
Translational Approaches with Antioxidant Phytochemicals against Alcohol-Mediated Oxidative Stress, Gut Dysbiosis, Intestinal Barrier Dysfunction, and Fatty Liver Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Gut Microbiome
2.1. Microbiome Present in Many Tissues
2.2. Gut Microbiota Altered by Aging, Disease States, and Environmental Factors
3. Oxidative Alcohol Metabolism and Progression to Alcoholic Liver Disease
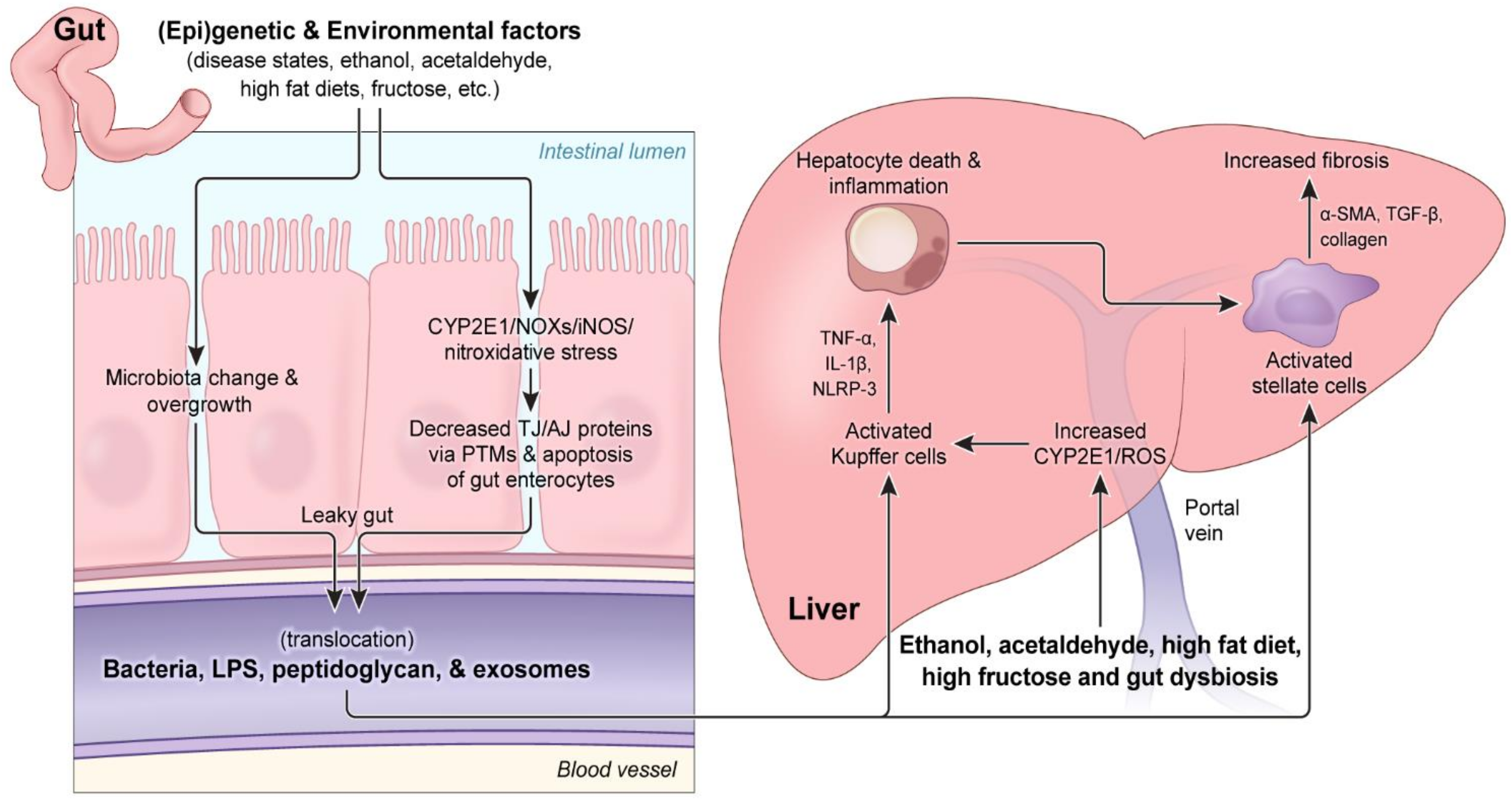
4. The Mechanisms of Alcohol-Mediated Gut Dysbiosis, Intestinal Barrier Dysfunction, and Consequences
4.1. The Effect of Alcohol on the Amounts and Composition of Gut Microbiota
4.2. Mechanisms of Alcohol-Induced Damage to the Intestines, Resulting in Leaky Gut and Endotoxemia
4.3. Mechanisms of Gut Leakiness via Oxidative Stress and PTMs of Paracellular Proteins
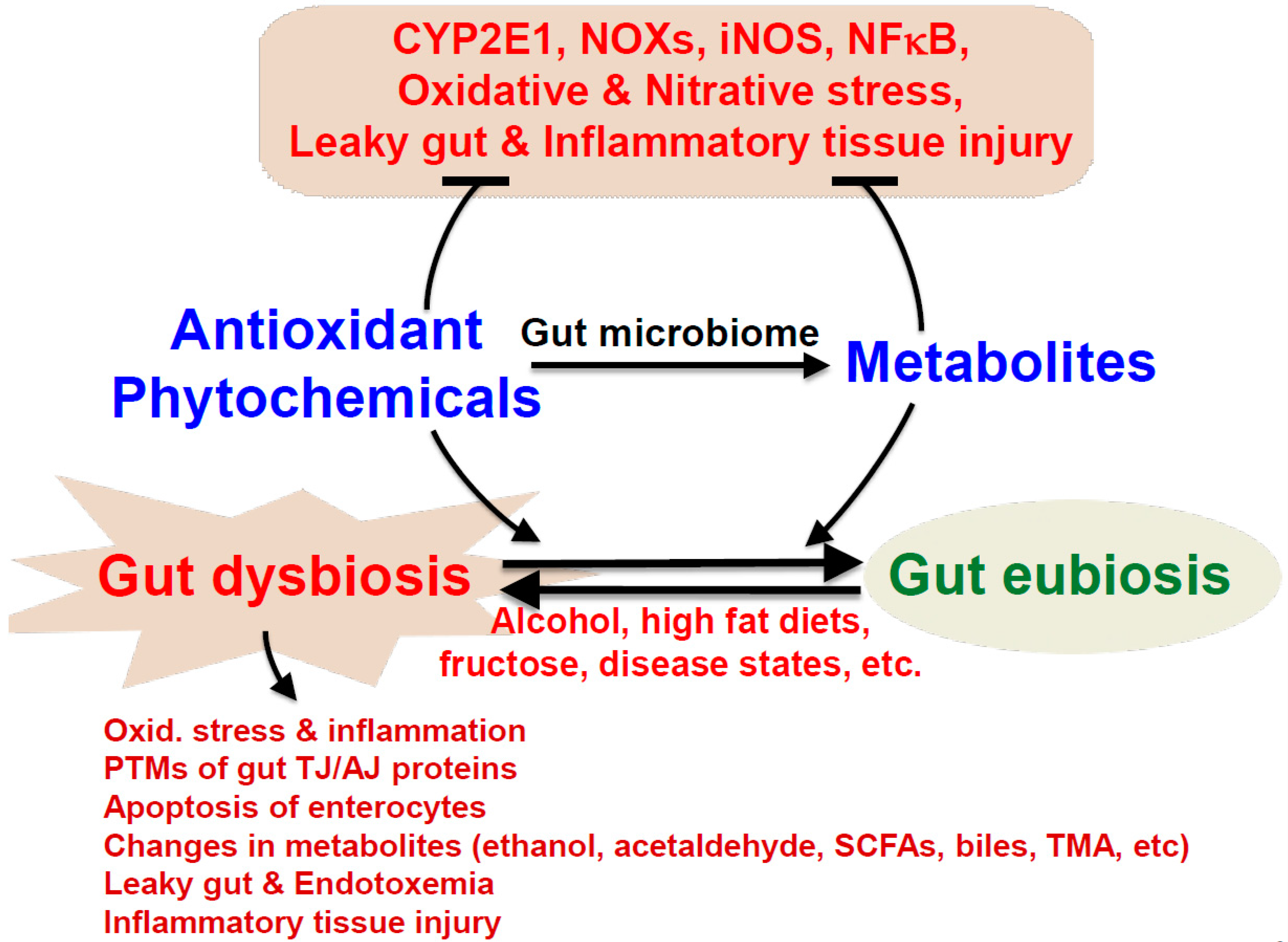
4.4. Crosstalk among Gut Dysbiosis, Intestinal Barrier Dysfunction, and ALD
5. The Antioxidant Properties, Metabolisms, and Health Benefits of Various Phytochemicals against Gut Dysbiosis, Intestinal Barrier Dysfunction, and Fatty Liver Disease
6. Translational Approaches and Therapeutics against Alcohol-Mediated Oxidative Stress, Gut Dysbiosis, Intestinal Barrier Dysfunction and Fatty Liver Disease
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekaboruah, E.; Suryavanshi, M.V.; Chettri, D.; Verma, A.K. Human microbiome: An academic update on human body site specific surveillance and its possible role. Arch. Microbiol. 2020, 202, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Howitt, M.R.; Garrett, W.S. Exploring host-microbiota interactions in animal models and humans. Genes Dev. 2013, 27, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Shanahan, F. The Colonic Microbiota and Colonic Disease. Curr. Gastroenterol. Rep. 2012, 14, 446–452. [Google Scholar] [CrossRef]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and Fungi of the Human Gut Microbiome: Correlations with Diet and Bacterial Residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Li, S.; Gan, R.-Y.; Zhou, T.; Xu, D.-P.; Li, H.-B. Impacts of Gut Bacteria on Human Health and Diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y.A. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Genet. 2016, 14, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Vasapolli, R.; Schütte, K.; Schulz, C.; Vital, M.; Schomburg, D.; Pieper, D.H.; Vilchez-Vargas, R.; Malfertheiner, P. Analysis of Transcriptionally Active Bacteria throughout the Gastrointestinal Tract of Healthy Individuals. Gastroenterology 2019, 157, 1081–1092.e3. [Google Scholar] [CrossRef] [Green Version]
- Zoetendal, E.G.; Raes, J.; Bogert, B.V.D.; Arumugam, M.; Booijink, C.C.G.M.; Troost, F.J.; Bork, P.; Wels, M.; De Vos, W.M.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Engstler, A.J.; Aumiller, T.; Degen, C.; Dürr, M.; Weiss, E.; Maier, I.B.; Schattenberg, J.M.; Jin, C.J.; Sellmann, C.; Bergheim, I. Insulin resistance alters hepatic ethanol metabolism: Studies in mice and children with non-alcoholic fatty liver disease. Gut 2016, 65, 1564–1571. [Google Scholar] [CrossRef]
- Elshaghabee, F.M.F.; Ebockelmann, W.; Emeske, D.; Vrese, M.E.; Ewalte, H.-G.; Eschrezenmeir, J.; Heller, K.J. Ethanol Production by Selected Intestinal Microorganisms and Lactic Acid Bacteria Growing under Different Nutritional Conditions. Front. Microbiol. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Kang, D.J.; Hylemon, P.B.; Gillevet, P.M.; Sartor, R.B.; Betrapally, N.S.; Kakiyama, G.; Sikaroodi, M.; Takei, H.; Nittono, H.; Zhou, H.; et al. Gut microbial composition can differentially regulate bile acid synthesis in humanized mice. Hepatol. Commun. 2017, 1, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bäumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nat. Cell Biol. 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubeda, C.; Djukovic, A.; Isaac, S. Roles of the intestinal microbiota in pathogen protection. Clin. Transl. Immunol. 2017, 6, e128. [Google Scholar] [CrossRef]
- Petra, A.I.; Panagiotidou, S.; Hatziagelaki, E.; Stewart, J.M.; Conti, P.; Theoharides, T.C. Gut-Microbiota-Brain Axis and Its Effect on Neuropsychiatric Disorders With Suspected Immune Dysregulation. Clin. Ther. 2015, 37, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Hyland, N.P.; Cryan, J.F. Microbe-host interactions: Influence of the gut microbiota on the enteric nervous system. Dev. Biol. 2016, 417, 182–187. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Wang, M.; Monaco, M.H.; Donovan, S.M. Impact of early gut microbiota on immune and metabolic development and function. Semin. Fetal Neonatal Med. 2016, 21, 380–387. [Google Scholar] [CrossRef]
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; De Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.G.; Fitzgerald, G.F.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ruiz, S.; Sanchez-Carrillo, S.; Ciordia, S.; Mena, M.C.; Méndez-García, C.; Rojo, D.; Bargiela, R.; Zubeldia-Varela, E.; Martínez-Martínez, M.; Barbas, C.; et al. Functional microbiome deficits associated with ageing: Chronological age threshold. Aging Cell 2019, 19, e13063. [Google Scholar] [CrossRef]
- Urban, R.J.; Pyles, R.B.; Stewart, C.J.; Ajami, N.; Randolph, M.K.; Durham, W.J.; Danesi, C.P.; Dillon, E.L.; Summons, M.J.R.; Singh, C.K.; et al. Altered Fecal Microbiome Years after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Angoa-Pérez, M.; Zagorac, B.; Anneken, J.H.; Briggs, D.I.; Winters, A.D.; Greenberg, J.M.; Ahmad, M.; Theis, K.R.; Kuhn, D.M. Repetitive, mild traumatic brain injury results in a progressive white matter pathology, cognitive deterioration, and a transient gut microbiota dysbiosis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cremonini, E.; Wang, Z.; Bettaieb, A.; Adamo, A.M.; Daveri, E.; Mills, D.A.; Kalanetra, K.M.; Haj, F.G.; Karakas, S.; Oteiza, P.I. (-)-Epicatechin protects the intestinal barrier from high fat diet-induced permeabilization: Implications for steatosis and insulin resistance. Redox Biol. 2018, 14, 588–599. [Google Scholar] [CrossRef]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrocola, R.; Ferrocino, I.; Liberto, E.; Chiazza, F.; Cento, A.S.; Collotta, D.; Querio, G.; Nigro, D.; Bitonto, V.; Cutrin, J.C.; et al. Fructose liquid and solid formulations differently affect gut integrity, microbiota composition and related liver toxicity: A comparative in vivo study. J. Nutr. Biochem. 2018, 55, 185–199. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.; Motti, M.L.; Meccariello, R. ω-3 and ω-6 Polyunsaturated Fatty Acids, Obesity and Cancer. Nutrients 2020, 12, 2751. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef]
- Araújo, J.R.; Tomas, J.; Brenner, C.; Sansonetti, P.J. Impact of high-fat diet on the intestinal microbiota and small intestinal physiology before and after the onset of obesity. Biochimie 2017, 141, 97–106. [Google Scholar] [CrossRef]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A.; et al. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014, 8, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Abu Halaka, D.; Hayouka, Z.; Tirosh, O. High-Fat Diet Induced Alteration of Mice Microbiota and the Functional Ability to Utilize Fructooligosaccharide for Ethanol Production. Front. Cell. Infect. Microbiol. 2020, 10, 376. [Google Scholar] [CrossRef]
- Do, M.H.; Lee, E.; Oh, M.-J.; Kim, Y.; Park, H.-Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, J.C.; Bode, C.; Heidelbach, R.; Dürr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34. [Google Scholar]
- Baraona, E.; Julkunen, R.; Tannenbaum, L.; Lieber, C.S. Role of intestinal bacterial overgrowth in ethanol production and metabolism in rats. Gastroenterology 1986, 90, 103–110. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, H.; Bai, Y.; Zhi, F. Gut dysbiosis-derived exosomes trigger hepatic steatosis by transiting HMGB1 from intestinal to liver in mice. Biochem. Biophys. Res. Commun. 2019, 509, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. Curr. Rev. 2017, 38, 147–161. [Google Scholar]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A. Metabolism of Ethanol and Some Associated Adverse Effects on the Liver and the Stomach. Recent Dev. Alcohol. 1998, 14, 7–40. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.G. Nonoxidative ethanol metabolism: Formation of fatty acid ethyl esters by cholesterol esterase. Proc. Natl. Acad. Sci. USA 1982, 79, 3954–3957. [Google Scholar] [CrossRef] [Green Version]
- Heier, C.; Xie, H.; Zimmermann, R. Nonoxidative ethanol metabolism in humans—from biomarkers to bioactive lipids. IUBMB Life 2016, 68, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.H.G. Clinical Pharmacokinetics of Ethanol. Clin. Pharmacokinet. 1987, 13, 273–292. [Google Scholar] [CrossRef]
- CDC. Alcohol Use and Your Health. Centers of Disease Control and Prevention. 2020. Available online: https://www.cdc.gov/alcohol/fact-sheets/alcohol-use.htm#:~:text=Over%20time%2C%20excessive%20alcohol%20use,liver%20disease%2C%20and%20digestive%20problems.&text=Cancer%20of%20the%20breast%2C%20mouth,esophagus%2C%20liver%2C%20and%20colon (accessed on 17 December 2020).
- NIAAA. Alcohol Facts and Statistics. The National Institute on Alcohol Abuse and Alcoholism (NIAAA). 2020. Available online: https://www.niaaa.nih.gov/sites/default/files/AlcoholFactsAndStats.pdf (accessed on 17 December 2020).
- Zakhari, S. Alcohol Metabolism and Epigenetics Changes. Alcohol Res. Curr. Rev. 2013, 35, 6–16. [Google Scholar]
- Lieber, C.S. Microsomal Ethanol-Oxidizing System (MEOS): The First 30 Years (1968–1998)—A Review. Alcohol. Clin. Exp. Res. 1999, 23, 991–1007. [Google Scholar] [CrossRef]
- Song, B.J.; Gelboin, H.V.; Park, S.S.; Yang, C.S.; Gonzalez, F.J. Complementary DNA and protein sequences of ethanol-inducible rat and human cytochrome P-450s. Transcriptional and post-transcriptional regulation of the rat enzyme. J. Biol. Chem. 1986, 261, 16689–16697. [Google Scholar] [CrossRef]
- Roberts, B.J.; Song, B.J.; Soh, Y.; Park, S.S.; Shoaf, S.E. Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. J. Biol. Chem. 1995, 270, 29632–29635. [Google Scholar] [CrossRef] [Green Version]
- Song, B.J. Ethanol-inducible cytochrome P450 (CYP2E1): Biochemistry, molecular biology and clinical relevance: 1996 update. Alcohol. Clin. Exp. Res. 1996, 20, 138–146. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 27–42. [Google Scholar] [CrossRef]
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.; French, S.W.; Morgan, T.R. Production of a cytochrome P450 2E1 transgenic mouse and initial evaluation of alcoholic liver damage. Hepatology 2002, 36, 122–134. [Google Scholar] [CrossRef]
- Cederbaum, A.I. CYP2E1 potentiates toxicity in obesity and after chronic ethanol treatment. Drug Metab. Drug Interact. 2012, 27, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Kathirvel, E.; Morgan, K.; French, S.W.; Morgan, T.R. Overexpression of liver-specific cytochrome P4502E1 impairs hepatic insulin signaling in a transgenic mouse model of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.-H.; Hood, B.L.; Kim, B.-J.; Hardwick, J.P.; Conrads, T.P.; Veenstra, T.D.; Song, B.J. Inactivation of oxidized andS-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology 2006, 44, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-J.; Hood, B.L.; Aragon, R.A.; Hardwick, J.P.; Conrads, T.P.; Veenstra, T.D.; Song, B.J. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics 2006, 6, 1250–1260. [Google Scholar] [CrossRef]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Akbar, M.; Abdelmegeed, M.A.; Byun, K.; Lee, B.; Yoon, S.K.; Hardwick, J.P. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014, 3, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galligan, J.J.; Smathers, R.L.; Shearn, C.T.; Fritz, K.S.; Backos, D.S.; Jiang, H.; Franklin, C.C.; Orlicky, D.J.; MacLean, K.N.; Petersen, D.R. Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease. J. Toxicol. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Song, B.-J.; Abdelmegeed, M.A.; Cho, Y.-E.; Akbar, M.; Rhim, J.S.; Song, M.-K.; Hardwick, J.P. Contributing Roles of CYP2E1 and Other Cytochrome P450 Isoforms in Alcohol-Related Tissue Injury and Carcinogenesis. Adv. Exp. Med. Biol. 2019, 1164, 73–87. [Google Scholar] [CrossRef]
- Seitz, H.K. The role of cytochrome P4502E1 in the pathogenesis of alcoholic liver disease and carcinogenesis. Chem. Interact. 2020, 316, 108918. [Google Scholar] [CrossRef]
- Szabo, G. Gut–Liver Axis in Alcoholic Liver Disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Bataller, R. Liver Fibrosis in Alcoholic Liver Disease. Semin. Liver Dis. 2015, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M. Developmental Morphogens & Recovery from Alcoholic Liver Disease. Results Probl. Cell Differ. 2018, 1032, 145–151. [Google Scholar] [CrossRef]
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021, 18, 73–91. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of Liver Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Doorn, J.A.; Hurley, T.D.; Petersen, D.R. Inhibition of Human Mitochondrial Aldehyde Dehydrogenase by 4-Hydroxynon-2-enal and 4-Oxonon-2-enal†. Chem. Res. Toxicol. 2006, 19, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Abdelmegeed, M.A.; Yoo, S.-H.; Kim, B.-J.; Jo, S.A.; Jo, I.; Moon, K.-H. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. J. Proteom. 2011, 74, 2691–2702. [Google Scholar] [CrossRef] [Green Version]
- Fritz, K.S.; Galligan, J.J.; Hirschey, M.D.; Verdin, E.; Petersen, D.R. Mitochondrial Acetylome Analysis in a Mouse Model of Alcohol-Induced Liver Injury Utilizing SIRT3 Knockout Mice. J. Proteome Res. 2012, 11, 1633–1643. [Google Scholar] [CrossRef] [Green Version]
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.-J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 1–21. [Google Scholar] [CrossRef]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic Liver Disease: The Gut Microbiome and Liver Cross Talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal Microbiome: Alcohol Effects on the Composition of Intestinal Microbiota. Alcohol Res. 2015, 37, 223–236. [Google Scholar]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 1–14. [Google Scholar] [CrossRef]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef]
- Mutlu, E.; Keshavarzian, A.; Engen, P.; Forsyth, C.B.; Sikaroodi, M.; Gillevet, P. Intestinal Dysbiosis: A Possible Mechanism of Alcohol-Induced Endotoxemia and Alcoholic Steatohepatitis in Rats. Alcohol. Clin. Exp. Res. 2009, 33, 1836–1846. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef] [PubMed]
- Queipo-Ortuño, M.I.; Boto-Ordóñez, M.; Murri, M.; Gomez-Zumaquero, J.M.; Clemente-Postigo, M.; Estruch, R.; Diaz, F.C.; Andrés-Lacueva, C.; Tinahones, F.J. Influence of red wine polyphenols and ethanol on the gut microbiota ecology and biochemical biomarkers. Am. J. Clin. Nutr. 2012, 95, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, R.; Zhou, Q.; Liu, L.; Huang, F.; Deng, Y.; Ma, Y.; Wei, Z.; Tang, X.; Zhang, M. Lychee (Litchi chinensis Sonn.) Pulp Phenolic Extract Provides Protection against Alcoholic Liver Injury in Mice by Alleviating Intestinal Microbiota Dysbiosis, Intestinal Barrier Dysfunction, and Liver Inflammation. J. Agric. Food Chem. 2017, 65, 9675–9684. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Shirakawa, H.; Lu, N.-S.; Peng, H.-C.; Xiao, Q.; Yang, S.-C. Impacts of fish oil on the gut microbiota of rats with alcoholic liver damage. J. Nutr. Biochem. 2020, 86, 108491. [Google Scholar] [CrossRef] [PubMed]
- Litwinowicz, K.; Choroszy, M.; Waszczuk, E. Changes in the composition of the human intestinal microbiome in alcohol use disorder: A systematic review. Am. J. Drug Alcohol Abus. 2019, 46, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruya, A.; Kuwahara, A.; Saito, Y.; Yamaguchi, H.; Tsubo, T.; Suga, S.; Inai, M.; Aoki, Y.; Takahashi, S.; Tsutsumi, E.; et al. Ecophysiological consequences of alcoholism on human gut microbiota: Implications for ethanol-related pathogenesis of colon cancer. Sci. Rep. 2016, 6, 27923. [Google Scholar] [CrossRef] [Green Version]
- Bjørkhaug, S.T.; Aanes, H.; Neupane, S.P.; Bramness, J.G.; Malvik, S.; Henriksen, C.; Skar, V.; Medhus, A.W.; Valeur, J. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes 2019, 10, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciocan, D.; Voican, C.S.; Wrzosek, L.; Hugot, C.; Rainteau, D.; Humbert, L.; Cassard, A.-M.; Perlemuter, G. Bile acid homeostasis and intestinal dysbiosis in alcoholic hepatitis. Aliment. Pharmacol. Ther. 2018, 48, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Jesudian, A.B. Small Intestinal Bacterial Overgrowth in Patients with Cirrhosis. J. Clin. Exp. Hepatol. 2019, 9, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Gurwara, S.; Dai, A.; Ajami, N.J.; Graham, D.Y.; White, D.L.; Chen, L.; Jang, A.; Chen, E.; El-Serag, H.B.; Petrosino, J.F.; et al. Alcohol use alters the colonic mucosa–associated gut microbiota in humans. Nutr. Res. 2020, 83, 119–128. [Google Scholar] [CrossRef]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J.; et al. Metagenomic Analyses of Alcohol Induced Pathogenic Alterations in the Intestinal Microbiome and the Effect of Lactobacillus rhamnosus GG Treatment. PLoS ONE 2013, 8, e53028. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyfors, S.; Salaspuro, V.; Jousimies-Somer, H.; Siitonen, A.; Salaspuro, M.; Heine, R. Ethanol oxidation and acetaldehyde production in vitro by human intestinal strains of Escherichia coli under aerobic, microaerobic, and anaerobic conditions. Scand. J. Gastroenterol. 1999, 34, 967–973. [Google Scholar]
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2010, 53, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Ya-E, Z.; Ji-Dong, W.; Yu-Fan, L.; Ying, Z.; Ya-Lun, S.; Meng-Yu, M.; Rui-Ling, Z. Comparison of Microbial Diversity and Composition in Jejunum and Colon of the Alcohol-dependent Rats. J. Microbiol. Biotechnol. 2018, 28, 1883–1895. [Google Scholar] [CrossRef]
- Addolorato, G.; Ponziani, F.R.; Dionisi, T.; Mosoni, C.; Vassallo, G.A.; Sestito, L.; Petito, V.; Picca, A.; Marzetti, E.; Tarli, C.; et al. Gut microbiota compositional and functional fingerprint in patients with alcohol use disorder and alcohol-associated liver disease. Liver Int. 2020, 40, 878–888. [Google Scholar] [CrossRef]
- Harnisch, J.P.; Tronca, E.; Nolan, C.M.; Turck, M.; Holmes, K.K. Diphtheria among Alcoholic Urban Adults. Ann. Intern. Med. 1989, 111, 71–82. [Google Scholar] [CrossRef]
- Cericco, M.; Iglicki, F.; Guillaumont, M.P.; Schmitt, J.L.; Dupas, J.L.; Capron, J.P. [Corynebacterium xerosis endocarditis associated with alcoholic cirrhosis]. Gastroentérologie Clin. Biol. 1996, 20, 514. [Google Scholar]
- Tsuruya, A.; Kuwahara, A.; Saito, Y.; Yamaguchi, H.; Tenma, N.; Inai, M.; Takahashi, S.; Tsutsumi, E.; Suwa, Y.; Totsuka, Y.; et al. Major Anaerobic Bacteria Responsible for the Production of Carcinogenic Acetaldehyde from Ethanol in the Colon and Rectum. Alcohol Alcohol. 2016, 51, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2017, 67, 891–901. [Google Scholar] [CrossRef]
- Halsted, C.H.; Robles, E.A.; Mezey, E. Distribution of ethanol in the human gastrointestinal tract. Am. J. Clin. Nutr. 1973, 26, 831–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elamin, E.E.; Masclee, A.; Dekker, J.; Jonkers, D. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhari, S. Overview: How Is Alcohol Metabolized by the Body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Bode, C.; Bode, J.C. Alcohol’s role in gastrointestinal tract disorders. Alcohol Health Res. World 1997, 21, 76–83. [Google Scholar]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef] [Green Version]
- Thoo, L.; Noti, M.; Krebs, P. Keep calm: The intestinal barrier at the interface of peace and war. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hammer, A.M.; Khan, O.M.; Morris, N.L.; Li, X.; Movtchan, N.V.; Cannon, A.R.; Choudhry, M.A. The Effects of Alcohol Intoxication and Burn Injury on the Expression of Claudins and Mucins in the Small and Large Intestines. Shock 2016, 45, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J. Chronic ethanol feeding affects intestinal mucus lipid composition and glycosylation in rats. Ann. Nutr. Metab. 2002, 46, 38–44. [Google Scholar] [CrossRef]
- Slomiany, A.; Piotrowski, E.; Piotrowski, J.; Slomiany, B.L. Impact of ethanol on innate protection of gastric mucosal epithelial surfaces and the risk of injury. J. Physiol. Pharmacol. 2000, 51, 433–447. [Google Scholar] [PubMed]
- Qin, X.; Deitch, E.A. Dissolution of lipids from mucus: A possible mechanism for prompt disruption of gut barrier function by alcohol. Toxicol. Lett. 2015, 232, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.-P.; Wu, C.-W.; Lee, S.-P.; Ho, J.-L.; Lee, S.-L.; Nieh, S.; Yin, S.-J. Expression Pattern, Ethanol-Metabolizing Activities, and Cellular Localization of Alcohol and Aldehyde Dehydrogenases in Human Small Intestine. Alcohol. Clin. Exp. Res. 2012, 36, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.J.; Shoaf, S.; Jeong, K.; Song, B.-J. Induction of CYP2E1 in Liver, Kidney, Brain and Intestine During Chronic Ethanol Administration and Withdrawal: Evidence That CYP2E1 Possesses a Rapid Phase Half-Life of 6 Hours or Less. Biochem. Biophys. Res. Commun. 1994, 205, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-E.; Song, B.-J. Pomegranate prevents binge alcohol-induced gut leakiness and hepatic inflammation by suppressing oxidative and nitrative stress. Redox Biol. 2018, 18, 266–278. [Google Scholar] [CrossRef]
- Cho, Y.-E.; Yu, L.-R.; Abdelmegeed, M.A.; Yoo, S.-H.; Song, B.-J. Apoptosis of enterocytes and nitration of junctional complex proteins promote alcohol-induced gut leakiness and liver injury. J. Hepatol. 2018, 69, 142–153. [Google Scholar] [CrossRef]
- Salaspuro, M. Bacteriocolonic Pathway for Ethanol Oxidation: Characteristics and Implications. Ann. Med. 1996, 28, 195–200. [Google Scholar] [CrossRef]
- Koivisto, T.; Salaspuro, M. Aldehyde Dehydrogenases of the Rat Colon: Comparison with Other Tissues of the Alimentary Tract and the Liver. Alcohol. Clin. Exp. Res. 1996, 20, 551–555. [Google Scholar] [CrossRef]
- Nosova, T.; Jousimies-Somer, H.; Jokelainen, K.; Heine, R.; Salaspuro, M. Acetaldehyde production and metabolism by human indigenous and probiotic lactobacillus and bifidobacterium strains. Alcohol Alcohol. 2000, 35, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, D.; Carryl, O.R.; Tran, M.X.; Krohn, R.L.; Bagchi, D.J.; Garg, A.; Bagchi, M.; Mitra, S.; Stohs, S.J. Stress, diet and alcohol-induced oxidative gastrointestinal mucosal injury in rats and protection by bismuth subsalicylate. J. Appl. Toxicol. 1998, 18, 3–13. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Bishehsari, F.; Magno, E.; Swanson, G.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and Gut-Derived Inflammation. Alcohol Res. 2017, 38, 163–171. [Google Scholar]
- Asai, K.; Buurman, W.A.; Reutelingsperger, C.P.M.; Schutte, B.; Kaminishi, M. Low concentrations of ethanol induce apoptosis in human intestinal cells. Scand. J. Gastroenterol. 2003, 38, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Klingensmith, N.J.; Fay, K.T.; Lyons, J.D.; Chen, C.-W.; Otani, S.; Liang, Z.; Chihade, D.B.; Burd, E.M.; Ford, M.L.; Coopersmith, C.M. Chronic Alcohol Ingestion Worsens Survival and Alters Gut Epithelial Apoptosis and CD8+ T Cell Function After Pseudomonas Aeruginosa Pneumonia-Induced Sepsis. Shock 2019, 51, 453–463. [Google Scholar] [CrossRef]
- Lambert, J.C.; Zhou, Z.; Wang, L.; Song, Z.; McClain, C.J.; Kang, Y.J. Prevention of Alterations in Intestinal Permeability Is Involved in Zinc Inhibition of Acute Ethanol-Induced Liver Damage in Mice. J. Pharmacol. Exp. Ther. 2003, 305, 880–886. [Google Scholar] [CrossRef]
- Lambert, J.C.; Zhou, Z.; Wang, L.; Song, Z.; McClain, C.J.; Kang, Y.J. Preservation of Intestinal Structural Integrity by Zinc Is Independent of Metallothionein in Alcohol-Intoxicated Mice. Am. J. Pathol. 2004, 164, 1959–1966. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.-H.; Yun, J.-W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.-J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free. Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresci, G.A. The Gut Microbiome: A New Frontier for Alcohol Investigation. Alcohol. Clin. Exp. Res. 2015, 39, 947–949. [Google Scholar] [CrossRef] [Green Version]
- Gyongyosi, B.; Cho, Y.; Lowe, P.; Calenda, C.D.; Iracheta-Vellve, A.; Satishchandran, A.; Ambade, A.; Szabo, G. Alcohol-induced IL-17A production in Paneth cells amplifies endoplasmic reticulum stress, apoptosis, and inflammasome-IL-18 activation in the proximal small intestine in mice. Mucosal Immunol. 2019, 12, 930–944. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Neunlist, M.; Van Landeghem, L.; Mahé, M.M.; Derkinderen, P.; Varannes, S.B.D.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2012, 10, 90–100. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, J.; Chang, B.; Wang, B.; Zhang, D.; Wang, B. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol. Med. Rep. 2014, 9, 2352–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; Van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of Ethanol and Acetaldehyde on Tight Junction Integrity: In Vitro Study in a Three Dimensional Intestinal Epithelial Cell Culture Model. PLoS ONE 2012, 7, e35008. [Google Scholar] [CrossRef] [Green Version]
- Mir, H.; Meena, A.S.; Chaudhry, K.K.; Shukla, P.K.; Gangwar, R.; Manda, B.; Padala, M.K.; Shen, L.; Turner, J.R.; Dietrich, P.; et al. Occludin deficiency promotes ethanol-induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2016, 1860, 765–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, K.J.; Rao, R.K. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am. J. Physiol. Liver Physiol. 2001, 280, G1280–G1288. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.K.; Basuroy, S.; Rao, V.U.; Karnaky, K.J., Jr.; Gupta, A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem. J. 2002, 368(Pt. 2), 471–481. [Google Scholar] [CrossRef]
- Kale, G.; Naren, A.P.; Sheth, P.; Rao, R.K. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem. Biophys. Res. Commun. 2003, 302, 324–329. [Google Scholar] [CrossRef]
- Samak, G.; Aggarwal, S.; Rao, R.K. ERK is involved in EGF-mediated protection of tight junctions, but not adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers. Am. J. Physiol. Liver Physiol. 2011, 301, G50–G59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Ye, T.J.; DeCaro, E.; Buehler, B.; Stahl, Z.; Bonavita, G.; Daniels, M.; You, M. Intestinal SIRT1 Deficiency Protects Mice from Ethanol-Induced Liver Injury by Mitigating Ferroptosis. Am. J. Pathol. 2020, 190, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörfel, M.J.; Huber, O. Modulation of Tight Junction Structure and Function by Kinases and Phosphatases Targeting Occludin. J. Biomed. Biotechnol. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Reiche, J.; Huber, O. Post-translational modifications of tight junction transmembrane proteins and their direct effect on barrier function. Biochim. Biophys. Acta (BBA)—Biomembr. 2020, 1862, 183330. [Google Scholar] [CrossRef] [PubMed]
- Sheth, P.; Seth, A.; Atkinson, K.J.; Gheyi, T.; Kale, G.; Giorgianni, F.; Desiderio, D.M.; Li, C.; Naren, A.; Rao, R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem. J. 2007, 402, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basuroy, S.; Sheth, P.; Mansbach, C.M.; Rao, R.K. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: Protection by EGF and l-glutamine. Am. J. Physiol. Liver Physiol. 2005, 289, G367–G375. [Google Scholar] [CrossRef] [PubMed]
- Dunagan, M.; Chaudhry, K.; Samak, G.; Rao, R.K. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. Am. J. Physiol. Liver Physiol. 2012, 303, G1356–G1364. [Google Scholar] [CrossRef] [Green Version]
- Haorah, J.; Heilman, D.; Knipe, B.; Chrastil, J.; Leibhart, J.; Ghorpade, A.; Miller, D.W.; Persidsky, Y. Ethanol-Induced Activation of Myosin Light Chain Kinase Leads to Dysfunction of Tight Junctions and Blood-Brain Barrier Compromise. Alcohol. Clin. Exp. Res. 2005, 29, 999–1009. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Jonkers, D.M. Chapter 14—Effects of Acetaldehyde on Intestinal Barrier Function. In Molecular Aspects of Alcohol and Nutrition; Patel, V.B., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 171–186. [Google Scholar]
- Chaudhry, K.K.; Chaudhry, K.K.; Samak, G.; Shukla, P.K.; Mir, H.; Gangwar, R.; Manda, B.; Isse, T.; Kawamoto, T.; Salaspuro, M.; et al. ALDH2 Deficiency Promotes Ethanol-Induced Gut Barrier Dysfunction and Fatty Liver in Mice. Alcohol. Clin. Exp. Res. 2015, 39, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Elamin, E.; Masclee, A.; Troost, F.; Dekker, J.; Jonkers, D. Cytotoxicity and metabolic stress induced by acetaldehyde in human intestinal LS174T goblet-like cells. Am. J. Physiol. Liver Physiol. 2014, 307, G286–G294. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Forsyth, C.B.; Farhadi, A.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Nitric Oxide-Mediated Intestinal Injury Is Required for Alcohol-Induced Gut Leakiness and Liver Damage. Alcohol. Clin. Exp. Res. 2009, 33, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, C.B.; Voigt, R.M.; Keshavarzian, A. Intestinal CYP2E1: A mediator of alcohol-induced gut leakiness. Redox Biol. 2014, 3, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Zhang, L.; Forsyth, C.B.; Shaikh, M.; Song, S.; Keshavarzian, A. The Role of miR-212 and iNOS in Alcohol-Induced Intestinal Barrier Dysfunction and Steatohepatitis. Alcohol. Clin. Exp. Res. 2015, 39, 1632–1641. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Role of snail activation in alcohol-induced iNOS-mediated disruption of intestinal epithelial cell permeability. Alcohol. Clin. Exp. Res. 2011, 35, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.-M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Abdelmegeed, M.A.; Song, B.-J. Preventive effects of indole-3-carbinol against alcohol-induced liver injury in mice via antioxidant, anti-inflammatory, and anti-apoptotic mechanisms: Role of gut-liver-adipose tissue axis. J. Nutr. Biochem. 2018, 55, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Wan, P.; Zhu, X.-D.; Ren, Y.-P.; Wang, C.; Yan, R.-W.; Guo, Y.; Bai, A.-P. Inhibition of NADPH oxidase activities ameliorates DSS-induced colitis. Biochem Pharmacol 2018, 158, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High-fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G453–G462. [Google Scholar] [CrossRef] [PubMed]
- Samak, G.; Gangwar, R.; Meena, A.S.; Rao, R.G.; Shukla, P.K.; Manda, B.; Narayanan, D.; Jaggar, J.H.; Rao, R. Calcium Channels and Oxidative Stress Mediate a Synergistic Disruption of Tight Junctions by Ethanol and Acetaldehyde in Caco-2 Cell Monolayers. Sci. Rep. 2016, 6, 38899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elamin, E.; Masclee, A.; Troost, F.; Pieters, H.-J.; Keszthelyi, D.; Aleksa, K.; Dekker, J.; Jonkers, D. Ethanol Impairs Intestinal Barrier Function in Humans through Mitogen Activated Protein Kinase Signaling: A Combined In Vivo and In Vitro Approach. PLoS ONE 2014, 9, e107421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, K.E.; Turner, J.R. Myosin light chain kinase: Pulling the strings of epithelial tight junction function. Ann. N. Y. Acad. Sci. 2012, 1258, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Zhao, Y.; McClain, C.J.; Kang, Y.J.; Zhou, Z. Inactivation of hepatocyte nuclear factor-4α mediates alcohol-induced downregulation of intestinal tight junction proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G643–G651. [Google Scholar] [CrossRef] [Green Version]
- Elamin, E.; Masclee, A.; Juuti-Uusitalo, K.; Van Ijzendoorn, S.; Troost, F.; Pieters, H.-J.; Dekker, J.; Jonkers, D. Fatty Acid Ethyl Esters Induce Intestinal Epithelial Barrier Dysfunction via a Reactive Oxygen Species-Dependent Mechanism in a Three-Dimensional Cell Culture Model. PLoS ONE 2013, 8, e58561. [Google Scholar] [CrossRef] [Green Version]
- Stärkel, P.; Schnabl, B. Bidirectional Communication between Liver and Gut during Alcoholic Liver Disease. Semin. Liver Dis. 2016, 36, 331–339. [Google Scholar] [CrossRef]
- Besten, G.D.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef]
- Urdaneta, V.; Casadesús, J. Interactions between Bacteria and Bile Salts in the Gastrointestinal and Hepatobiliary Tracts. Front. Med. 2017, 4, 163. [Google Scholar] [CrossRef] [PubMed]
- Korsten, M.A.; Matsuzaki, S.; Feinman, L.; Lieber, C.S. High Blood Acetaldehyde Levels after Ethanol Administration. N. Engl. J. Med. 1975, 292, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.; Lewis, S.A.; Sureshchandra, S.; Doratt, B.; Grant, K.A.; Messaoudi, I. Chronic ethanol consumption alters lamina propria leukocyte response to stimulation in a region-dependent manner. FASEB J. 2019, 33, 7767–7777. [Google Scholar] [CrossRef] [Green Version]
- Ling, X.; Linglong, P.; Weixia, D.; Hong, W. Protective Effects of Bifidobacterium on Intestinal Barrier Function in LPS-Induced Enterocyte Barrier Injury of Caco-2 Monolayers and in a Rat NEC Model. PLoS ONE 2016, 11, e0161635. [Google Scholar] [CrossRef] [PubMed]
- Akiba, Y.; Maruta, K.; Takajo, T.; Narimatsu, K.; Said, H.; Kato, I.; Kuwahara, A.; Kaunitz, J.D. Lipopolysaccharides transport during fat absorption in rodent small intestine. Am. J. Physiol. Liver Physiol. 2020, 318, G1070–G1087. [Google Scholar] [CrossRef]
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Schäfer, C.; Parlesak, A.; Schütt, C.; Bode, J.C.; Bode, C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble cd14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol Alcohol. 2002, 37, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Liangpunsakul, S.; Toh, E.; Ross, R.A.; Heathers, L.E.; Chandler, K.; Oshodi, A.; McGee, B.; Modlik, E.; Linton, T.; Mangiacarne, D.; et al. Quantity of alcohol drinking positively correlates with serum levels of endotoxin and markers of monocyte activation. Sci. Rep. 2017, 7, 4462. [Google Scholar] [CrossRef]
- Gao, B.; Emami, A.; Nath, S.; Schnabl, B. Microbial Products and Metabolites Contributing to Alcohol-Related Liver Disease. Mol. Nutr. Food Res. 2020, e2000023. [Google Scholar] [CrossRef]
- Fleming, S.; Toratani, S.; Shea-Donohue, T.; Kashiwabara, Y.; Vogel, S.N.; Metcalf, E.S. Pro- and anti-inflammatory gene expression in the murine small intestine and liver after chronic exposure to alcohol. Alcohol Clin. Exp. Res. 2001, 25, 579–589. [Google Scholar] [CrossRef]
- Veazey, R.S.; Amedee, A.; Wang, X.; Kaack, M.B.; Porretta, C.; Dufour, J.; Welsh, D.; Happel, K.; Pahar, B.; Molina, P.E.; et al. Chronic Binge Alcohol Administration Increases Intestinal T-Cell Proliferation and Turnover in Rhesus Macaques. Alcohol. Clin. Exp. Res. 2015, 39, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like receptors as targets in chronic liver diseases. Gut 2009, 58, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Ahmad, M.F.; Nagy, L.E.; Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 2019, 70, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, J.-B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leitemoreira, A.F. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, L.; Wang, X.; Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch. Toxicol. 2012, 86, 1337–1348. [Google Scholar] [CrossRef]
- Kawaratani, H.; Tsujimoto, T.; Douhara, A.; Takaya, H.; Moriya, K.; Namisaki, T.; Noguchi, R.; Yoshiji, H.; Fujimoto, M.; Fukui, H. The Effect of Inflammatory Cytokines in Alcoholic Liver Disease. Mediat. Inflamm. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yang, Y.; Ren, L.; Shao, T.; Li, F.; Zhao, C.; Liu, L.; Zhang, H.; McClain, C.J.; Feng, W. Activation of autophagy attenuates EtOH-LPS-induced hepatic steatosis and injury through MD2 associated TLR4 signaling. Sci. Rep. 2017, 7, 9292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, S.; Nobili, V.; Alisi, A. Toll-like receptor-mediated signaling cascade as a regulator of the inflammation network during alcoholic liver disease. World J. Gastroenterol. 2014, 20, 16443–16451. [Google Scholar] [CrossRef]
- Gandhi, C.R. Pro- and Anti-fibrogenic Functions of Gram-Negative Bacterial Lipopolysaccharide in the Liver. Front. Med. 2020, 7, 130. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I.; Yang, L.; Wang, X.; Wu, D. CYP2E1 Sensitizes the Liver to LPS- and TNF α-Induced Toxicity via Elevated Oxidative and Nitrosative Stress and Activation of ASK-1 and JNK Mitogen-Activated Kinases. Int. J. Hepatol. 2012, 2012, 582790. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, A.I.; Lu, Y.; Wang, X.; Wu, D. Synergistic Toxic Interactions Between CYP2E1, LPS/TNF? and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury. Results Probl. Cell Differ. 2014, 815, 145–172. [Google Scholar] [CrossRef]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef] [Green Version]
- Cubero, F.J.; Nieto, N. Arachidonic acid stimulates TNFα production in Kupffer cells via a reactive oxygen species-pERK1/2-Egr1-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G228–G239. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Jeong, J.-M.; Kim, S.J.; Seo, W.H.; Kim, M.-H.; Choi, W.-M.; Yoo, W.; Lee, J.-H.; Shim, Y.-R.; Yi, H.-S.; et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat. Commun. 2017, 8, 2247. [Google Scholar] [CrossRef]
- Yamashina, S.; Takei, Y.; Ikejima, K.; Enomoto, N.; Kitamura, T.; Sato, N. Ethanol-Induced Sensitization to Endotoxin in Kupffer Cells Is Dependent upon Oxidative Stress. Alcohol. Clin. Exp. Res. 2005, 29, 246S–250S. [Google Scholar] [CrossRef]
- Shen, Z.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Le, L.; Murr, M.M.; Peng, Y.; You, M. Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-α production in cultured macrophage cell lines. Am. J. Physiol. Liver Physiol. 2009, 296, G1047–G1053. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Kono, H.; Yin, M.; Nakagami, M.; Uesugi, T.; Arteel, G.E.; Gabele, E.; Rusyn, I.; Yamashina, S.; Froh, M. The role of kupffer cell oxidant production in early ethanol-induced liver disease1, 2 1Guest Editor: Arthur Cederbaum 2This article is part of a series of reviews on “Alcohol, Oxidative Stress and Cell Injury”. The full list of papers may be found on the homepage of the journal. Free Radic. Biol. Med. 2001, 31, 1544–1549. [Google Scholar]
- Sun, Q.; Zhang, W.; Zhong, W.; Sun, X.; Zhou, Z. Pharmacological inhibition of NOX4 ameliorates alcohol-induced liver injury in mice through improving oxidative stress and mitochondrial function. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 2912–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Nagy, L.E.; Park, P.-H. Globular Adiponectin Inhibits Ethanol-Induced Reactive Oxygen Species Production through Modulation of NADPH Oxidase in Macrophages: Involvement of Liver Kinase B1/AMP-Activated Protein Kinase Pathway. Mol. Pharmacol. 2014, 86, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Dong, L.; Dang, X.; Liu, Y.; Jiang, J.; Wang, Y.; Lu, X.; Guo, X. Effect of chlorogenic acid on LPS-induced proinflammatory signaling in hepatic stellate cells. Inflamm. Res. 2013, 62, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Longato, L.; Petersen, D.R.; Ronis, M.; Chen, W.C.; Wands, J.R.; De La Monte, S.M. Limited Therapeutic Effect of N-Acetylcysteine on Hepatic Insulin Resistance in an Experimental Model of Alcohol-Induced Steatohepatitis. Alcohol. Clin. Exp. Res. 2011, 35, 2139–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingeo, G.; Brito, A.; Samouda, H.; Iddir, M.; La Frano, M.R.; Bohn, T. Phytochemicals as modifiers of gut microbial communities. Food Funct. 2020, 11, 8444–8471. [Google Scholar] [CrossRef] [PubMed]
- Holst, B.; Williamson, G. Nutrients and phytochemicals: From bioavailability to bioefficacy beyond antioxidants. Curr. Opin. Biotechnol. 2008, 19, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Tsao, R. Chemistry and Biochemistry of Dietary Polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef]
- Luca, S.V.; Macovei, I.; Bujor, A.; Miron, A.; Skalicka-Woźniak, K.; Aprotosoaie, A.C.; Trifan, A. Bioactivity of dietary polyphenols: The role of metabolites. Crit. Rev. Food Sci. Nutr. 2020, 60, 626–659. [Google Scholar] [CrossRef] [PubMed]
- Summerlin, N.; Soo, E.; Thakur, S.; Qu, Z.; Jambhrunkar, S.; Popat, A. Resveratrol nanoformulations: Challenges and opportunities. Int. J. Pharm. 2015, 479, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Mouhid, L.; Corzo-Martínez, M.; Torres, C.; Vázquez, L.; Reglero, G.; Fornari, T.; De Molina, A.R. ImprovingIn VivoEfficacy of Bioactive Molecules: An Overview of Potentially Antitumor Phytochemicals and Currently Available Lipid-Based Delivery Systems. J. Oncol. 2017, 2017, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín, L.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Bioavailability of Dietary Polyphenols and Gut Microbiota Metabolism: Antimicrobial Properties. BioMed Res. Int. 2015, 2015, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Murota, K.; Nakamura, Y.; Uehara, M. Flavonoid metabolism: The interaction of metabolites and gut microbiota. Biosci. Biotechnol. Biochem. 2018, 82, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koudoufio, M.; Desjardins, Y.; Feldman, F.; Spahis, S.; Delvin, E.; Levy, E. Insight into Polyphenol and Gut Microbiota Crosstalk: Are Their Metabolites the Key to Understand Protective Effects against Metabolic Disorders? Antioxidants 2020, 9, 982. [Google Scholar] [CrossRef]
- Chaplin, A.; Carpéné, C.; Mercader, J. Resveratrol, Metabolic Syndrome, and Gut Microbiota. Nutrients 2018, 10, 1651. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Machin, L.; Monzote, L.; Sharifi-Rad, J.; Ezzat, S.M.; Salem, M.A.; Merghany, R.M.; El Mahdy, N.M.; Kılıç, C.S.; Sytar, O.; et al. Therapeutic Potential of Quercetin: New Insights and Perspectives for Human Health. ACS Omega 2020, 5, 11849–11872. [Google Scholar] [CrossRef]
- Williamson, G.; Clifford, M.N. Role of the small intestine, colon and microbiota in determining the metabolic fate of polyphenols. Biochem. Pharmacol. 2017, 139, 24–39. [Google Scholar] [CrossRef] [Green Version]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: A review. Eur. J. Nutr. 2015, 54, 325–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Possemiers, S.; Bolca, S.; Verstraete, W.; Heyerick, A. The intestinal microbiome: A separate organ inside the body with the metabolic potential to influence the bioactivity of botanicals. Fitoterapia 2011, 82, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.; Abdollahi, M. The Role of Dietary Polyphenols in the Management of Inflammatory Bowel Disease. Curr. Pharm. Biotechnol. 2015, 16, 196–210. [Google Scholar] [CrossRef]
- Godos, J.; Currenti, W.; Angelino, D.; Mena, P.; Castellano, S.; Caraci, F.; Galvano, F.; Del Rio, D.; Ferri, R.; Grosso, G. Diet and Mental Health: Review of the Recent Updates on Molecular Mechanisms. Antioxidants 2020, 9, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C. Interactions of gut microbiota with dietary polyphenols and consequences to human health. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Quintanar, L.; Roa, R.I.L.; Quintero-Fabián, S.; Sánchez-Sánchez, M.A.; Vizmanos, B.; Ortuño-Sahagún, D. Phytochemicals That Influence Gut Microbiota as Prophylactics and for the Treatment of Obesity and Inflammatory Diseases. Mediat. Inflamm. 2018, 2018, 1–18. [Google Scholar] [CrossRef]
- Liu, H.; Liu, M.; Fu, X.; Zhang, Z.; Zhu, L.; Zheng, X.; Liu, J. Astaxanthin Prevents Alcoholic Fatty Liver Disease by Modulating Mouse Gut Microbiota. Nutrients 2018, 10, 1298. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Carres, L.; Hughes, M.D.; Steele, C.N.; Ponder, M.A.; Davy, K.P.; Neilson, A.P. Use of dietary phytochemicals for inhibition of trimethylamine N-oxide formation. J. Nutr. Biochem. 2021, 108600. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef]
- Peiyuan, H.; Zhiping, H.; Chengjun, S.; Chunqing, W.; Bingqing, L.; Imam, M.U. Resveratrol Ameliorates Experimental Alcoholic Liver Disease by Modulating Oxidative Stress. Evid.-Based Complement. Altern. Med. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Meenu, M.; Liu, H.; Xu, B. A Concise Review on the Molecular Structure and Function Relationship of β-Glucan. Int. J. Mol. Sci. 2019, 20, 4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandran, M.; Chen, J.; Chung, S.S.M.; Xu, B. A critical review on the impacts of β-glucans on gut microbiota and human health. J. Nutr. Biochem. 2018, 61, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, X.; Lou, Y. Interactions of tea polyphenols with intestinal microbiota and their implication for cellular signal conditioning mechanism. J. Food Biochem. 2019, 43, e12953. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Fortinguerra, S.; Caudullo, G.; Buriani, A. Deciphering the Role of Polyphenols in Sports Performance: From Nutritional Genomics to the Gut Microbiota toward Phytonutritional Epigenomics. Nutrients 2020, 12, 1265. [Google Scholar] [CrossRef]
- Mopuri, R.; Islam, M.S. Medicinal plants and phytochemicals with anti-obesogenic potentials: A review. Biomed Pharmacother. 2017, 89, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; De Stefani, D.; De Vivo, I.; Scapagnini, G. Polyphenols as Caloric Restriction Mimetics Regulating Mitochondrial Biogenesis and Mitophagy. Trends Endocrinol. Metab. 2020, 31, 536–550. [Google Scholar] [CrossRef]
- Rothenberg, D.O.; Zhou, C.; Zhang, L. A Review on the Weight-Loss Effects of Oxidized Tea Polyphenols. Molecules 2018, 23, 1176. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224. [Google Scholar] [CrossRef]
- Kitagawa, R.; Kon, K.; Uchiyama, A.; Arai, K.; Yamashina, S.; Kuwahara-Arai, K.; Kirikae, T.; Ueno, T.; Ikejima, K. Rifaximin prevents ethanol-induced liver injury in obese KK-A(y) mice through modulation of small intestinal microbiota signature. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G707–G715. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Gangarapu, V.; Ince, A.T.; Baysal, B.; Kayar, Y.; Kılıç, U.; Gök, Ö.; Uysal, Ö.; Şenturk, H. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 840–845. [Google Scholar] [CrossRef]
- Harputluoglu, M.M.M.; Demirel, U.; Gul, M.; Temel, I.; Gursoy, S.; Selcuk, E.B.; Aladag, M.; Bilgic, Y.; Gunduz, E.; Seckin, Y. Effects of Rifaximin on Bacterial Translocation in Thioacetamide-Induced Liver Injury in Rats. Inflammation 2012, 35, 1512–1517. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.-C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.-H.; Jang, S.; Gonzalez, F.J.; Song, B.-J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Liu, Y.; Hu, S.; You, Y.; Wen, J.; Li, W.; Wang, Y. Probiotics for Alleviating Alcoholic Liver Injury. Gastroenterol. Res. Pract. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Puchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C.; et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2017, 66, 806–815. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Gavis, E.A.; Kassam, Z.; Sikaroodi, M.; Gillevet, P.M. Long-term Outcomes of Fecal Microbiota Transplantation in Patients With Cirrhosis. Gastroenterology 2019, 156, 1921–1923.e3. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Salzman, N.H.; Acharya, C.; Sterling, R.K.; White, M.B.; Gavis, E.A.; Fagan, A.; Hayward, M.; Holtz, M.L.; Matherly, S.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Gavis, E.A.; Fagan, A.; Wade, J.B.; Thacker, L.R.; Fuchs, M.; Patel, S.; Davis, B.; Meador, J.; Puri, P.; et al. A Randomized Clinical Trial of Fecal Microbiota Transplant for Alcohol Use Disorder. Hepatology 2020. [Google Scholar] [CrossRef]
- Nanji, A.A.; Khettry, U.; Sadrzadeh, S.M.H. Lactobacillus Feeding Reduces Endotoxemia and Severity of Experimental Alcoholic Liver (Disease). Exp. Biol. Med. 1994, 205, 243–247. [Google Scholar] [CrossRef]
- Konkit, M.; Kim, K.; Kim, J.-H.; Kim, W. Protective effects of Lactococcus chungangensis CAU 28 on alcohol-metabolizing enzyme activity in rats. J. Dairy Sci. 2018, 101, 5713–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, R.; Rappa, F.; Macaluso, F.; Bavisotto, C.C.; Sangiorgi, C.; Di Paola, G.; Tomasello, G.; Di Felice, V.; Marcianò, V.; Farina, F.; et al. Alcoholic Liver Disease: A Mouse Model Reveals Protection by Lactobacillus fermentum. Clin. Transl. Gastroenterol. 2016, 7, e138. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.; Jeon, K.; Moon, S.; Lee, K.; Kim, W.-K.; Jeong, H.; Cha, K.H.; Lim, M.Y.; Kang, W.; Kweon, M.-N.; et al. Roseburia spp. Abundance Associates with Alcohol Consumption in Humans and Its Administration Ameliorates Alcoholic Fatty Liver in Mice. Cell Host Microbe 2020, 27, 25–40.e6. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Meena, A.S.; Manda, B.; Gomes-Solecki, M.; Dietrich, P.; Dragatsis, I.; Rao, R. Lactobacillus plantarum prevents and mitigates alcohol-induced disruption of colonic epithelial tight junctions, endotoxemia, and liver damage by an EGF receptor-dependent mechanism. FASEB J. 2018, 32, 6274–6292. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.B.; Chang, M.H.; Lee, J.H.; Heo, W.; Kim, J.K.; Pan, J.H.; Kim, Y.J.; Kim, J.H. The Edible Insect Gryllus bimaculatus Protects against Gut-Derived Inflammatory Responses and Liver Damage in Mice after Acute Alcohol Exposure. Nutrients 2019, 11, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Forsyth, C.B.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Oats Supplementation Prevents Alcohol-Induced Gut Leakiness in Rats by Preventing Alcohol-Induced Oxidative Tissue Damage. J. Pharmacol. Exp. Ther. 2009, 329, 952–958. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-R.; Chen, Y.-L.; Peng, H.-C.; Lu, Y.-A.; Chuang, H.-L.; Chang, H.-Y.; Wang, H.-Y.; Su, Y.-J.; Yang, S.-C. Fish Oil Reduces Hepatic Injury by Maintaining Normal Intestinal Permeability and Microbiota in Chronic Ethanol-Fed Rats. Gastroenterol. Res. Pract. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.; Ma, L.-J.; Wang, M.; Liu, C.; Yang, R.; Su, H.; Yang, Y.; Wan, J.-B. Oxidation of fish oil exacerbates alcoholic liver disease by enhancing intestinal dysbiosis in mice. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Moon, K.-H.; Olsson, N.U.; Salem, N. Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J. Hepatol. 2008, 49, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; McClain, C.J.; Cave, M.; Kang, Y.J.; Zhou, Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. Liver Physiol. 2010, 298, G625–G633. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Zhou, Z.; Kang, Y.J. Suppression of Fas-Mediated Signaling Pathway is Involved in Zinc Inhibition of Ethanol-Induced Liver Apoptosis. Exp. Biol. Med. 2003, 228, 406–412. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, X.; Lambert, J.C.; Saari, J.T.; Kang, Y.J. Metallothionein-Independent Zinc Protection from Alcoholic Liver Injury. Am. J. Pathol. 2002, 160, 2267–2274. [Google Scholar] [CrossRef] [Green Version]
- Lou, Z.; Wang, J.; Chen, Y.; Xu, C.; Chen, X.; Shao, T.; Zhang, K.; Pan, H. Linderae radix ethanol extract attenuates alcoholic liver injury via attenuating inflammation and regulating gut microbiota in rats. Braz. J. Med Biol. Res. 2019, 52, e7628. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, K.; Sun, L.; Cheng, B.; Qiao, S.; Dai, H.; Shi, W.; Ma, J.; Liu, H. Therapeutic manipulation of gut microbiota by polysaccharides of Wolfiporia cocos reveals the contribution of the gut fungi-induced PGE2 to alcoholic hepatic steatosis. Gut Microbes 2020, 12, 1830693. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Yan, N.; Wang, P.; Xia, Y.; Hao, H.; Wang, G.; Gonzalez, F.J. Herbal drug discovery for the treatment of nonalcoholic fatty liver disease. Acta Pharm. Sin. B 2020, 10, 3–18. [Google Scholar] [CrossRef]
- Eom, T.; Ko, G.; Kim, K.C.; Kim, J.-S.; Unno, T. Dendropanax morbifera Leaf Extracts Improved Alcohol Liver Injury in Association with Changes in the Gut Microbiota of Rats. Antioxidants 2020, 9, 911. [Google Scholar] [CrossRef]
- Peterson, C.T.; Denniston, K.; Chopra, D. Therapeutic Uses of Triphala in Ayurvedic Medicine. J. Altern. Complement. Med. 2017, 23, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Cota, D.; Mishra, S.; Shengule, S. Arjunarishta alleviates experimental colitis via suppressing proinflammatory cytokine expression, modulating gut microbiota and enhancing antioxidant effect. Mol. Biol. Rep. 2020, 47, 1–11. [Google Scholar] [CrossRef]
- Samuelson, D.R.; Gu, M.; Shellito, J.E.; Molina, P.E.; Taylor, C.M.; Luo, M.; Welsh, D.A. Intestinal Microbial Products From Alcohol-Fed Mice Contribute to Intestinal Permeability and Peripheral Immune Activation. Alcohol. Clin. Exp. Res. 2019, 43, 2122–2133. [Google Scholar] [CrossRef]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin Supplementation Protects Mice from Acute Ethanol-Induced Gut Injury. Alcohol. Clin. Exp. Res. 2014, 38, 1489–1501. [Google Scholar] [CrossRef]
- Jiang, X.-W.; Li, Y.-T.; Ye, J.-Z.; Lv, L.-X.; Yang, L.-Y.; Bian, X.-Y.; Wu, W.-R.; Wu, J.-J.; Shi, D.; Wang, Q.; et al. New strain of Pediococcus pentosaceus alleviates ethanol-induced liver injury by modulating the gut microbiota and short-chain fatty acid metabolism. World J. Gastroenterol. 2020, 26, 6224–6240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-H.; Xin, F.-Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.-L.; Cui, A.; Liu, Z.; et al. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.M.; Ecton, K.E.; Trikha, S.R.J.; Wrigley, S.D.; Thomas, K.N.; Battson, M.L.; Wei, Y.; Johnson, S.A.; Weir, T.L.; Gentile, C.L. Microbial metabolite indole-3-propionic acid supplementation does not protect mice from the cardiometabolic consequences of a Western diet. Am. J. Physiol. Liver Physiol. 2020, 319, G51–G62. [Google Scholar] [CrossRef]
- Wei, Z.; Shen, P.; Cheng, P.; Lu, Y.; Wang, A.; Sun, Z. Gut Bacteria Selectively Altered by Sennoside A Alleviate Type 2 Diabetes and Obesity Traits. Oxidative Med. Cell. Longev. 2020, 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Hernández-Arriaga, A.; Kehm, R.; Sánchez, V.; Jin, C.J.; Nier, A.; Baumann, A.; Camarinha-Silva, A.; Bergheim, I. Metformin attenuates the onset of non-alcoholic fatty liver disease and affects intestinal microbiota and barrier in small intestine. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, S.; Razazan, A.; Nagpal, R.; Jain, S.; Wang, B.; Mishra, S.P.; Wang, S.; Justice, J.; Ding, J.; McClain, D.A.; et al. Metformin Reduces Aging-Related Leaky Gut and Improves Cognitive Function by Beneficially Modulating Gut Microbiome/Goblet Cell/Mucin Axis. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2020, 75, e9–e21. [Google Scholar] [CrossRef] [PubMed]
- Ichim, T.E.; Kesari, S.; Shafer, K. Protection from chemotherapy- and antibiotic-mediated dysbiosis of the gut microbiota by a probiotic with digestive enzymes supplement. Oncotarget 2018, 9, 30919–30935. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.I.; Kang, K.S. N-acetylcysteine modulates lipopolysaccharide-induced intestinal dysfunction. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Circadian rhythms: A regulator of gastrointestinal health and dysfunction. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 411–424. [Google Scholar] [CrossRef]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [Green Version]
- Potì, F.; Santi, D.; Spaggiari, G.; Zimetti, F.; Zanotti, I. Polyphenol Health Effects on Cardiovascular and Neurodegenerative Disorders: A Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 351. [Google Scholar] [CrossRef] [Green Version]
- Simini, B. Serge Renaud: From French paradox to Cretan miracle. Lancet 2000, 355, 48. [Google Scholar] [CrossRef]
- Ferrières, J. The French paradox: Lessons for other countries. Heart 2004, 90, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xiao, Y.-Y. Grape Phytochemicals and Associated Health Benefits. Crit. Rev. Food Sci. Nutr. 2013, 53, 1202–1225. [Google Scholar] [CrossRef]
- Cremonini, E.; Fraga, C.G.; Oteiza, P.I. (–)-Epicatechin in the control of glucose homeostasis: Involvement of redox-regulated mechanisms. Free. Radic. Biol. Med. 2019, 130, 478–488. [Google Scholar] [CrossRef]
- Wu, X.; Li, C.; Xing, G.; Qi, X.; Ren, J. Resveratrol Downregulates Cyp2e1 and Attenuates Chemically Induced Hepatocarcinogenesis in SD Rats. J. Toxicol. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free. Radic. Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, M.A.; Jang, S.; Banerjee, A.; Hardwick, J.P.; Song, B.-J. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free. Radic. Biol. Med. 2013, 60, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Anhê, F.F.; Pilon, G.; Roy, D.; Desjardins, Y.; Levy, E.; Marette, A. Triggering Akkermansia with dietary polyphenols: A new weapon to combat the metabolic syndrome? Gut Microbes 2016, 7, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.K.; Kamath, P.S.; Gores, G.J.; Shah, V.H. Alcoholic Hepatitis: Current Challenges and Future Directions. Clin. Gastroenterol. Hepatol. 2014, 12, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Henry, L. Contribution of Alcoholic and Nonalcoholic Fatty Liver Disease to the Burden of Liver-Related Morbidity and Mortality. Gastroenterology 2016, 150, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Lourens, S.; Sunjaya, D.B.; Singal, A.; Liangpunsakul, S.; Puri, P.; Sanyal, A.; Ren, X.; Gores, G.J.; Radaeva, S.; Chalasani, N. Acute Alcoholic Hepatitis: Natural History and Predictors of Mortality Using a Multicenter Prospective Study. Mayo Clin. Proc. Innov. Qual. Outcomes 2017, 1, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G. Clinical Trial Design for Alcoholic Hepatitis. Semin. Liver Dis. 2017, 37, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Shah, V.H. Current trials and novel therapeutic targets for alcoholic hepatitis. J. Hepatol. 2019, 70, 305–313. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballway, J.W.; Song, B.-J. Translational Approaches with Antioxidant Phytochemicals against Alcohol-Mediated Oxidative Stress, Gut Dysbiosis, Intestinal Barrier Dysfunction, and Fatty Liver Disease. Antioxidants 2021, 10, 384. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030384
Ballway JW, Song B-J. Translational Approaches with Antioxidant Phytochemicals against Alcohol-Mediated Oxidative Stress, Gut Dysbiosis, Intestinal Barrier Dysfunction, and Fatty Liver Disease. Antioxidants. 2021; 10(3):384. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030384
Chicago/Turabian StyleBallway, Jacob W., and Byoung-Joon Song. 2021. "Translational Approaches with Antioxidant Phytochemicals against Alcohol-Mediated Oxidative Stress, Gut Dysbiosis, Intestinal Barrier Dysfunction, and Fatty Liver Disease" Antioxidants 10, no. 3: 384. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030384