
Structural Insights into a Bifunctional Peptide Methionine Sulfoxide Reductase MsrA/B Fusion Protein from Helicobacter pylori

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cloning, Expression, and Purification
2.2. Crystallization and Data Collection
2.3. Kinetic Analysis of MsrAB with an Enzymatic Assay
2.4. Structure Determination and Refinement
2.5. Inflection Temperature (Ti) Measurement
2.6. Size-Exclusion Chromatography with Multiangle Light Scattering (SEC-MALS)
3. Results and Discussion
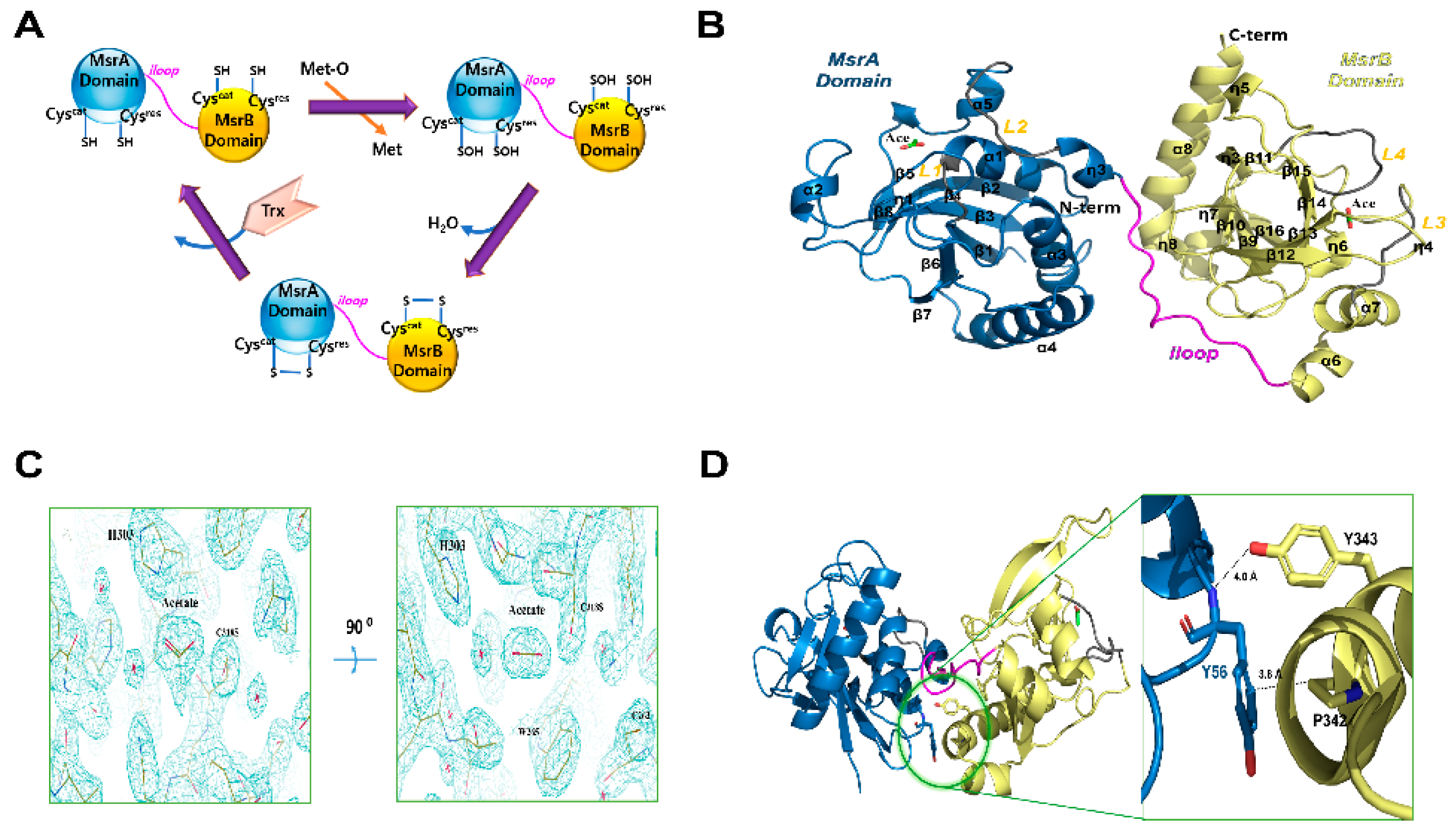
3.1. Overall Structures of HpMsrABC44S/C318S
3.2. Active Sites of HpMsrABC44S/C318S
3.3. Biochemical and Kinetic Analysis of HpMsrAB
3.4. Structural Comparison with Other Fusion MsrAB Proteins
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhandayuthapani, S.; Jagannath, C.; Nino, C.; Saikolappan, S.; Sasindran, S.J. Methionine sulfoxide reductase B (MsrB) of Mycobacterium smegmatis plays a limited role in resisting oxidative stress. Tuberculosis 2009, 89, S26–S32. [Google Scholar] [CrossRef] [Green Version]
- Achilli, C.; Ciana, A.; Minetti, G. The discovery of methionine sulfoxide reductase enzymes: An historical account and future perspectives. Biofactors 2015, 41, 135–152. [Google Scholar] [CrossRef]
- Jiang, B.; Moskovitz, J. The functions of the mammalian methionine sulfoxide reductase system and related diseases. Antioxidants 2018, 7, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-Y. The methionine sulfoxide reduction system: Selenium utilization and methionine sulfoxide reductase enzymes and their functions. Antioxid. Redox Signal. 2013, 19, 958–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Hartke, A.; La Sorda, M.; Posteraro, B.; Laplace, J.-M.; Auffray, Y.; Sanguinetti, M. Role of methionine sulfoxide reductases A and B of Enterococcus faecalis in oxidative stress and virulence. Infect. Immun. 2010, 78, 3889–3897. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.S.; Hashino, M.; Suzuki, J.; Uda, A.; Watanabe, K.; Shimizu, T.; Watarai, M. Contribution of methionine sulfoxide reductase B (MsrB) to Francisella tularensis infection in mice. FEMS Microbiol. Lett. 2017, 364, 2. [Google Scholar] [CrossRef] [Green Version]
- Moskovitz, J.; Rahman, M.A.; Strassman, J.; Yancey, S.O.; Kushner, S.R.; Brot, N.; Weissbach, H. Escherichia coli peptide methionine sulfoxide reductase gene: Regulation of expression and role in protecting against oxidative damage. J. Bacteriol. 1995, 177, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Moskovitz, J.; Poston, J.M.; Berlett, B.S.; Nosworthy, N.J.; Szczepanowski, R.; Stadtman, E.R. Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J. Biol. Chem. 2000, 275, 14167–14172. [Google Scholar] [CrossRef] [Green Version]
- Moskovitz, J.; Singh, V.K.; Requena, J.; Wilkinson, B.J.; Jayaswal, R.K.; Stadtman, E.R. Purification and characterization of methionine sulfoxide reductases from mouse and Staphylococcus aureus and their substrate stereospecificity. Biochem. Biophys. Res. Commun. 2002, 290, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Moskovitz, J.; Berlett, B.S.; Poston, J.M.; Stadtman, E.R. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo. Proc. Natl. Acad. Sci. USA 1997, 94, 9585–9589. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, M.-J.; Moskovitz, J.; Salamini, F.; Bartels, D. Reverse genetic approaches in plants and yeast suggest a role for novel, evolutionarily conserved, selenoprotein-related genes in oxidative stress defense. Mol. Genet. Genom. 2002, 267, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Delaye, L.; Becerra, A.; Orgel, L.; Lazcano, A. Molecular evolution of peptide methionine sulfoxide reductases (MsrA and MsrB): On the early development of a mechanism that protects against oxidative damage. J. Mol. Evol. 2007, 64, 15–32. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Kumar, R.A.; Koc, A.; Sun, Z.; Gladyshev, V.N. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc. Natl. Acad. Sci. USA 2002, 99, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Geer, L.Y.; Domrachev, M.; Lipman, D.J.; Bryant, S.H. CDART: Protein homology by domain architecture. Genome Res. 2002, 12, 1619–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Shin, Y.J.; Lee, W.H.; Kim, H.Y.; Hwang, K.Y. Structural and kinetic analysis of an MsrA–MsrB fusion protein from Streptococcus pneumoniae. Mol. Microbiol. 2009, 72, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, A.-r.; Kim, M.-J.; Kwak, G.-H.; Son, J.; Hwang, K.Y.; Kim, H.-Y. Essential role of the linker region in the higher catalytic efficiency of a bifunctional MsrA–MsrB fusion protein. Biochemistry 2016, 55, 5117–5127. [Google Scholar] [CrossRef]
- Chen, B.; Markillie, L.M.; Xiong, Y.; Mayer, M.U.; Squier, T.C. Increased catalytic efficiency following gene fusion of bifunctional methionine sulfoxide reductase enzymes from Shewanella oneidensis. Biochemistry 2007, 46, 14153–14161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wizemann, T.M.; Moskovitz, J.; Pearce, B.J.; Cundell, D.; Arvidson, C.G.; So, M.; Weissbach, H.; Brot, N.; Masure, H.R. Peptide methionine sulfoxide reductase contributes to the maintenance of adhesins in three major pathogens. Proc. Natl. Acad. Sci. USA 1996, 93, 7985–7990. [Google Scholar] [CrossRef] [Green Version]
- Lowther, W.T.; Brot, N.; Weissbach, H.; Matthews, B.W. Structure and mechanism of peptide methionine sulfoxide reductase, an “anti-oxidation” enzyme. Biochemistry 2000, 39, 13307–13312. [Google Scholar] [CrossRef]
- Lowther, W.T.; Weissbach, H.; Etienne, F.; Brot, N.; Matthews, B.W. The mirrored methionine sulfoxide reductases of Neisseria gonorrhoeae pilB. Nat. Struct. Biol. 2002, 9, 348–352. [Google Scholar] [CrossRef]
- Lee, E.H.; Kwak, G.-H.; Kim, M.-J.; Kim, H.-Y.; Hwang, K.Y. Structural analysis of 1-Cys type selenoprotein methionine sulfoxide reductase A. Arch. Biochem. Biophys. 2014, 545, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rouhier, N.; Kauffmann, B.; Tete-Favier, F.; Palladino, P.; Gans, P.; Branlant, G.; Jacquot, J.-P.; Boschi-Muller, S. Functional and structural aspects of poplar cytosolic and plastidial type a methionine sulfoxide reductases. J. Biol. Chem. 2007, 282, 3367–3378. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.B.; Benglis Jr, D.M.; Dhandayuthapani, S.; Hart, P.J. Structure of Mycobacterium tuberculosis methionine sulfoxide reductase A in complex with protein-bound methionine. J. Bacteriol. 2003, 185, 4119–4126. [Google Scholar] [CrossRef] [Green Version]
- Ranaivoson, F.M.; Neiers, F.; Kauffmann, B.; Boschi-Muller, S.; Branlant, G.; Favier, F. Methionine sulfoxide reductase B displays a high level of flexibility. J. Mol. Biol. 2009, 394, 83–93. [Google Scholar] [CrossRef]
- Alamuri, P.; Maier, R.J. Methionine sulfoxide reductase in Helicobacter pylori: Interaction with methionine-rich proteins and stress-induced expression. J. Bacteriol. 2006, 188, 5839–5850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Berlett, B.S.; Moskovitz, J.; Mosoni, L.; Stadtman, E.R. Methionine residues may protect proteins from critical oxidative damage. Mech. Ageing Dev. 1999, 107, 323–332. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Moskovitz, J.; Berlett, B.S.; Levine, R.L. Cyclic oxidation and reduction of protein methionine residues is an important antioxidant mechanism. Oxyg. Nitrogen Radic. Cell Inj. Dis. 2002, 37, 3–9. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276. [Google Scholar] [CrossRef]
- Minetti, G.; Balduini, C.; Brovelli, A. Reduction of DABS-L-methionine-dl-sulfoxide by protein methionine sulfoxide reductase from polymorphonuclear leukocytes: Stereospecificity towards the l-sulfoxide. Ital. J. Biochem. 1994, 43, 273–283. [Google Scholar]
- Kim, H.-Y. Glutaredoxin serves as a reductant for methionine sulfoxide reductases with or without resolving cysteine. Acta Biochim. Biophys Sin. 2012, 44, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Kim, J.-R. Thioredoxin as a reducing agent for mammalian methionine sulfoxide reductases B lacking resolving cysteine. Biochem. Biophys. Res. Commun. 2008, 371, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Project, C.C. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Brot, N.; Collet, J.-F.; Johnson, L.C.; Jönsson, T.J.; Weissbach, H.; Lowther, W.T. The thioredoxin domain of Neisseria gonorrhoeae PilB can use electrons from DsbD to reduce downstream methionine sulfoxide reductases. J. Biol. Chem. 2006, 281, 32668–32675. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HpMsrABC44SC318S | |
|---|---|
| Data collection | |
| Wavelength (Å) | 1.00000 |
| Resolution (Å) | 30–2.2 (2.24–2.20) a |
| Space group | P1 |
| Unit cell parameter | a = 52.125, b = 72.505, c = 82.230 α = 93.16°, β = 93.80°, γ = 111.08° |
| Observed/unique reflections | 158,134/53,424 |
| Redundancy | 3.1 (2.3) |
| Completeness (%) | 94.1 (86.6) |
| I/σ | 29.7 (5.32) |
| Rmerge (%) b | 6.3 (17.9) |
| Refinement | |
| Resolution (Å) | 30.00–2.2 |
| Rwork/Rfree (%) c | 18.8/21.4 |
| Average B-factor (Å2) | 39.42 |
| Root-mean-square-deviations | |
| Bond length (Å) | 0.006 |
| Bond angle (°) | 0.759 |
| Ramachandran favored (%) | 97.7 |
| Ramachandran outliers (%) | 0.0 |
| PDB entry | 5FA9 |
| Form | Substrate | Km (mM) | kcat (min−1) | kcat/Km (mM−1 min−1) |
|---|---|---|---|---|
| WT | Met-S-O | 0.17 ± 0.06 | 10.2 ± 1.3 | 60 ± 7 |
| Met-R-O | 0.05 ± 0.02 | 9.4 ± 0.4 | 188 ± 8 | |
| MsrA_D | Met-S-O | 0.25 ± 0.09 | 2.2 ± 0.3 | 8.8 ± 1.2 |
| MsrB_D | Met-R-O | 1.7 ± 0.1 | 4.8 ± 0.3 | 2.8 ± 0.2 |
| E193A | Met-S-O | 0.14 ± 0.06 | 6.5 ± 0.8 | 46 ± 6 |
| Met-R-O | 0.06 ± 0.02 | 4.7 ± 0.3 | 78 ± 5 | |
| D197A | Met-S-O | 0.15 ± 0.04 | 6.1 ± 0.9 | 41 ± 6 |
| Met-R-O | 0.06 ± 0.02 | 4.4 ± 0.3 | 73 ± 5 | |
| E193A/D197A | Met-S-O | 0.12 ± 0.05 | 5.7 ± 0.6 | 47 ± 5 |
| Met-R-O | 0.05 ± 0.02 | 3.0 ± 0.3 | 60 ± 6 | |
| E339A | Met-S-O | 0.04 ± 0.01 | 3.5 ± 0.1 | 88 ± 3 |
| Met-R-O | 0.02 ± 0.01 | 2.7 ± 0.4 | 135 ± 25 | |
| Y343F | Met-S-O | 0.12 ± 0.02 | 5.8 ± 0.2 | 48 ± 2 |
| Met-R-O | 0.03 ± 0.02 | 5.2 ± 0.7 | 173 ± 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Lee, K.; Park, S.-H.; Kwak, G.-H.; Kim, M.S.; Kim, H.-Y.; Hwang, K.Y. Structural Insights into a Bifunctional Peptide Methionine Sulfoxide Reductase MsrA/B Fusion Protein from Helicobacter pylori. Antioxidants 2021, 10, 389. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030389
Kim S, Lee K, Park S-H, Kwak G-H, Kim MS, Kim H-Y, Hwang KY. Structural Insights into a Bifunctional Peptide Methionine Sulfoxide Reductase MsrA/B Fusion Protein from Helicobacter pylori. Antioxidants. 2021; 10(3):389. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030389
Chicago/Turabian StyleKim, Sulhee, Kitaik Lee, Sun-Ha Park, Geun-Hee Kwak, Min Seok Kim, Hwa-Young Kim, and Kwang Yeon Hwang. 2021. "Structural Insights into a Bifunctional Peptide Methionine Sulfoxide Reductase MsrA/B Fusion Protein from Helicobacter pylori" Antioxidants 10, no. 3: 389. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030389