioxidative reactions in renal damage. Exogenous (environmental factors such as air and water pollution, smoking, drugs, and radiation) and endogenous (normal metabolic processes in living organisms) sources of oxidative stress produce reactive oxygen species (ROS). Endogenously, ROS are generated as products of biochemical reactions in the mitochondria (electron-transport system; ETS), plasma membrane, cytoplasm (including peroxisomes and lysozymes), and the membrane of the endoplasmic reticulum. The mitochondrial ETS, adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, myeloperoxidase, and endothelial nitric oxide synthase (eNOS) are the main sources of cellular ROS formation. An important reaction in free radical formation is the Fenton and Fenton-like reactions to produce ROS in which Fe2+ and Cu+ react with H2O2 to form OH•, respectively. To protect and repair the molecular injury caused by ROS, cells use a defense system composed of enzymatic antioxidants, including superoxide dismutase (SOD), catalase, and peroxidase, and nonenzymatic antioxidants made by the glutathione system. The main site of O2•− generation is the inner mitochondrial membrane during ETS-processes. The decomposition of H2O2 into water and oxygen is done by SOD, the glutathione system, and catalase, in that order. Excess ROS causes lipid peroxidation, nitro-oxidation, glyco-oxidation, and oxidative DNA damage, which can together cause protein alterations, DNA damage, cellular senescence, and apoptosis. All of those changes eventually lead to glomerulosclerosis and tubulointerstitial fibrosis.
Figure 1.
Schematic overview of endogenous sources of oxidative stress and antioxidative reactions in renal damage. Exogenous (environmental factors such as air and water pollution, smoking, drugs, and radiation) and endogenous (normal metabolic processes in living organisms) sources of oxidative stress produce reactive oxygen species (ROS). Endogenously, ROS are generated as products of biochemical reactions in the mitochondria (electron-transport system; ETS), plasma membrane, cytoplasm (including peroxisomes and lysozymes), and the membrane of the endoplasmic reticulum. The mitochondrial ETS, adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, myeloperoxidase, and endothelial nitric oxide synthase (eNOS) are the main sources of cellular ROS formation. An important reaction in free radical formation is the Fenton and Fenton-like reactions to produce ROS in which Fe2+ and Cu+ react with H2O2 to form OH•, respectively. To protect and repair the molecular injury caused by ROS, cells use a defense system composed of enzymatic antioxidants, including superoxide dismutase (SOD), catalase, and peroxidase, and nonenzymatic antioxidants made by the glutathione system. The main site of O2•− generation is the inner mitochondrial membrane during ETS-processes. The decomposition of H2O2 into water and oxygen is done by SOD, the glutathione system, and catalase, in that order. Excess ROS causes lipid peroxidation, nitro-oxidation, glyco-oxidation, and oxidative DNA damage, which can together cause protein alterations, DNA damage, cellular senescence, and apoptosis. All of those changes eventually lead to glomerulosclerosis and tubulointerstitial fibrosis.


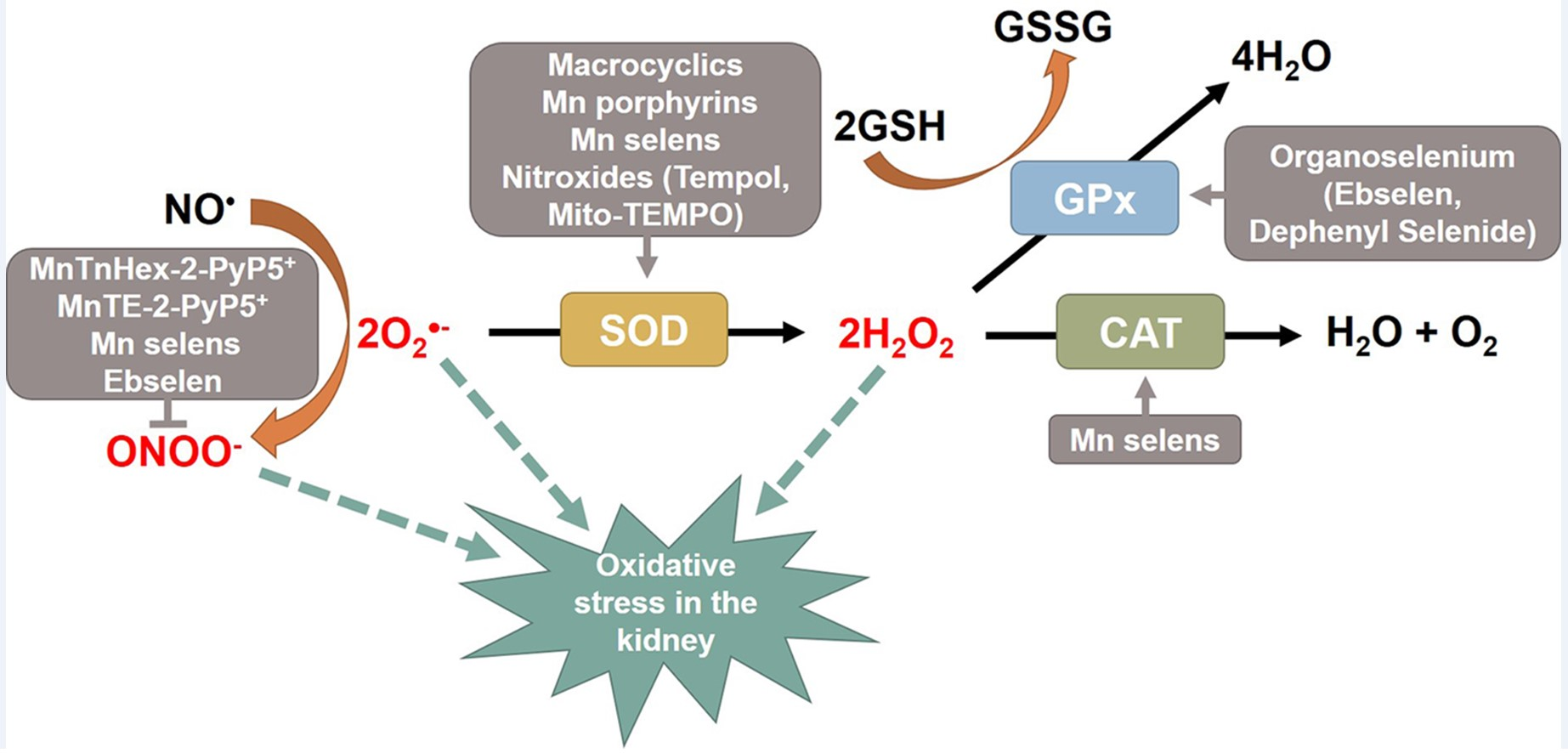{kind=link}
{kind=link}
Table 1.
Summary of in vitro and in vivo trials of catalytic antioxidants in kidney diseases.
| Groups | Structures | Compounds | Diseases | Models | Species or Cells | Ref. |
|---|---|---|---|---|---|---|
| SOD mimics: metal-based | Macrocyclics | M40403 | AKI | Gentamycin | Rats | [97] |
| Mn Porphyrins | AEOL10112 (MnTM-2-PyP5+) | DKD | Streptozotocin | Rats | [120,121] | |
| AEOL10113 (MnTE-2-PyP5+) | AKI | Lipopolysaccharide | Mice | [119] | ||
| MnTM-4-PyP5+ | AKI | I/R injury | Rats, Mice | [113,114,115,116] | ||
| Cecal ligation and puncture | Mice | [118] | ||||
| CKD | Unilateral nephrectomy | Mice | [117] | |||
| MnTnHex-2-PyP5+ | AKI | I/R injury | Rats | [110] | ||
| Non-SOD mimics: metal-based | Mn Porphyrins | AEOL10201 (MnTBAP3-) | AKI | I/R injury | Rats | [126,127] |
| Cisplatin | Mice | [128] | ||||
| CKD | Albumin | Mice | [129] | |||
| Aldosterone | Mice/HK-2 cells | [130] | ||||
| 5/6 nephrectomy | Mice/mPT cells | [131] | ||||
| SOD/CAT mimics: metal-based | Mn Salens | EUK-134 | AKI | I/R injury | Rats | [134,135] |
| Paraquat | NRK-52E cells | [136] | ||||
| Lipopolysaccharide | Pigs | [138] | ||||
| EUK-8 | AKI | Lipopolysaccharide | Rats | [137] | ||
| EUK-118, EUK-134 | CKD | Uremic media | Endothelial cells | [139] | ||
| SOD mimics: Non-metal-based | Nitroxides | Tempol | AKI | Paraquat | NRK-52E cells | [136] |
| I/R injury | Rats | [142,143] | ||||
| Lipopolysaccharide | Rats | [144] | ||||
| DKD | Streptozotocin | Rats | [145,146,147] | |||
| KK/Ta-Akita mice | Mice | [42] | ||||
| Obesity | ZSF1 rats | Rats | [148] | |||
| HTN | Spontaneously hypertensive rats | Rats | [149,150] | |||
| Dahl salt-resistant rats | Rats | [152] | ||||
| Unilateral renal artery stenosis | Rats | [153] | ||||
| Angiotensin II | Rats | [154] | ||||
| Fructose | Rats | [151] | ||||
| CKD | 5/6 nephrectomy | Rats | [155] | |||
| 5/6 nephrectomy + IS | Rats | [161] | ||||
| Mito-TEMPO | AKI | Cecal ligation and puncture | Mice | [158] | ||
| CKD | 5/6 nephrectomy + IS | Rats | [161] | |||
| 5/6 nephrectomy | Mice | [160] | ||||
| Aldosterone | Mice | [159] | ||||
| GPx mimics | Organoselenium | Ebselen | AKI | Cisplatin | Rats, Mice, LLC-PK1 cells | [163,164,165,166] |
| Gentamycin | Rats | [167] | ||||
| I/R injury | Rats | [168] | ||||
| Radiocontrast | Rats | [169] | ||||
| Gentamycin | Rats | [167] | ||||
| CKD | Uremic media | Endothelial cells | [139] | |||
| DKD | Akita mice | Mice | [172] | |||
| ApoE/GPx1 dKO | Mice | [76,171] | ||||
| Zucker diabetic fat | Rats | [170] | ||||
| Diphenyl diselenide | AKI | Cisplatin | Rats | [178] | ||
| Mercuric chloride | Mice | [176] | ||||
| Glycerol | Rats | [175] | ||||
| DKD | Streptozotocin | Rats | [177] |
Abbreviations: AKI; acute kidney injury, ApoE; Apolipoprotein E, CAT; catalase, CKD; chronic kidney disease, DKD; diabetic kidney disease, dKO; double knock out, GPx; glutathione peroxidase, HK-2; human kidney-2, HTN; hypertension, I/R; ischemia/reperfusion, IS; indoxyl sulfate, LLC-PK1; Epithelial-like pig kidney1, Mn; manganese, mPT; mouse proximal tubule, NRK-52E; normal rat kidney-52E, SOD; superoxide dismutase. AEOL series, EUK series, and M series are currently being developed by Aeolus Pharmaceuticals, Eukarion, and Metaphore Pharmaceuticals, respectively.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hong, Y.A.; Park, C.W. Catalytic Antioxidants in the Kidney. Antioxidants 2021, 10, 130. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010130
AMA Style
Hong YA, Park CW. Catalytic Antioxidants in the Kidney. Antioxidants. 2021; 10(1):130. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010130
Chicago/Turabian StyleHong, Yu Ah, and Cheol Whee Park. 2021. "Catalytic Antioxidants in the Kidney" Antioxidants 10, no. 1: 130. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010130
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.