
Harnessing the Benefits of Endogenous Hydrogen Sulfide to Reduce Cardiovascular Disease

Carlyle Fraser Heart Center, Division of Cardiothoracic Surgery, Department of Surgery, Emory University School of Medicine, Atlanta, GA 30322, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2021, 10(3), 383; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030383
Submission received: 3 February 2021
/
Revised: 26 February 2021
/
Accepted: 1 March 2021
/
Published: 4 March 2021
(This article belongs to the Special Issue Protective Effects of H2S and H2S-Releasing Drugs in Cardiovascular Diseases)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cardiovascular disease is the leading cause of death in the U.S. While various studies have shown the beneficial impact of exogenous hydrogen sulfide (H2S)-releasing drugs, few have demonstrated the influence of endogenous H2S production. Modulating the predominant enzymatic sources of H2S—cystathionine-β-synthase, cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase—is an emerging and promising research area. This review frames the discussion of harnessing endogenous H2S within the context of a non-ischemic form of cardiomyopathy, termed diabetic cardiomyopathy, and heart failure. Also, we examine the current literature around therapeutic interventions, such as intermittent fasting and exercise, that stimulate H2S production.
1. Introduction
Hydrogen sulfide (H2S) is an endogenously produced gaseous signaling molecule and is critical for the regulation of cardiovascular homeostasis [1,2]. It is mostly produced enzymatically in mammalian species by three enzymes in the cysteine biosynthesis pathway: cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST). New enzymes are emerging as important contributors to H2S production. Apart from these enzymes, H2S can also be produced from sulfur reservoirs (i.e., sulfane sulfur). Clinically, there is a negative association between heart failure, diabetes and H2S, as evidenced by the findings that lower circulating H2S levels are detected in plasma samples taken from diabetic and heart failure patients [3,4,5]. Therapeutic strategies aimed at increasing the levels of H2S are protective in models of acute myocardial ischemia-reperfusion injury and heart failure [6,7,8,9]. Numerous studies have shown the key role of H2S in maintaining cardiovascular homeostasis, but many of these studies rely on exogenous H2S donors. More studies that provide novel insight into endogenous H2S dynamics can help investigators understand the role of H2S in heart physiology.
In this review, we discuss the current understanding of H2S and its role in diabetic cardiomyopathy, heart failure and interventional therapies, such as intermittent fasting and exercise physiology. While significant advances have been made, the field has not fully understood how H2S-driven mechanisms regulate cardiac metabolism and help the heart adapt to physiological stresses (e.g., exercise). One beneficial mechanism is the H2S-mediated modification of cysteine residues by a process termed protein persulfidation or S-sulfhydration (PSSH), which is a reportedly prevalent modification that targets several metabolic enzymes and alters their function [10,11]. Harnessing this specific endogenous redox mechanism to rapidly modulate critical physiological and pathological signaling pathways is an exciting idea that may help to develop novel therapeutic strategies to reduce the burden of cardiovascular disease.
2. Overview of Hydrogen Sulfide
2.1. The Molecule and Post-Translational Modification
Originally described as a toxicant, H2S has emerged as an essential gaseous molecule that is both clinically and physiologically relevant to the heart. While the chemistry of this molecule is reviewed elsewhere (see Filipovic et al. (2018)), in this section we will briefly revisit important properties [12].
H2S is a volatile, water-soluble molecule with the bond dissociation energy similar to thiols (90 kcal/mol vs 92 kcal/mol, respectively). This property indicates that H2S does not readily decompose, making it a good signaling molecule [13]. Although H2S is membrane diffusible with a diffusion capacity from 0.5–10 cm/s, as a reactive electrophile, intracellular factors may regulate its travel [14]. Autooxidation involving metal catalysts and oxygen that can extinguish the reactivity of the molecule and transition it into alternative oxidation states has been reported [13]. Importantly, H2S not only reacts with other oxidants, such as nitric oxide, but can modify oxidized thiols (e.g., sulfenic acid) to form PSSH [12,15,16]. The post-translational modification of proteins is the subject of active research to understand the molecular and physiological consequences.
For a protein to be S-sulfhydrated, the oxidation of H2S or the targeted thiol is required [14]. Though PSSH cannot shield proteins from oxidative damage, PSSH has a higher bond dissociation energy than other oxidative modifications, such as S-nitrosation (PSSH: 70 kcal/mol vs. 31–32 kcal/mol) [15]. This may allow PSSH to serve as a more stable signaling modification [17,18,19]. While significant advances in proteomics have identified several proteins to be dynamically S-sulfhydrated under various physiological conditions, more studies are needed to further understand the impact of PSSH and its regulation of cardiac metabolism and disease [20,21,22,23].
2.2. Mechanisms of Hydrogen Sulfide Production
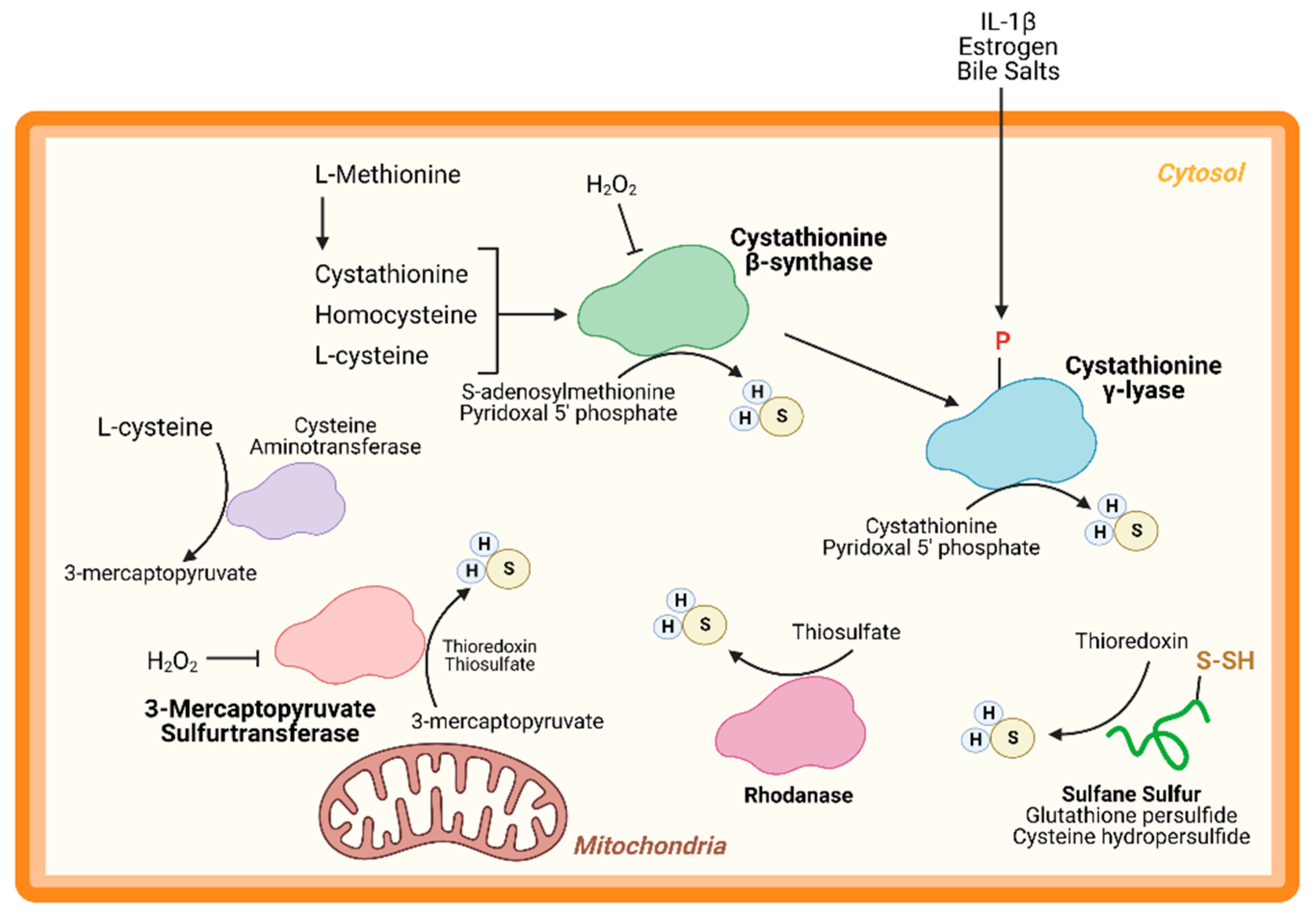
An important node for H2S regulation is in its production. L-cysteine, generated from the cysteine biosynthesis pathway, is the fundament substrate for enzymatic H2S production. Three enzymes are primarily responsible for H2S generation: CBS, CSE, and 3-MST (Figure 1). Both CBS and CSE utilize cystathionine in a pyridoxal 5′ phosphate (PLP)-dependent reaction. On the other hand, in the cysteine catabolism pathway, L-cysteine is converted into 3-mercaptopyruvate by cysteine aminotransferase in a PLP-dependent manner and is then metabolized by 3-MST into pyruvate and H2S. Apart from these enzymes, others are thought to participate in transsulfuration and facilitate H2S production. Together, these enzymes form key nodes to produce and regulate endogenous H2S production. While the enzymology has been extensively studied, few studies have shown the role of these enzymes (except CSE) in cardiac biology.
CBS is a key enzyme in the reverse transsulfuration pathway that regulates the flux of sulfur from the methionine cycle to the cysteine and glutathione biosynthesis pathways. In humans, CBS is a PLP-dependent tetramer that condenses cysteine and homocysteine into cystathionine with a heme co-factor to release H2S (Figure 1). CBS can also condense cysteines to produce H2S and lanthionine. Its activity is limited by the concentration of cysteines and is regulated by S-adenosylmethionine [24,25]. Oxidative stress conditions can lead to a truncated, S-adenosylmethionine-insensitive form of CBS that has increased activity, yet other studies suggest that the oxidation of its N-terminal heme group or CXXC oxidoreductase motif may inhibit the enzyme [26,27,28]. More studies are needed to understand the role of CBS in cardiac biology and disease. While few, if any, studies have proven a cardioprotective role for CBS, mutations in this protein can lead to hyperhomocysteinemia and an increased risk for cardiovascular disease along with accelerated atherosclerosis [29].
CSE is the predominant H2S-generating enzyme in the cardiovascular system and its ability to remediate cardiac injury is under active investigation [30]. Like CBS, human CSE exists in a PLP-bound, tetrameric state that, apart from H2S, yields α-ketobutyrate, pyruvate, and ammonia (Figure 1) [31]. CSE is also sensitive to homocysteine levels [28]. CSE is a key player in protection from various diseases, including heart disease. Several studies have demonstrated that CSE inhibition or genetic deficiency increased infarct size and enhanced transaortic constriction (TAC)-induced heart failure severity [32,33]. Conversely, cardiac-specific CSE overexpression blunted TAC-induced heart failure progression [34]. Also, CSE and PSSH was shown to decrease in the aged heart [15]. Taken together, these studies demonstrate that CSE is an integral component of both cardiovascular disease and disease risk.
Recently, CSE was found to be phosphorylated during atherosclerosis, leading to a decrease in H2S production [35]. Conversely, 17β-estradiol, the active form of estrogen, stimulates H2S production by CSE phosphorylation (Human Ser56) in a cyclic guanosine monophosphate/protein kinase G-dependent mechanism [36]. Also, bile salts stimulate CSE phosphorylation in endothelial cells [37]. This finding shows that H2S produced by CSE can be regulated by post-translational modifications (Figure 1). CSE has been shown to translocate to the mitochondria during endoplasmic reticulum stress and helps to maintain adenosine triphosphate (ATP) production. Furthermore, this translocation is Tom20 dependent and may help to regulate cystathionine levels in the mitochondria [38].
3-MST is a homodimeric, PLP-independent enzyme that receives 3-mercaptopyrvuate from cysteines metabolized by a PLP-mediated reaction via cysteine aminotransferase (CAT/GOT). Also, in the brain and kidneys, in a PLP-independent manner, D-cysteine can contribute to 3-mercaptopyruvate production via D-amino acid oxidase [39]. Upon binding, 3-MST converts 3-mercaptopyruvate into sulfane sulfur, then either thioredoxin or a thiol-based reductant liberates H2S and pyruvate (Figure 1). While CBS and CSE are predominately cytosolic, 3-MST is reportedly found in both the cytosol and mitochondria, suggesting that 3-MST may be a key protein that regulates mitochondrial protein PSSH.
While phosphorylation, or other post-translational modifications have not yet been reported, or investigated, 3-MST is reportedly regulated in two ways: oxidative stress and dimerization (Figure 1). Oxidative stress from hydrogen peroxide inhibited 3-MST activity in cultured hepatoma cells, suggesting that disease which stimulate excess oxidation may suppress H2S production from 3-MST [40]. Furthermore, redox-sensitive cysteines within 3-MST can govern its dimerization [41,42,43]. Oxidation of 3-MST can induce dimerization rendering the enzyme inactive, however—evident from treatment with reducing agents and shielding cysteines from oxidation with an alkylating agent—3-MST monomerization maintains H2S production [41,43].
While CBS, CSE, and 3-MST are the most widely studied enzymes, others have also been identified. Rhodanese, or thiosulfurtransferase, participates in a transsulfuration reaction by catalyzing the transfer of sulfur from thiosulfate to thiol or cyanide to form persulfides or thiocyanate, respectively (Figure 1) [44]. This protein closely resembles 3-MST and may be functionally cooperative [45,46]. Interestingly, 3-MST knockout mice are reported to have a 3-fold increase in rhodanese expression that may be an adaptive response to the loss of 3-MST, however, the consequences of excessive rhodanese in these mice are not yet understood [47]. Conversely, mutation of key residues in rhodanese decreased its activity and increased 3-MST activity [48,49]. While the role of 3-MST in cardiac homeostasis and disease has been recently reported, to our knowledge, no study has yet to investigate the importance of rhodanese in cardiac biology.
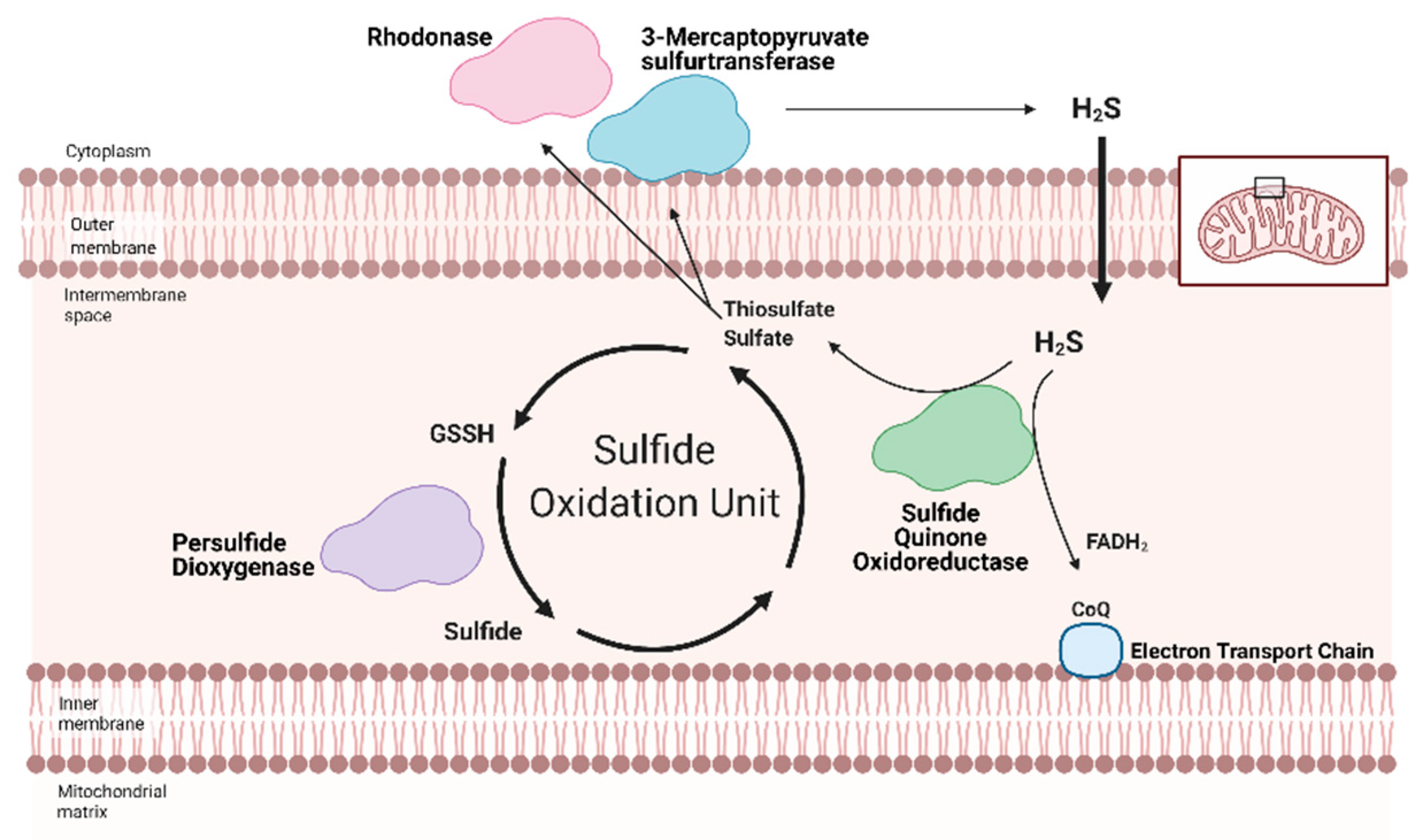
Sulfane sulfurs are H2S reservoirs bound to proteins (e.g., PSSH) or other molecules and are important sources of H2S. Cysteine hydropersulfides and glutathione persulfides (GSSH) are produced endogenously and display properties for maintaining intracellular homeostasis (Figure 2) [50]. PSSH are reduced, or de-sulfhydrated, by thioredoxin to release H2S [23,51]. Sulfate and sulfite are also important H2S reservoirs. GSSH is reduced to sulfite by persulfide dioxygenase, then further oxidized to by sulfide oxidase (SO). Through sulfite production, at least with SO deficiency, GOT can also produce H2S by catalyzing the deamination of cysteine sulfinic acid, the first product of oxidative cysteine metabolism [52]. While there have been advances in H2S biology, more studies are needed to fully comprehend the complexity of this redox molecule, especially regarding its role in cardiac metabolism.
3. Hydrogen Sulfide in Heart Metabolism and Cardiac Disease
3.1. Overview
The heart relies on fatty acids to generate energy, while other organs utilize glucose as their primary energy substrate [53,54]. The energy demand of the heart requires the use of fatty acids, because this substrate yield more ATP molecules than glycolysis [55]. Long-chain fatty acids are transported into cells by fatty acid translocase, CD36 [56]. Once inside the cell, fatty acid oxidation (FAO) is initiated with the transport of long-chain fatty acids that are modified by long-chain acyl-co-enzyme A (CoA) synthetase (LACS) and carnitine palmotyltransferase 1 (CPT1). LACS cleaves an acyl-CoA molecule and delivers it to CPT1 [57].
CPT1 is a rate-limiting enzyme of FAO that controls the entry of acyl-CoA into the mitochondria. In cardiomyocytes, CPT1b is the predominant isoform, accounting for 98% of CPT1 activity, while in other organs CPT1a (e.g., liver) and CPT1c (neurons) are more active [58,59,60,61,62]. CPT1 converts acyl-CoA into acylcarnitine and transports the product across the outer mitochondrial membrane. Then, carnitine acylcarnitine translocase (CACT) allows acylcarnitine to traverse the inner mitochondrial membrane and react with CPT2. With CPT2, acylcarnitine is converted back into acyl-CoA and enters the tricarboxylic acid (TCA) cycle [57].
Briefly, the TCA cycle is amongst the most well-studied and most important metabolic cycles in biology. The churn begins with the condensation of acyl-CoA into citrate, then the cycle carries citrate to oxaloacetate. Along the way, the TCA cycle produces two essential reducing equivalents—nicotinamide adenosine dinucleotide + hydrogen (NADH) and flavin adenosine dinucleotide + 2 hydrogen (FADH2)—along with other metabolites that participate in other metabolic systems. Both NADH and FADH2 feed electrons into the Electron Transport Chain that ultimately reduces oxygen and drives the migration of hydrogen over the mitochondrial membrane, power ATP synthase, which generates ATP (Figure 2). From one C18 fatty acid, FAO can yield 120 ATP molecules versus the same amount of ATP from three glucose molecules [55].
The heart beats ceaselessly throughout a human’s, or animal’s, life and generates pressures needed to overcome vascular resistance. This process requires enough energy to sustain homeostasis for both the cardiomyocytes and the other cells in the body. FAO gives the heart the energy it needs to function, however disease can the shift the metabolic substrate used and often forces the heart to utilize glycolysis instead of FAO [63]. H2S may be a way to preserve cardiac metabolism and remediate cardiovascular disease.
3.2. Hydrogen Sulfide Regulation Integrated with Mitochondrial Respiration
Redox homeostasis requires both the production of reactive molecules that can alter cellular function and a means of controlling that production. H2S regulation is intimately linked with metabolism and mitochondrial respiration (as reviewed by Paul et al. (2021)) [64]. Although H2S can suppress metabolism at high doses, recent studies show that low dose H2S is a metabolic stimulator, in part by providing electrons for mitochondrial respiration [65,66,67,68]. At low concentrations, H2S can serve as an inorganic electron donor—as it does in microorganisms—to Coenzyme Q with the help of sulfide quinone oxidoreductase (SQR) [64,69].
Along with persulfide dioxygenase and rhodanese, SQR is a key component of the Sulfide Oxidation Unit, which is responsible for the catabolism of free H2S (Figure 2) [69,70]. H2S is oxidized by SQR to yield two electrons that are transferred to FAD, then Coenzyme Q, which reduces Complex III [71]. This reaction also produces thiosulfate or sulfate that reacts with reduced glutathione to form GSSH. This new persulfide on glutathione is then oxidized to sulfide by persulfide dioxygenase. Sulfide can be further metabolized by either sulfide oxidase to sulfate or rhodanese to a thiosulfate. Thiosulfate can be catabolized back into H2S by 3-MST or rhodanese [72,73]. The cyclical nature of these reactions not only controls the toxic accumulation of H2S but may help to maintain a physiologic pool of sulfur that can be used for important redox reactions and other essential molecular processes.
Various cardiac diseases are linked to decreased H2S production and availability. Under active investigation are mechanisms that regulate H2S production and the ways they are impaired during disease. Below, we focus on two cardiac diseases well understood to suppress H2S production and availability, and their linked to H2S-mediated metabolic regulation: diabetic cardiomyopathy and heart failure.
3.3. Diabetic Cardiomyopathy and Hydrogen Sulfide
The prevalence of obesity, insulin resistance, diabetes, and dyslipidemia are increasing worldwide. People who have these disorders, collectively called metabolic syndrome, are twice as likely to develop heart failure and have worse prognoses after cardiovascular disease development [74]. In the U.S., obesity has become a major risk factor for metabolic syndrome development. From 2013–2019, 34% of overall children (2–19 years old) and 38% of adults in the U.S. were obese. Notably, the highest prevalence was amongst Hispanics [75]. Studies implicate “Western” diets, or high-fat diets (HFD), as a key driver of obesity [76]. Most patients with metabolic syndrome have hypertriglyceridemia and increased plasma levels of fatty acids. These lipids are taken into the heart, where due to an overload in storage capacity and utilization, accumulate and become lipotoxic to the cardiomyocytes. This induces a non-ischemic form of cardiomyopathy termed lipotoxic or diabetic cardiomyopathy (DCM) that can progress to heart failure [74,77].
Metabolic syndrome is strongly associated with a significant decrease in H2S availability and protein PSSH [3,4]. This syndrome leads to DCM due to the accumulation of lipids that become toxic. Oxidative stress and inflammation merge with toxic lipid accumulation to form more oxidants that overwhelm the metabolic system of the heart and kill the irreplaceable cardiomyocytes [74]. Cell death then leads to pathologic hypertrophy and can decompensate into heart failure [78,79]. This non-ischemic form of cardiomyopathy is linked to dysregulated metabolism and suppressed H2S production.
Clinically, there is a negative association between diabetes and H2S, evident from the findings that lower circulating H2S levels are detected in plasma samples taken from diabetic patients [3,4]. Preclinical models, specifically HFD-fed mice or fatty acid-treated cell models, have replicated these findings. Recent animal work showed that lost H2S bioavailability (e.g., lowered CSE expression, reduced protein PSSH, and impaired H2S production) contributes to the etiology of metabolic syndrome [80,81,82,83,84,85,86]. Additionally, cell models treated with toxic lipids, such as palmitate, reported impaired H2S production and reduced protein PSSH [87]. Many studies have shown that exogenous H2S can protect against excessive heart injury, including DCM [88,89,90]. While H2S donors are a promising form of therapy, stimulating endogenous H2S production is an innovative strategy that may improve target specificity.
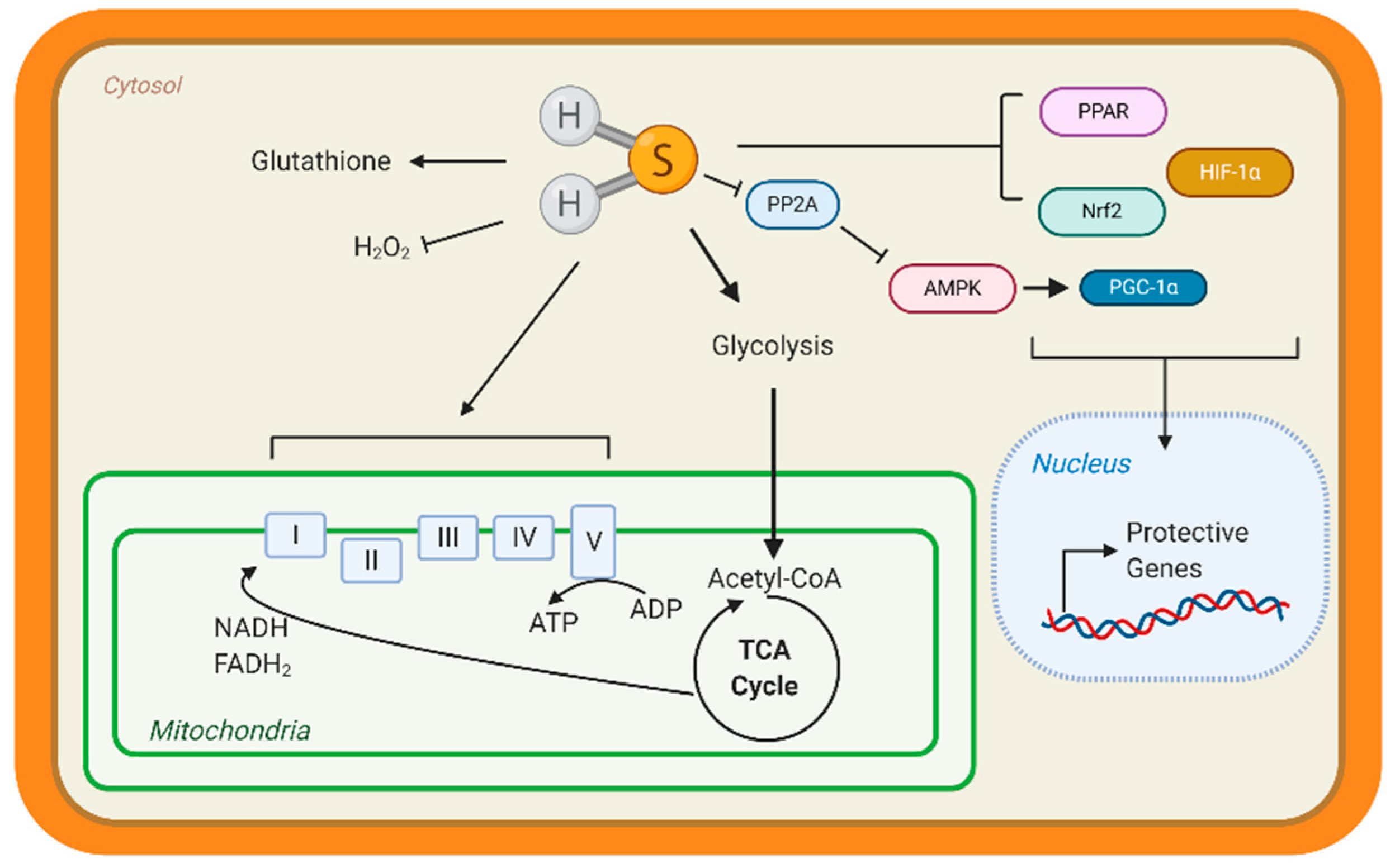
Few studies have studied the underlying mechanisms that govern H2S-mediated regulation of metabolism; however, this is an active area of research. H2S can not only donate to the mitochondrial respiration, but it can also modify ATP production. Under stress conditions, mitochondrial CSE translocation was shown to increase mitochondrial H2S and enhanced ATP production in smooth muscle cells [38]. ATP5A1, a subunit of ATP synthase, is also known to be S-sulfhydrated in a CSE-dependent manner [91]. In streptozotocin-induced, diabetic rats, 3-mercaptopyruvate—the substrate for 3-MST—was injected intraperitonially and elevated circulating H2S, along with stimulating oxygen consumption from liver mitochondria [92]. These studies suggest that elevating mitochondrial H2S may be a protective adaptation to regulate bioenergetics. Apart from mitochondrial respiration, our group has shown that H2S can stimulate carbohydrate and lipid metabolism. Through the activation of adenosine monophosphate kinase (AMPK) by S-sulfhydrating and inhibiting scaffolding protein phosphatase 2A (PP2A), the heart upregulated metabolic genes (Figure 1) [93]. Recently, Bithi et al. (2019) described a cardiac and endothelial S-sulfhydryl proteome [20,94]. Several proteins involved in metabolism were identified, yet the impact of PSSH on enzymatic activity and cardiac metabolism remains to be understood.
A way that H2S can regulate metabolism is by modulating transcription factors activation, specifically those responsible for the upregulation of metabolic genes (Figure 3). Peroxisome proliferator-activated receptors (PPAR) are major transcription factors that control the expression of FAO and uptake genes in all organs [95,96]. Both PPAR-δ and PPAR-γ are modified and activated by H2S, inducing lipid metabolism [97,98]. Our group has shown that peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α, a key co-regulator of PPAR signaling is induced by H2S treatment through the stimulation of AMPK [93]. Hypoxia-inducible factor (HIF)-1α is an essential transcription factor that stimulates the expression of glycolytic genes in response to hypoxia or oxidation [99]. Under normoxic conditions, CBS-dependent H2S inhibits prolyl hydroxylase 2, which helps target HIF-1α for degradation, thus allowing HIF-1α to accumulate and induce gene expression [100,101]. These important proteins regulate the expression of metabolic genes demonstrating that H2S can even regulate metabolism at the transcriptional level.
Few studies have focused on the impact that dysregulated H2S can have on cardiac metabolism and disease. The field needs more innovative experiments and approaches that capture the dynamics of H2S production and signaling to understand the beneficial nature of this gaseous molecule. Harnessing this endogenous donor pool may help reduce the burden of cardiovascular disease by adding specificity to the existing body of exogenous H2S donors. A way to study these dynamics may be to study the underlying molecular mechanism in the development of metabolic syndrome and the interventions that reduce cardiovascular disease risk from metabolic disorders.
3.4. Heart Failure
According to the annual American Heart Association statistics, about 6.2 million Americans over the age of 20 were affected with heart failure between 2013–2016, and that number is expected to rise with an aging population. Heart failure is projected to increase by 46% between 2012–2030 [102]. Racial, ethnic, and gender disparities exist with heart failure. In the Health, Aging and Body Composition Study, the risk of developing heart failure the elderly black population—attributed to modifiable risk factors, such as smoking and blood pressure—was about 68% compared to 49% for the elderly white population [103]. The Hispanic population, which also has a high cardiometabolic risk—as previously described—also has a high risk of developing heart failure [104,105]. While women tend to have more co-morbidities, such as diabetes and high blood pressure, heart failure mortality prognosis is better for women compared to men [106,107]. Reducing the burden of heart failure in the aging population is imperative and molecular insights may help to develop preventative interventions that can lower heart failure risk.
Heart failure—a consequence of DCM—is characterized as a significant loss of cardiac output due to a substantial alteration in heart structure [108]. The etiology of heart failure can come from various sources, mostly from events that injure the heart and results in the death of irreplaceable cardiomyocytes. As previously described, DCM is a non-ischemic form of cardiomyopathy and over time results in heart failure, however ischemic injury also reduces cardiac function. Myocardial infarctions—cardiac muscle death usually resulting from an occlusion of the coronary arteries which feed the heart—and subsequent reperfusion is an example of ischemic injury that can lead to heart failure [109]. The cardiomyocytes of the injured heart swell and undergoes pathological hypertrophy to compensate for a loss in cardiac output, however this adaptation is unsustainable [110,111]. Over time, the muscle degrades, the heart scars, and decompensates into heart failure [78,79].
Heart failure manifests as symptoms of dyspnea (labored breathing) and pulmonary congestion (i.e., edema) among others, resulting from significant physiological alterations [108]. A consequence of reduced cardiac output is a drop in blood pressure. This stimulates the brain to increase β-adrenergic signaling by releasing more norepinephrine and epinephrine from the adrenal glands. Meanwhile, the kidneys initiate the renin-angiotensin system to induce vasoconstriction, which can lead to kidney disease [112,113]. Both adaptations increase blood pressure to healthy levels, attempting to elevate organ perfusion and compensating for the loss of cardiac output. However, the heart is not as it once was; this “high” blood pressure further strains the weakened heart, killing more cardiomyocytes [110,114].
The role of endogenous H2S production and regulation is not completely understood in heart failure. Heart failure can be categorized into heart failure with reduced or preserved ejection fraction (HFrPF or HFpPF, respectively) [115,116]. While reports are emerging that focus on HFpPF, a large body of literature has described the molecular mechanisms involved in HFrPF. In severe, end-stage heart failure patients, there is a marked reduction in circulation H2S that is reproduced in pressure-induced heart failure animal models, such as TAC [5]. Furthermore, several studies have shown that pharmacological donors of H2S, such as sodium sulfide, SG-1003, among others, preserve cardiac function in pressure-induced and ischemia-induced heart failure animal models [9,117,118,119]. Heart failure in CSE deficient mice was more severe compared to WT controls [34]. These studies strongly implicate H2S as a key mechanism of heart failure.
In animal models, heart failure can be induced by pressure overload with TAC—banding of the aorta to simulate excess arterial pressure. Also, surgical occlusion of a coronary artery—cardiac vessels that feed the heart—can cause ischemia. Our group showed that sodium sulfide protected hearts from ischemic injury in a Nuclear factor E2-related factor 2 (Nrf2)-dependent manner (Figure 3). H2S-mediated activation of this transcription factor, which is responsible for upregulating antioxidant responses, sustained proteosome activity, thus eliminating dysfunctional proteins and preserving cellular homeostasis [119,120]. Moreover, H2S stimulated pro-angiogenic factors, such as nitric oxide bioavailability and vascular endothelial growth factors, increasing cardiac vascular density [117]. In culture cardiomyocytes, H2S attenuated hypertrophy and stimulated glycolysis by upregulating glucose transporter 4, along with increasing pyruvate kinase and succinate dehydrogenase activity [121]. Also, age-dependent cardioprotection against ischemia-reperfusion injury was reported in young 3-MST knock out mice, while 18-month-old mice showed hypertension and cardiac hypertrophy [122]. Taken together, H2S donors preserve cardiac structure and function, however endogenous H2S—evident from CSE deficient mice—also contributes to protection.
While H2S donors remain a vital strategy to efficently deliver this potent and protective molecules, we propose that exploiting the endogenous, cardiac H2S system can also be an integral and specific way of remediating heart failure and other cardiovascular diseases. Historically, the benefits of H2S of heart disease were demonstrated by donors, because—until recently—few stimulators of endogenous H2S were known. These H2S donors are non-specific because they release H2S throughout the body or an organ. H2S-producing enzymes are naturally positioned throughout and likely target specific, redox-sensitive proteins initiate a cardiac stress response, thus confer protection against disease. Targeting these H2S enzymes and modifying their activity—without genetic manipulation, such as removal or overexpression—may be a way to develop targeted therapeutics. However, few studies have investigated endogenous H2S regulation during disease. Of the few reports, cultured hepatocytes supplemented with 3-mercaptopyruvate—the substrate for 3-MST—stimulated 3-MST- and SQR-dependent H2S production. This treatment increased mitochondrial bioenergetics, yet the experiments were not set in the context of disease [123]. The role of endogenous H2S in the heart remains to be fully understood, yet there is clearly a role for H2S enzymes, such as 3-MST, in maintaining cardiac homeostasis during disease. Physiological stimulators of cardiac H2S are emerging, and these stimulators are well-known interventional strategies, which reduce the risk of cardiovascular disease.
4. Physiological Stimulators of Hydrogen Sulfide Production
4.1. Overview of Cardiovascular Disease Risk
Evaluating cardiovascular disease risk is an important metric that can help predict the likelihood of developing cardiovascular disease and is influence by a number of modifiable risk factors. Body weight, insulin sensitivity (i.e., diabetes), low-density lipoprotein (LDL) cholesterol, and physical inactivity, along with cigarette smoking, are well-known risk factors that can affect cardiovascular disease risk [124]. Because they are considered modifiable risk factors, interventions can significantly help to prevent, or exacerbate, the development of cardiovascular disease and reduce the public health burden of these diseases. In this section, we review two interventions that are linked to stimulating H2S production. The molecular mechanisms that confer the cardiovascular benefits of intermittent fasting and exercise are emerging and H2S is intimately associated with these intervention strategies.
4.2. Intermittent Fasting and Hydrogen Sulfide
The ability of intermittent fasting (IF) to reduce cardiovascular disease risk, especially from metabolic diseases is well-described and extends beyond reducing body weight [125,126,127,128,129]. There are three different methods of IF: (1) alternate-day fasting (ADF), where feeding is restricted for one day, then ad libitum feeding another day; (2) Modified ADF (MADF), such as with “IF 5:2”, feeding is ad libitum for 5 days, then restricted for 2 days [130]. Finally, (3) time-restricted feeding (TRF) is where feeding is ad libitum for a few hours, then fasting occurs for another few hours [131,132]. TRF is commonly done with overnight fasting models. These methods are often coupled with caloric restriction, which limits the energy obtained from ad libitum by a certain percentage [131]. In human and animal models studies have aimed at assessing the benefits of IF on cardiovascular disease prevention and remediation.
IF strategies can reduce cardiovascular disease risk and several studies have demonstrated this effect (as reviewed by Crupi et al. (2020)) [133,134]. Apart from weight loss, caloric restriction reduced cardiovascular disease risk markers, such as LDL cholesterol in nonobese subjects [126]. IF can also help reduce cardiovascular disease risk in at-risk populations, such as pre-diabetic, overweight men. In a randomized study, patients were grouped into a TRE or control group. The investigators concluded that TRE, particularly early morning restriction, improved insulin sensitivity, blood pressure, and oxidative stress, despite no change in body weight [135]. While diet adherence is a challenge, a study found that self-administered ADF reduced blood pressure, heart rate, arterial pulse pressure, and body fat over a long period of time [136]. More research is needed to understand the mechanisms that contribute to the benefits of IF.
An important feature of intermittent fasting is metabolic switching. In other organs, such as the liver, reduced caloric intake activates lipolysis from adipose tissues, allowing for FAO [137,138]. This metabolic switch allows for greater metabolic flexibility and more energy production [139]. From FAO, acyl-CoA feeds the TCA cycle, but when acyl-CoA concentrations exceed citrate synthase activity, ketogenesis occurs [140]. These ketone bodies, apart from participating in metabolic signaling, are used as energy substrates [141]. Ketone bodies, specifically β-hydroxybutyrate, activate cellular stress defenses—for instance, antioxidant and anti-inflammation—improving disease resistance [142,143]. The effect of IF on cardiac metabolism remains to be investigated, however insight from other organs can help us understand how the heart may respond.
The significance of ketogenesis on cardiac health, especially during disease is emerging in the field, due, in part, to its role in IF. While ketone bodies cannot provide as much energy as FAO, they can yield more ATP for less oxygen than glycolysis and can help prevent mitochondrial uncoupling—referred to as the “thrifty fuel” hypothesis” [54]. β-hydroxybutyrate—elevated by IF and heart failure—is a potent repressor of oxidative stress, and infusion was shown to enhance cardiac output in animals and HFrEF patients [142,144,145]. Recently, HFpEF patients were reported to have less β-hydroxybutyrate—a ketone body that accounts for about 75% of circulating ketones—however in a HFpEF animal model, β-hydroxybutyrate treatment reduced fibrosis, lung edema and other HFpEF markers [146]. While in human U937 monocytes, β-hydroxybutyrate had no effect on CSE expression or activity, hyperketonemia from acetoacetate decreased H2S production [147]. Since IF increases β-hydroxybutyrate and given the protective ability of this ketone, IF may be a beneficial practice to help reduce cardiovascular disease risk. While a direct link between ketone metabolism and H2S in the heart has not yet been made, more studies are needed to show this association.
IF is an important stimulator of H2S production in various organs, but few studies have examined cardiac H2S production. From redox biology, we can deduce that H2S may be a way to fine tune cardiac metabolism. Gao et al. (2020) described that under oxidative stress conditions, excess H2S can stimulate a thiol switch from S-glutathioinylation to PSSH. According to the article, this switch creates a new redox active thiol [22]. Further studies are needed to determine if this event can be a product of metabolic status, but this may be possible. Dietary caloric restriction enhanced H2S production and protein PSSH in various organs in a CSE-dependent mechanism [20,148]. Also, a hyperhomocysteinemia model—CSE and CBS deletion—abrogated fasting-induced protection from myocardial ischemic injury [149]. These studies suggest that H2S may be a key mediator of the benefits afforded from IF.
4.3. Exercise and Hydrogen Sulfide
In addition to IF, exercise also modulates cardiovascular diseases risk factors [150,151,152]. Physical inactivity is a risk factor for cardiovascular disease and exercise is a potent preventative factor that can reduce the risk of heart disease [153,154]. A recent study showed that physically inactive behaviors, established during childhood, can continue through adulthood and impact the cardiovascular disease risk. The study also found that even a little bit of physical activity can lower disease risk [155]. Establishing an exercise routine even with older adults can be beneficial. In 2004, Brach et al. found that in men and women age 70–79, acute (20–30 min), moderate exercise improves physical functions [156]. Exercise can be categorized by intensity. Moderate- to high-intensity, aerobic training is known to benefit cardiac health, especially for those with the risk factors for cardiovascular disease [157,158,159]. Coupled with IF, or caloric restriction, exercise can greatly improve heart health. Many of the underlying mechanisms, specifically relating to H2S production, are still under active investigation [158].
As opposed to pathological hypertrophy in heart failure, exercise induces physiological hypertrophy. This is a beneficial adaptation that increases cardiac output and allows for greater muscle perfusion, raising oxygen delivery [160]. Exercise stimulates skeletal muscle metabolism, specially by raise glycolytic flux from glycogen stores depletion and higher insulin sensitivity. These metabolic changes also alter muscular pH and helps to liberate oxygen bound to hemoglobin. Exercise also raises respiratory ventilation that can match elevated blood perfusion, which allows the removal of excess carbon dioxide—generated from increased muscle metabolism—from the blood and the infusion of more oxygen. These adaptations, while temporary, can have lasting benefits on cardiovascular function.
Preclinical, animal models can provide insight into the beneficial molecular mechanisms of exercise. Apart from intensity, exercise can also be categorized by modality, frequency, and duration [161]. An example of an exercise modality includes dynamic exercise, which employs various muscle groups, along with static and resistive training [162]. Investigators often us treadmill running, voluntary wheel running, or swimming to study the benefits of dynamic exercise, because these experiments require multiple muscle through throughout the animal’s body [163,164]. A model to assess the benefits of resistive training. Briefly, for this method, the animal is suspended and positioned on its hind legs, then electrical stimulation coerces the muscle to flex and the animal squats against a weight [165,166]. All have their advantages and disadvantages (as reviewed by Feng et al. (2019)), but they have allowed researchers to study the molecular benefits of exercise.
Few studies have explored the direct mechanisms that regulate H2S production during exercise, but these few studies suggested that exercise stimulates H2S. These studies also demonstrated that H2S dynamics are an essential component of exercise physiology. Several studies have shown that sedentary patients with varying intensity and frequency of training modified homocysteine concentrations—a substate for CBS, which can initiate the reverse transsulfuration pathway and trigger H2S production [167,168,169,170,171]. Animal models have shown that exercise training elevates the expression of H2S-generating enzymes, such as CSE and 3-MST. Moderate-intensity exercise in aged rats increase CSE and 3-MST expression in the heart, thus increasing H2S availability, while lowering oxidative stress and fibrosis [172]. In ovariectomized rats, which have reduced CSE expression, exercise augmented the damage induced by heart failure and restored CSE expression [173]. Although H2S production was not evaluated, these results suggest a role of H2S by altering the flux of metabolites in the reverse transsulfuration pathway.
Coupling exercise with dietary status is often used by researchers to study H2S production, which is likely a more clinically relevant design to examine the molecular pathways involved with preventing cardiovascular disease [174,175,176]. In mice fed an HFD for 20 weeks, exercise attenuated metabolic syndrome development and blunted cardiac dysfunction. In the exercise/HFD-fed group, left ventricular H2S increased, along with CBS and CSE upregulation [152]. Additionally, endothelial CSE influences exercise capacity. Genetic CSE overexpression and deletion, respectively, improves and impairs running distance with a treadmill exercise test [177]. Insight can also be gleamed from non-cardiac tissue. Exercise training of HFD-fed mice enhanced CBS, CSE, and 3-MST expression and H2S bioavailability in liver tissue, while elevating antioxidant capacity [178]. In a model of chronic kidney disease, where sections of the kidney were removed from rats, exercise training for 8-weeks not only reduced oxidative stress, but also increased H2S levels [179]. These studies show that exercise is an important intervention strategy to protect the heart from cardiovascular disease linked to metabolic syndrome, in part by stimulating cardiac H2S production and that this mechanism is necessary to sustain exercise capacity.
5. Conclusions
Interventions and therapeutics to reduce the burden of cardiovascular disease are essential for improving public health. While H2S donors are potent treatments for cardiac injury, stimulating endogenous H2S production may be a novel and natural strategy to prevent cardiovascular disease. Several studies have shown that heart disease negatively impact H2S dynamics in the heart, and this impairment is linked to bioenergetics. Preventative strategies proven to reduce the risk of cardiovascular disease developing—such as intermittent fasting and exercise—are shown to stimulate and regulate H2S production. In turn, H2S can regulate bioenergetics that beneficially promote cardiac health. Further study of these dynamics in the heart may unlock a new perspective on cardiac metabolism and may help to remediate cardiovascular disease.
Author Contributions
Conceptualization, K.M.C. and J.W.C.; Writing—Original draft preparation, K.M.C.; Writing—Review and editing, K.M.C. and J.W.C.; visualization, K.M.C.; supervision, J.W.C.; funding acquisition, J.W.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health, grant numbers R01HL136915-02S1, R01DK115213 and R01HL136915.
Acknowledgments
The authors acknowledge the following website used to generate the figure: https://www.biorender.com (accessed on 24 February 2021).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 3-MST: | 3-mercaptopyruvate sulfutransferase |
| ADF: | Alternate-day fasting |
| ADP: | Adenosine diphosphate |
| AMPK: | Adenosine monophosphate kinase |
| ATP: | Adenosine triphosphate |
| CACT: | Carnitine acylcarnitine translocase |
| CAT/GOT: | Cysteine aminotransferase |
| CBS: | Cystathionine-β-synthase |
| CoA: | Co-enzyme A |
| CPT: | Carnitine palmotyltransferase |
| CSE: | Cystathionine-γ-lyase |
| Cyt c: | Cytochrome c |
| DCM: | Diabetic cardiomyopathy |
| FADH2: | Flavin adenosine dinucleotide + 2 hydrogen |
| FAO: | Fatty acid oxidation |
| GSSH: | Glutathione persulfide |
| H2O2: | Hydrogen peroxide |
| H2S: | Hydrogen Sulfide |
| HFD: | High-fat Diet |
| HFpEF: | Heart failure with preserved ejection fraction |
| HFrEF: | Heart failure with reduced ejection fraction |
| HIF-1α: | Hypoxia inducible factor 1α |
| IF: | Intermittent Fasting |
| LACS: | Long-chain Acyl-CoA Synthatase |
| LDL: | Low-density lipoprotein |
| MADF: | Modified alternate-day fasting |
| NADH: | Nicotinamide adenosine dinucleotide + hydrogen |
| Nrf2: | Nuclear factor E2-related factor 2 |
| PGC-1α: | Peroxisome proliferator-activated receptor γ coactivator-1α |
| PLP: | Pyridoxal 5’ phosphate |
| PP2A: | Protein phosphatase 2A |
| PPAR: | Peroxisome proliferator-activated receptors |
| SO: | Sulfide oxidase |
| SQR: | Sulfide quinone oxidoreductase |
| PSSH: | S-sulfhydration |
| TAC: | Transaortic constriction |
| TCA: | Tricarboxylic acid |
| TRF: | Time-restricted feeding |
References
- Doeller, J.E.; Isbell, T.S.; Benavides, G.; Koenitzer, J.; Patel, H.; Patel, R.P.; Lancaster, J.R., Jr.; Darley-Usmar, V.M.; Kraus, D.W. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal. Biochem. 2005, 341, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid. Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef]
- Whiteman, M.; Gooding, K.M.; Whatmore, J.L.; Ball, C.I.; Mawson, D.; Skinner, K.; Tooke, J.E.; Shore, A.C. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 2010, 53, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Polhemus, D.J.; Calvert, J.W.; Butler, J.; Lefer, D.J. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica 2014, 2014, 768607. [Google Scholar] [CrossRef] [Green Version]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Calvert, J.W.; Elston, M.; Nicholson, C.K.; Gundewar, S.; Jha, S.; Elrod, J.W.; Ramachandran, A.; Lefer, D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 2010, 122, 11–19. [Google Scholar] [CrossRef]
- Mishra, P.K.; Tyagi, N.; Sen, U.; Givvimani, S.; Tyagi, S.C. H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H451–H456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Polhemus David, J.; Lefer David, J. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ. Res. 2018, 123, 590–600. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lancaster, J.R., Jr. Chemical foundations of hydrogen sulfide biology. Nitric Oxide Biol. Chem. 2013, 35, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchoam, D.; Cuevasanta, E.; Möller, M.N.; Alvarez, B. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxidants 2019, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e13. [Google Scholar] [CrossRef]
- Eberhardt, M.; Dux, M.; Namer, B.; Miljkovic, J.; Cordasic, N.; Will, C.; Kichko, T.I.; de la Roche, J.; Fischer, M.; Suárez, S.A.; et al. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 2014, 5, 4381. [Google Scholar] [CrossRef] [PubMed]
- Bartberger, M.D.; Mannion, J.D.; Powell, S.C.; Stamler, J.S.; Houk, K.N.; Toone, E.J. S−N Dissociation Energies of S-Nitrosothiols: On the Origins of Nitrosothiol Decomposition Rates. J. Am. Chem. Soc. 2001, 123, 8868–8869. [Google Scholar] [CrossRef]
- Benson, S.W. Thermochemistry and kinetics of sulfur-containing molecules and radicals. Chem. Rev. 1978, 78, 23–35. [Google Scholar] [CrossRef]
- Iciek, M.; Kowalczyk-Pachel, D.; Bilska-Wilkosz, A.; Kwiecień, I.; Górny, M.; Włodek, L. S-sulfhydration as a cellular redox regulation. Biosci. Rep. 2015, 36, e00304. [Google Scholar] [CrossRef] [PubMed]
- Bithi, N.; Link, C.; Wang, R.; Willard, B.; Hine, C. Dietary restriction transforms the protein sulfhydrome in a tissue-specific and cystathionine γ-lyase-dependent manner. bioRxiv 2019, 869271. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Liu, K.; He, J.; Tian, C.; Yu, X.; Yang, J. Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid. Redox Signal. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Li, L.; Parisien, M.; Wu, J.; Bederman, I.; Gao, Z.; Krokowski, D.; Chirieleison, S.M.; Abbott, D.W.; Wang, B.; et al. Discovery of a redox thiol switch: Implications for cellular energy metabolism. Mol. Cell. Proteom. 2020, 19, 852–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dóka, É.; Pader, I.; Bíró, A.; Johansson, K.; Cheng, Q.; Ballagó, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef] [Green Version]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martínez-Cruz, L.A. Structural insight into the molecular mechanism of allosteric activation of human cystathionine β-synthase by S-adenosylmethionine. Proc. Natl. Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Evande, R.; Kabil, Ö.; Ojha, S.; Taoka, S. Reaction mechanism and regulation of cystathionine β-synthase. Biochim. Biophys. Acta Proteins Proteom. 2003, 1647, 30–35. [Google Scholar] [CrossRef]
- Zou, C.-G.; Banerjee, R. Tumor Necrosis Factor-α-induced Targeted Proteolysis of Cystathionine β-Synthase Modulates Redox Homeostasis. J. Biol. Chem. 2003, 278, 16802–16808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taoka, S.; Ohja, S.; Shan, X.; Kruger, W.D.; Banerjee, R. Evidence for Heme-mediated Redox Regulation of Human Cystathionine β-Synthase Activity. J. Biol. Chem. 1998, 273, 25179–25184. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Banerjee, R. PLP-dependent H(2)S biogenesis. Biochim. Biophys. Acta 2011, 1814, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Li, H.; Ge, J. A cardioprotective insight of the cystathionine γ-lyase/hydrogen sulfide pathway. Int. J. Cardiol. Heart Vasc. 2015, 7, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Collins, R.; Huang, S.; Holmberg-Schiavone, L.; Anand, G.S.; Tan, C.-H.; van-den-Berg, S.; Deng, L.-W.; Moore, P.K.; Karlberg, T.; et al. Structural Basis for the Inhibition Mechanism of Human Cystathionine γ-Lyase, an Enzyme Responsible for the Production of H2S. J. Biol. Chem. 2009, 284, 3076–3085. [Google Scholar] [CrossRef] [Green Version]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.Z.; Wang, Z.J.; Ho, P.; Loke, Y.Y.; Zhu, Y.C.; Huang, S.H.; Tan, C.S.; Whiteman, M.; Lu, J.; Moore, P.K. Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J. Appl. Physiol. 2007, 102, 261–268. [Google Scholar] [CrossRef]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G., Sr.; Gojon, G., Jr.; et al. H₂S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibli, S.I.; Hu, J.; Sigala, F.; Wittig, I.; Heidler, J.; Zukunft, S.; Tsilimigras, D.I.; Randriamboavonjy, V.; Wittig, J.; Kojonazarov, B.; et al. Cystathionine γ Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation 2019, 139, 101–114. [Google Scholar] [CrossRef]
- Xu, X.; Yan, Q.; Liu, X.; Li, P.; Li, X.; Chen, Y.; Simoncini, T.; Liu, J.; Zhu, D.; Fu, X. 17β-Estradiol nongenomically induces vascular endothelial H2S release by promoting phosphorylation of cystathionine γ-lyase. J. Biol. Chem. 2019, 294, 15577–15592. [Google Scholar] [CrossRef]
- Renga, B.; Bucci, M.; Cipriani, S.; Carino, A.; Monti, M.C.; Zampella, A.; Gargiulo, A.; d’Emmanuele di Villa Bianca, R.; Distrutti, E.; Fiorucci, S. Cystathionine γ-lyase, a H2S-generating enzyme, is a GPBAR1-regulated gene and contributes to vasodilation caused by secondary bile acids. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H114–H126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [Green Version]
- Módis, K.; Asimakopoulou, A.; Coletta, C.; Papapetropoulos, A.; Szabo, C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem. Biophys. Res. Commun. 2013, 433, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N. Regulation of mercaptopyruvate sulfurtransferase activity via intrasubunit and intersubunit redox-sensing switches. Antioxid. Redox Signal. 2013, 19, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N.; Yoshii, T.; Abe, Y.; Matsumura, T. Thioredoxin-dependent enzymatic activation of mercaptopyruvate sulfurtransferase. An intersubunit disulfide bond serves as a redox switch for activation. J. Biol. Chem. 2007, 282, 1561–1569. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, N.; Katayama, A. Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J. Biol. Chem. 2005, 280, 34569–34576. [Google Scholar] [CrossRef] [Green Version]
- Billaut-Laden, I.; Allorge, D.; Crunelle-Thibaut, A.; Rat, E.; Cauffiez, C.; Chevalier, D.; Houdret, N.; Lo-Guidice, J.-M.; Broly, F. Evidence for a functional genetic polymorphism of the human thiosulfate sulfurtransferase (Rhodanese), a cyanide and H2S detoxification enzyme. Toxicology 2006, 225, 1–11. [Google Scholar] [CrossRef]
- Cipollone, R.; Ascenzi, P.; Visca, P. Common themes and variations in the rhodanese superfamily. Iubmb Life 2007, 59, 51–59. [Google Scholar] [CrossRef]
- Nakajima, T. Roles of Sulfur Metabolism and Rhodanese in Detoxification and Anti-Oxidative Stress Functions in the Liver: Responses to Radiation Exposure. Med. Sci. Monit. 2015, 21, 1721–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, N.; Tanaka, M.; Tanaka, Y.; Ito, T. Novel Characterization of Antioxidant Enzyme, 3-Mercaptopyruvate Sulfurtransferase-Knockout Mice: Overexpression of the Evolutionarily-Related Enzyme Rhodanese. Antioxidants 2019, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, N.; Okazaki, T.; Nishino, T. Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 1995, 270, 16230–16235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2S and polysulfide production and possible SOx production. Br. J. Pharm. 2018, 175, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, T.; Motohashi, H.; Ihara, H.; Akaike, T. Enzymatic Regulation and Biological Functions of Reactive Cysteine Persulfides and Polysulfides. Biomolecules 2020, 10, 1245. [Google Scholar] [CrossRef]
- Wedmann, R.; Onderka, C.; Wei, S.; Szijártó, I.A.; Miljkovic, J.L.; Mitrovic, A.; Lange, M.; Savitsky, S.; Yadav, P.K.; Torregrossa, R.; et al. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016, 7, 3414–3426. [Google Scholar] [CrossRef] [Green Version]
- Mellis, A.T.; Misko, A.L.; Arjune, S.; Liang, Y.; Erdélyi, K.; Ditrói, T.; Kaczmarek, A.T.; Nagy, P.; Schwarz, G. The role of glutamate oxaloacetate transaminases in sulfite biosynthesis and H2S metabolism. Redox Biol. 2020, 38, 101800. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone metabolism in the failing heart. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef] [PubMed]
- Goetzman, E.S. Chapter 10—Modeling Disorders of Fatty Acid Metabolism in the Mouse. In Progress in Molecular Biology and Translational Science; Chang, K.T., Min, K.-T., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 100, pp. 389–417. [Google Scholar]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef]
- Weis, B.C.; Cowan, A.T.; Brown, N.; Foster, D.W.; McGarry, J.D. Use of a selective inhibitor of liver carnitine palmitoyltransferase I (CPT I) allows quantification of its contribution to total CPT I activity in rat heart. Evidence that the dominant cardiac CPT I isoform is identical to the skeletal muscle enzyme. J. Biol. Chem. 1994, 269, 26443–26448. [Google Scholar] [CrossRef]
- Brown, N.F.; Weis, B.C.; Husti, J.E.; Foster, D.W.; McGarry, J.D. Mitochondrial carnitine palmitoyltransferase I isoform switching in the developing rat heart. J. Biol. Chem. 1995, 270, 8952–8957. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Roa-Mansergas, X.; Fadó, R.; Atari, M.; Mir, J.F.; Muley, H.; Serra, D.; Casals, N. CPT1C promotes human mesenchymal stem cells survival under glucose deprivation through the modulation of autophagy. Sci. Rep. 2018, 8, 6997. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation-like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.C.; Woon, T.C.; Nicholls, P.; Peterson, J.; Greenwood, C.; Thomson, A.J. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem. J. 1984, 224, 591–600. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Miljkovic, J.L.; Bostelaar, T.; Adhikari, B.; Yadav, P.K.; Steiger, A.K.; Torregrossa, R.; Pluth, M.D.; Whiteman, M.; Banerjee, R.; et al. Cytochrome c Reduction by H2S Potentiates Sulfide Signaling. ACS Chem. Biol. 2018, 13, 2300–2307. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Baeza-Garza, C.D.; Logan, A.; Rosa, T.; Wedmann, R.; Prime, T.A.; Martin, J.L.; Saeb-Parsy, K.; Krieg, T.; Filipovic, M.R.; et al. Assessment of H(2)S in vivo using the newly developed mitochondria-targeted mass spectrometry probe MitoA. J. Biol. Chem. 2017, 292, 7761–7773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [Green Version]
- Marutani, E.; Ichinose, F. Emerging pharmacological tools to control hydrogen sulfide signaling in critical illness. Intensive Care Med. Exp. 2020, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H. Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signal. 2014, 20, 783–793. [Google Scholar] [CrossRef] [Green Version]
- Olson, K.R.; Deleon, E.R.; Gao, Y.; Hurley, K.; Sadauskas, V.; Batz, C.; Stoy, G.F. Thiosulfate: A readily accessible source of hydrogen sulfide in oxygen sensing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R592–R603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Benjamin Emelia, J.; Muntner, P.; Alonso, A.; Bittencourt Marcio, S.; Callaway Clifton, W.; Carson April, P.; Chamberlain Alanna, M.; Chang Alexander, R.; Cheng, S.; Das Sandeep, R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [Green Version]
- Van Gaal, L.F.; Mertens, I.L.; De Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef]
- Diwan, A.; Dorn, G.W., 2nd. Decompensation of cardiac hypertrophy: Cellular mechanisms and novel therapeutic targets. Physiology 2007, 22, 56–64. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Barr, L.A.; Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Calvert, J.W. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide 2015, 46, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, M.; Kwong Huat, B.T.; Hsu, A.; Whiteman, M.; Bhatia, M.; Moore, P.K. Streptozotocin-induced diabetes in the rat is associated with enhanced tissue hydrogen sulfide biosynthesis. Biochem. Biophys. Res. Commun. 2005, 333, 1146–1152. [Google Scholar] [CrossRef]
- Brancaleone, V.; Roviezzo, F.; Vellecco, V.; De Gruttola, L.; Bucci, M.; Cirino, G. Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br. J. Pharm. 2008, 155, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2012, 17, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Peake, B.F.; Nicholson, C.K.; Lambert, J.P.; Hood, R.L.; Amin, H.; Amin, S.; Calvert, J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1215–H1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Seweidy, M.M.; Sadik, N.A.; Shaker, O.G. Role of sulfurous mineral water and sodium hydrosulfide as potent inhibitors of fibrosis in the heart of diabetic rats. Arch. Biochem. Biophys. 2011, 506, 48–57. [Google Scholar] [CrossRef]
- Suzuki, K.; Olah, G.; Modis, K.; Coletta, C.; Kulp, G.; Gero, D.; Szoleczky, P.; Chang, T.; Zhou, Z.; Wu, L.; et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. USA 2011, 108, 13829–13834. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, J.; Sun, A.; Sun, Y.; Yu, X.; Liu, N.; Dong, S.; Yang, F.; Zhang, L.; Zhong, X.; et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Zhang, L.; Gao, Z.; Sun, X.; Yu, M.; Dong, S.; Wu, J.; Zhao, Y.; Xu, C.; Zhang, W.; et al. Exogenous H2S Protects Against Diabetic Cardiomyopathy by Activating Autophagy via the AMPK/mTOR Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 1168–1187. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Wu, J.; Zhang, L.; Gao, Z.; Sun, Y.; Yu, M.; Zhao, Y.; Dong, S.; Lu, F.; Zhang, W. Hydrogen Sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J. Cell. Mol. Med. 2017, 21, 3190–3203. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Li, Y.; Li, L.; Xu, S.; Feng, X.; Liu, S. Hydrogen Sulfide (H2S)-Releasing Compounds: Therapeutic Potential in Cardiovascular Diseases. Front. Pharmacol. 2018, 9, 1066. [Google Scholar] [CrossRef] [Green Version]
- Módis, K.; Ju, Y.; Ahmad, A.; Untereiner, A.A.; Altaany, Z.; Wu, L.; Szabo, C.; Wang, R. S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharm. Res. 2016, 113, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Coletta, C.; Módis, K.; Szczesny, B.; Brunyánszki, A.; Oláh, G.; Rios, E.C.; Yanagi, K.; Ahmad, A.; Papapetropoulos, A.; Szabo, C. Regulation of Vascular Tone, Angiogenesis and Cellular Bioenergetics by the 3-Mercaptopyruvate Sulfurtransferase/H2S Pathway: Functional Impairment by Hyperglycemia and Restoration by DL-α-Lipoic Acid. Mol. Med. 2015, 21, 1–14. [Google Scholar] [CrossRef]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Nicholson, C.K.; Koczor, C.A.; Wang, R.; Lewis, W.; Shiva, S.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell. Cardiol. 2018, 116, 29–40. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell S-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2020. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Dong, J.H.; Teng, X.; Jin, S.; Xue, H.M.; Liu, S.Y.; Guo, Q.; Shen, W.; Ni, X.C.; Wu, Y.M. Hydrogen sulfide improves endothelial dysfunction in hypertension by activating peroxisome proliferator-activated receptor delta/endothelial nitric oxide synthase signaling. J. Hypertens. 2018, 36, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Shi, X.; Wang, H.; Fan, J.; Feng, Y.; Lin, X.; Yang, J.; Cui, Q.; Tang, C.; Xu, G.; et al. Cystathionine γ lyase-hydrogen sulfide increases peroxisome proliferator-activated receptor γ activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes. Biochim. Biophys. Acta 2016, 1861, 419–429. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Prabhudesai, S.; Zhang, Y.; Rao, G.; Thirugnanam, K.; Hossen, M.N.; Dwivedi, S.K.D.; Ramchandran, R.; Mukherjee, P.; Bhattacharya, R. Cystathione β-synthase regulates HIF-1α stability through persulfidation of PHD2. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Budde, M.W.; Roth, M.B. Hydrogen sulfide increases hypoxia-inducible factor-1 activity independently of von Hippel-Lindau tumor suppressor-1 in C. elegans. Mol. Biol. Cell 2010, 21, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, A.; Georgiopoulou, V.; Kritchevsky, S.B.; Psaty, B.M.; Smith, N.L.; Newman, A.B.; Rodondi, N.; Satterfield, S.; Bauer, D.C.; Bibbins-Domingo, K.; et al. Epidemiology of incident heart failure in a contemporary elderly cohort: The health, aging, and body composition study. Arch. Intern. Med. 2009, 169, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Balfour, P.C., Jr.; Ruiz, J.M.; Talavera, G.A.; Allison, M.A.; Rodriguez, C.J. Cardiovascular Disease in Hispanics/Latinos in the United States. J. Lat./O Psychol. 2016, 4, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Vivo, R.P.; Krim, S.R.; Cevik, C.; Witteles, R.M. Heart failure in Hispanics. J. Am. Coll. Cardiol. 2009, 53, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarese, G.; D’Amario, D. Sex Differences in Heart Failure. Adv. Exp. Med. Biol. 2018, 1065, 529–544. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Arnott, C.; Beale, A.L.; Chandramouli, C.; Hilfiker-Kleiner, D.; Kaye, D.M.; Ky, B.; Santema, B.T.; Sliwa, K.; Voors, A.A. Sex differences in heart failure. Eur. Heart J. 2019, 40, 3859–3868c. [Google Scholar] [CrossRef]
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Curr. Heart Fail Rep. 2017, 14, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.; Larson, M.G.; Vasan, R.S.; Kannel, W.B.; Ho, K.K. The progression from hypertension to congestive heart failure. JAMA 1996, 275, 1557–1562. [Google Scholar] [CrossRef]
- Perrino, C.; Naga Prasad, S.V.; Mao, L.; Noma, T.; Yan, Z.; Kim, H.S.; Smithies, O.; Rockman, H.A. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Investig. 2006, 116, 1547–1560. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Gonçalves, D.; Henriques-Coelho, T.; Fonseca, H.; Ferreira, R.; Padrão, A.I.; Santa, C.; Vieira, S.; Silva, A.F.; Amado, F.; Leite-Moreira, A.; et al. Intermittent cardiac overload results in adaptive hypertrophy and provides protection against left ventricular acute pressure overload insult. J. Physiol. 2015, 593, 3885–3897. [Google Scholar] [CrossRef] [Green Version]
- Jessup, M.; Brozena, S. Heart failure. N. Engl. J. Med. 2003, 348, 2007–2018. [Google Scholar] [CrossRef]
- Gilbert, C.; Cherney, D.Z.; Parker, A.B.; Mak, S.; Floras, J.S.; Al-Hesayen, A.; Parker, J.D. Hemodynamic and neurochemical determinates of renal function in chronic heart failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R167–R175. [Google Scholar] [CrossRef] [Green Version]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Urmaliya, V.; Franchelli, G. A multidimensional sight on cardiac failure: Uncovered from structural to molecular level. Heart Fail. Rev. 2017, 22, 357–370. [Google Scholar] [CrossRef]
- Hsu, J.J.; Ziaeian, B.; Fonarow, G.C. Heart Failure With Mid-Range (Borderline) Ejection Fraction: Clinical Implications and Future Directions. Jacc. Heart Fail. 2017, 5, 763–771. [Google Scholar] [CrossRef]
- Polhemus, D.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Nicholson, C.K.; Lambert, J.P.; Barr, L.A.; Kuek, N.; Herszenhaut, D.; Tan, L.; Murohara, T.; Hansen, J.M.; Husain, A.; et al. Sodium Sulfide Attenuates Ischemic-Induced Heart Failure by Enhancing Proteasomal Function in an Nrf2-Dependent Manner. Circ. Heart Fail. 2016, 9, e002368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vomhof-Dekrey, E.E.; Picklo, M.J., Sr. The Nrf2-antioxidant response element pathway: A target for regulating energy metabolism. J. Nutr. Biochem. 2012, 23, 1201–1206. [Google Scholar] [CrossRef]
- Liang, M.; Jin, S.; Wu, D.-D.; Wang, M.-J.; Zhu, Y.-C. Hydrogen sulfide improves glucose metabolism and prevents hypertrophy in cardiomyocytes. Nitric Oxide Biol. Chem. 2015, 46, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Peleli, M.; Bibli, S.I.; Li, Z.; Chatzianastasiou, A.; Varela, A.; Katsouda, A.; Zukunft, S.; Bucci, M.; Vellecco, V.; Davos, C.H.; et al. Cardiovascular phenotype of mice lacking 3-mercaptopyruvate sulfurtransferase. Biochem. Pharmacol. 2020, 176, 113833. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Erdélyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 601–611. [Google Scholar] [CrossRef]
- Hajar, R. Risk Factors for Coronary Artery Disease: Historical Perspectives. Heart Views Off. J. Gulf Heart Assoc. 2017, 18, 109–114. [Google Scholar] [CrossRef]
- Malinowski, B.; Zalewska, K.; Węsierska, A.; Sokołowska, M.M.; Socha, M.; Liczner, G.; Pawlak-Osińska, K.; Wiciński, M. Intermittent Fasting in Cardiovascular Disorders-An Overview. Nutrients 2019, 11, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Villareal, D.T.; Weiss, E.P.; Racette, S.B.; Steger-May, K.; Klein, S.; Holloszy, J.O. Calorie restriction or exercise: Effects on coronary heart disease risk factors. A randomized, controlled trial. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E197–E202. [Google Scholar] [CrossRef] [PubMed]
- Bales, C.W.; Kraus, W.E. Caloric restriction: Implications for human cardiometabolic health. J. Cardiopulm. Rehabil. Prev. 2013, 33, 201–208. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, M.S.; Dong, T.A.; Ragazzo, G.; Dhindsa, D.S.; Mehta, A.; Sandesara, P.B.; Freeman, A.M.; Taub, P.; Sperling, L.S. From Fad to Fact: Evaluating the Impact of Emerging Diets on the Prevention of Cardiovascular Disease. Am. J. Med. 2020, 133, 1126–1134. [Google Scholar] [CrossRef]
- Sarro, A.; Payedimarri, A.B.; Concina, D.; Farsoni, M.; Piu, N.; Panella, M. The efficacy of fasting regimens on health outcomes: A systematic overview. Minerva Gastroenterol. E Dietol. 2020. [Google Scholar] [CrossRef]
- Park, J.; Seo, Y.G.; Paek, Y.J.; Song, H.J.; Park, K.H.; Noh, H.M. Effect of alternate-day fasting on obesity and cardiometabolic risk: A systematic review and meta-analysis. Metab. Clin. Exp. 2020, 111, 154336. [Google Scholar] [CrossRef] [PubMed]
- Halpern, B.; Mendes, T.B. Intermittent fasting for obesity and related disorders: Unveiling myths, facts, and presumptions. Arch. Endocrinol. Metab. 2021. [Google Scholar] [CrossRef]
- Świątkiewicz, I.; Woźniak, A.; Taub, P.R. Time-Restricted Eating and Metabolic Syndrome: Current Status and Future Perspectives. Nutrients 2021, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Crupi, A.N.; Haase, J.; Brandhorst, S.; Longo, V.D. Periodic and Intermittent Fasting in Diabetes and Cardiovascular Disease. Curr. Diabetes Rep. 2020, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- Di Daniele, N.; Marrone, G.; Di Lauro, M.; Di Daniele, F.; Palazzetti, D.; Guerriero, C.; Noce, A. Effects of Caloric Restriction Diet on Arterial Hypertension and Endothelial Dysfunction. Nutrients 2021, 13, 274. [Google Scholar] [CrossRef]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. 2018, 27, 1212–1221.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stekovic, S.; Hofer, S.J.; Tripolt, N.; Aon, M.A.; Royer, P.; Pein, L.; Stadler, J.T.; Pendl, T.; Prietl, B.; Url, J.; et al. Alternate Day Fasting Improves Physiological and Molecular Markers of Aging in Healthy, Non-obese Humans. Cell Metab. 2019, 30, 462–476.e6. [Google Scholar] [CrossRef] [PubMed]
- Yamaza, H.; Chiba, T.; Higami, Y.; Shimokawa, I. Lifespan extension by caloric restriction: An aspect of energy metabolism. Microsc. Res. Tech. 2002, 59, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- De Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, N.J.; Soni, S.; Takahara, S.; Ferdaoussi, M.; Al Batran, R.; Darwesh, A.M.; Levasseur, J.L.; Beker, D.; Vos, D.Y.; Schmidt, M.A.; et al. Chronically Elevating Circulating Ketones Can Reduce Cardiac Inflammation and Blunt the Development of Heart Failure. Circ. Heart Fail. 2020, 13, e006573. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Matsuura, T.R.; Silljé, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Gungor, N.; McVie, R.; Jain, S.K. Decreased cystathionine-γ-lyase (CSE) activity in livers of type 1 diabetic rats and peripheral blood mononuclear cells (PBMC) of type 1 diabetic patients. J. Biol. Chem. 2014, 289, 11767–11778. [Google Scholar] [CrossRef] [Green Version]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Treviño-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Nakano, S.; Ishii, I.; Shinmura, K.; Tamaki, K.; Hishiki, T.; Akahoshi, N.; Ida, T.; Nakanishi, T.; Kamata, S.; Kumagai, Y.; et al. Hyperhomocysteinemia abrogates fasting-induced cardioprotection against ischemia/reperfusion by limiting bioavailability of hydrogen sulfide anions. J. Mol. Med. 2015, 93, 879–889. [Google Scholar] [CrossRef]
- Chair, S.Y.; Zou, H.; Cao, X. Effects of Exercise Therapy for Adults With Coronary Heart Disease: A Systematic Review and Meta-analysis of Randomized Controlled Trials. J. Cardiovasc. Nurs. 2021, 36, 56–77. [Google Scholar] [CrossRef]
- Park, S.; Han, K.; Lee, S.; Kim, Y.; Lee, Y.; Kang, M.W.; Park, S.; Kim, Y.C.; Han, S.S.; Lee, H.; et al. Association Between Moderate-to-Vigorous Physical Activity and the Risk of Major Adverse Cardiovascular Events or Mortality in People With Various Metabolic Syndrome Status: A Nationwide Population-Based Cohort Study Including 6 Million People. J. Am. Heart Assoc. 2020, 9, e016806. [Google Scholar] [CrossRef]
- Kar, S.; Shahshahan, H.R.; Hackfort, B.T.; Yadav, S.K.; Yadav, R.; Kambis, T.N.; Lefer, D.J.; Mishra, P.K. Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxidants 2019, 8, 638. [Google Scholar] [CrossRef] [Green Version]
- Bijnen, F.C.; Caspersen, C.J.; Mosterd, W.L. Physical inactivity as a risk factor for coronary heart disease: A WHO and International Society and Federation of Cardiology position statement. Bull. World Health Organ. 1994, 72, 1–4. [Google Scholar] [PubMed]
- Giannuzzi, P.; Mezzani, A.; Saner, H.; Björnstad, H.; Fioretti, P.; Mendes, M.; Cohen-Solal, A.; Dugmore, L.; Hambrecht, R.; Hellemans, I.; et al. Physical activity for primary and secondary prevention. Position paper of the Working Group on Cardiac Rehabilitation and Exercise Physiology of the European Society of Cardiology. Eur. J. Cardiovasc. Prev. Rehabil. Off. J. Eur. Soc. Cardiol. Work. Groups Epidemiol. Prev. Card. Rehabil. Exerc. Physiol. 2003, 10, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.; Pahkala, K.; Heinonen, O.J.; Tammelin, T.H.; Pälve, K.; Hirvensalo, M.; Juonala, M.; Loo, B.M.; Magnussen, C.G.; Rovio, S.; et al. Physical inactivity from youth to adulthood and adult cardiometabolic risk profile. Prev. Med. 2021, 106433. [Google Scholar] [CrossRef] [PubMed]
- Brach, J.S.; Simonsick, E.M.; Kritchevsky, S.; Yaffe, K.; Newman, A.B. The association between physical function and lifestyle activity and exercise in the health, aging and body composition study. J. Am. Geriatr. Soc. 2004, 52, 502–509. [Google Scholar] [CrossRef]
- Kemmler, W.; Scharf, M.; Lell, M.; Petrasek, C.; von Stengel, S. High versus moderate intensity running exercise to impact cardiometabolic risk factors: The randomized controlled RUSH-study. Biomed. Res. Int. 2014, 2014, 843095. [Google Scholar] [CrossRef] [PubMed]
- Landaeta-Díaz, L.; Fernández, J.M.; Da Silva-Grigoletto, M.; Rosado-Alvarez, D.; Gómez-Garduño, A.; Gómez-Delgado, F.; López-Miranda, J.; Pérez-Jiménez, F.; Fuentes-Jiménez, F. Mediterranean diet, moderate-to-high intensity training, and health-related quality of life in adults with metabolic syndrome. Eur. J. Prev. Cardiol. 2013, 20, 555–564. [Google Scholar] [CrossRef]
- Tjønna, A.E.; Lee, S.J.; Rognmo, Ø.; Stølen, T.O.; Bye, A.; Haram, P.M.; Loennechen, J.P.; Al-Share, Q.Y.; Skogvoll, E.; Slørdahl, S.A.; et al. Aerobic interval training versus continuous moderate exercise as a treatment for the metabolic syndrome: A pilot study. Circulation 2008, 118, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Hellsten, Y.; Nyberg, M. Cardiovascular Adaptations to Exercise Training. Compr. Physiol. 2015, 6, 1–32. [Google Scholar] [CrossRef]
- Feng, R.; Wang, L.; Li, Z.; Yang, R.; Liang, Y.; Sun, Y.; Yu, Q.; Ghartey-Kwansah, G.; Sun, Y.; Wu, Y.; et al. A systematic comparison of exercise training protocols on animal models of cardiovascular capacity. Life Sci. 2019, 217, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Thu, V.T.; Kim, H.K.; Han, J. Acute and Chronic Exercise in Animal Models. Adv. Exp. Med. Biol. 2017, 999, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Gargiulo, G.; Pironti, G.; Franzone, A.; Scudiero, L.; De Laurentis, M.; Magliulo, F.; Ilardi, F.; Carotenuto, G.; Schiattarella, G.G.; et al. Cardiovascular effects of treadmill exercise in physiological and pathological preclinical settings. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1983–H1989. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wisloff, U.; Kemi, O.J. Animal models in the study of exercise-induced cardiac hypertrophy. Physiol. Res. 2010, 59, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Barauna, V.G.; Batista, M.L., Jr.; Costa Rosa, L.F.; Casarini, D.E.; Krieger, J.E.; Oliveira, E.M. Cardiovascular adaptations in rats submitted to a resistance-training model. Clin. Exp. Pharmacol. Physiol. 2005, 32, 249–254. [Google Scholar] [CrossRef]
- Melo, S.F.S.; da Silva Júnior, N.D.; Barauna, V.G.; Oliveira, E.M. Cardiovascular Adaptations Induced by Resistance Training in Animal Models. Int. J. Med. Sci. 2018, 15, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Duncan, G.E.; Perri, M.G.; Anton, S.D.; Limacher, M.C.; Martin, A.D.; Lowenthal, D.T.; Arning, E.; Bottiglieri, T.; Stacpoole, P.W. Effects of exercise on emerging and traditional cardiovascular risk factors. Prev. Med. 2004, 39, 894–902. [Google Scholar] [CrossRef]
- Pantelić, S.; Popović, M.; Miloradović, V.; Kostić, R.; Milanović, Z.; Bratić, M. Effects of Short-term Exercise Training on Cardiorespiratory Fitness of Male Adults with Myocardial Infarction. J. Phys. Ther. Sci. 2013, 25, 929–935. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, H.R.; Khoshnam, E.; Jahromi, M.K.; Khoshnam, M.S.; Karampour, E. The Effect of 12-Week of Aerobic Training on Homocysteine, Lipoprotein A and Lipid Profile Levels in Sedentary Middle-aged Men. Int. J. Prev. Med. 2014, 5, 1060–1066. [Google Scholar] [PubMed]
- Iglesias-Gutiérrez, E.; Egan, B.; Díaz-Martínez, Á.E.; Peñalvo, J.L.; González-Medina, A.; Martínez-Camblor, P.; O’Gorman, D.J.; Úbeda, N. Transient increase in homocysteine but not hyperhomocysteinemia during acute exercise at different intensities in sedentary individuals. PLoS ONE 2012, 7, e51185. [Google Scholar] [CrossRef] [Green Version]
- Deminice, R.; Ribeiro, D.F.; Frajacomo, F.T. The Effects of Acute Exercise and Exercise Training on Plasma Homocysteine: A Meta-Analysis. PLoS ONE 2016, 11, e0151653. [Google Scholar] [CrossRef]
- Ma, N.; Liu, H.M.; Xia, T.; Liu, J.D.; Wang, X.Z. Chronic aerobic exercise training alleviates myocardial fibrosis in aged rats through restoring bioavailability of hydrogen sulfide. Can. J. Physiol. Pharm. 2018, 96, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wang, Y.; Zhu, X.; Ni, X.; Lu, J. Exercise Increases Cystathionine-γ-lyase Expression and Decreases the Status of Oxidative Stress in Myocardium of Ovariectomized Rats. Int. Heart J. 2016, 57, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson-Meyer, D.E.; Heilbronn, L.K.; Redman, L.M.; Newcomer, B.R.; Frisard, M.I.; Anton, S.; Smith, S.R.; Alfonso, A.; Ravussin, E. Effect of calorie restriction with or without exercise on insulin sensitivity, beta-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care 2006, 29, 1337–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basilio, P.G.; Oliveira, A.P.C.; Castro, A.C.F.; Carvalho, M.R.; Zagatto, A.M.; Martinez, P.F.; Okoshi, M.P.; Okoshi, K.; Ota, G.E.; Reis, F.A.D.; et al. Intermittent Fasting Attenuates Exercise Training-Induced Cardiac Remodeling. Arq. Bras. Cardiol. 2020, 115, 184–193. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, J. Effects of Combined Exercise and Low Carbohydrate Ketogenic Diet Interventions on Waist Circumference and Triglycerides in Overweight and Obese Individuals: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 828. [Google Scholar] [CrossRef]
- Xia, H.; Li, Z.; Sharp, T.E., 3rd; Polhemus, D.J.; Carnal, J.; Moles, K.H.; Tao, Y.X.; Elrod, J.; Pfeilschifter, J.; Beck, K.F.; et al. Endothelial Cell Cystathionine γ-Lyase Expression Level Modulates Exercise Capacity, Vascular Function, and Myocardial Ischemia Reperfusion Injury. J. Am. Heart Assoc. 2020, 9, e017544. [Google Scholar] [CrossRef]
- Wang, B.; Zeng, J.; Gu, Q. Exercise restores bioavailability of hydrogen sulfide and promotes autophagy influx in livers of mice fed with high-fat diet. Can. J. Physiol. Pharm. 2017, 95, 667–674. [Google Scholar] [CrossRef]
- Seifi, B.; Sajedizadeh, A.; Kadkhodaee, M.; Ranjbaran, M. Long-term exercise restores hydrogen sulfide in the kidney and contributes to exercise benefits in 5/6 nephrectomized rats. Clin. Exp. Hypertens. 2019, 41, 87–91. [Google Scholar] [CrossRef]
Figure 1.
Hydrogen sulfide (H2S) Production. H2S is predominately produced by three enzymes—cystathionine-β-synthase, cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase—and can be generated from other processes, such as sulfide quinone oxidoreductase and sulfane sulfur reservoirs (e.g., glutathione/cysteine (hydro)persulfides). H2S can modify free cysteines on enzymes, a reaction known as S-sulfhydration, to alter metabolic pathways, such as glycolysis, and induce up-regulation of metabolic genes via transcription factor activation.
Figure 1.
Hydrogen sulfide (H2S) Production. H2S is predominately produced by three enzymes—cystathionine-β-synthase, cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase—and can be generated from other processes, such as sulfide quinone oxidoreductase and sulfane sulfur reservoirs (e.g., glutathione/cysteine (hydro)persulfides). H2S can modify free cysteines on enzymes, a reaction known as S-sulfhydration, to alter metabolic pathways, such as glycolysis, and induce up-regulation of metabolic genes via transcription factor activation.

Figure 2.
Hydrogen Sulfide (H2S) catabolized by the Sulfide Oxidation Unit. Free H2S can be removed from the cell either by storage in sulfane sulfur pools or the Sulfide Oxidation Unit. H2S catabolism is linked to mitochondrial respiration via sulfide quinone oxidoreductase (SQR). Nicotinamide adenosine dinucleotide + hydrogen (NADH) and flavin adenosine dinucleotide + 2 hydrogen (FADH2) reduce Complex I and II, respectively. The electrons travel across the Electron Transport Chain, through Complex III and cytochrome c (Cyt c) to ultimately reduce oxygen in Complex IV. This flow generates a hydrogen gradient that drives adenosine triphosphate (ATP) synthase and allows for ATP generation from adenosine dinucleotide (ADP). SQR, a key part of the Sulfide Oxidation Unit, extracts electrons from H2S and reduces co-enzyme Q (CoQ), which transfers the electrons to Complex III, contributing to ATP production. As a byproduct, SQR also produces thiosulfate and sulfate—the former can be converted back into H2S by 3-mercaptopyruvate sulfurtransferase. As part of the Sulfide Oxidation Unit, thiosulfate and sulfate react with glutathione to form glutathione persulfide (GSSH), which is then converted into sulfide and recycled back into thiosulfate. Adapted from “Electron Transport Chain”, by BioRender.com (accessed on 24 February 2021). Retrieved from https://app.biorender.com/biorender-templates(accessed on 24 February 2021).
Figure 2.
Hydrogen Sulfide (H2S) catabolized by the Sulfide Oxidation Unit. Free H2S can be removed from the cell either by storage in sulfane sulfur pools or the Sulfide Oxidation Unit. H2S catabolism is linked to mitochondrial respiration via sulfide quinone oxidoreductase (SQR). Nicotinamide adenosine dinucleotide + hydrogen (NADH) and flavin adenosine dinucleotide + 2 hydrogen (FADH2) reduce Complex I and II, respectively. The electrons travel across the Electron Transport Chain, through Complex III and cytochrome c (Cyt c) to ultimately reduce oxygen in Complex IV. This flow generates a hydrogen gradient that drives adenosine triphosphate (ATP) synthase and allows for ATP generation from adenosine dinucleotide (ADP). SQR, a key part of the Sulfide Oxidation Unit, extracts electrons from H2S and reduces co-enzyme Q (CoQ), which transfers the electrons to Complex III, contributing to ATP production. As a byproduct, SQR also produces thiosulfate and sulfate—the former can be converted back into H2S by 3-mercaptopyruvate sulfurtransferase. As part of the Sulfide Oxidation Unit, thiosulfate and sulfate react with glutathione to form glutathione persulfide (GSSH), which is then converted into sulfide and recycled back into thiosulfate. Adapted from “Electron Transport Chain”, by BioRender.com (accessed on 24 February 2021). Retrieved from https://app.biorender.com/biorender-templates(accessed on 24 February 2021).

Figure 3.
Hydrogen sulfide (H2S) Metabolic Targets in the Heart. Few studies have identified metabolic targets of H2S, but those that have been identified are key for cardiac metabolism. H2S can modify free cysteines on enzymes, a reaction known as S-sulfhydration, to alter metabolic pathways, such as glycolysis, and induce up-regulation of metabolic genes via transcription factor activation. Peroxisome proliferator-activated receptors (PPAR), hypoxia-inducible factor 1α (HIF-1α), and nuclear factor E2-related factor 2 (Nrf2) are transcription factors known to be S-sulfhydrated and regulate heart metabolism. H2S also modified the complexes of the Electron Transport Chain and can stimulate mitochondrial respiration. H2S is considered an antioxidant molecule because it can react with glutathione and hydrogen peroxide (H2O2). Finally, our group found that S-sulfhydration represses protein phosphatase 2A (PP2A) activity, allowing for the activation of adenosine monophosphate kinase (AMPK) and phosphorylation of the transcription factor peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) to induce gene expression.
Figure 3.
Hydrogen sulfide (H2S) Metabolic Targets in the Heart. Few studies have identified metabolic targets of H2S, but those that have been identified are key for cardiac metabolism. H2S can modify free cysteines on enzymes, a reaction known as S-sulfhydration, to alter metabolic pathways, such as glycolysis, and induce up-regulation of metabolic genes via transcription factor activation. Peroxisome proliferator-activated receptors (PPAR), hypoxia-inducible factor 1α (HIF-1α), and nuclear factor E2-related factor 2 (Nrf2) are transcription factors known to be S-sulfhydrated and regulate heart metabolism. H2S also modified the complexes of the Electron Transport Chain and can stimulate mitochondrial respiration. H2S is considered an antioxidant molecule because it can react with glutathione and hydrogen peroxide (H2O2). Finally, our group found that S-sulfhydration represses protein phosphatase 2A (PP2A) activity, allowing for the activation of adenosine monophosphate kinase (AMPK) and phosphorylation of the transcription factor peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) to induce gene expression.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Casin, K.M.; Calvert, J.W. Harnessing the Benefits of Endogenous Hydrogen Sulfide to Reduce Cardiovascular Disease. Antioxidants 2021, 10, 383. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030383
AMA Style
Casin KM, Calvert JW. Harnessing the Benefits of Endogenous Hydrogen Sulfide to Reduce Cardiovascular Disease. Antioxidants. 2021; 10(3):383. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030383
Chicago/Turabian StyleCasin, Kevin M., and John W. Calvert. 2021. "Harnessing the Benefits of Endogenous Hydrogen Sulfide to Reduce Cardiovascular Disease" Antioxidants 10, no. 3: 383. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030383
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.