
PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis

 , , , , , and
, , , , , and
Abstract
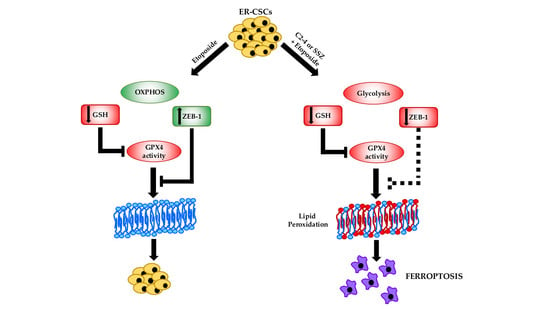
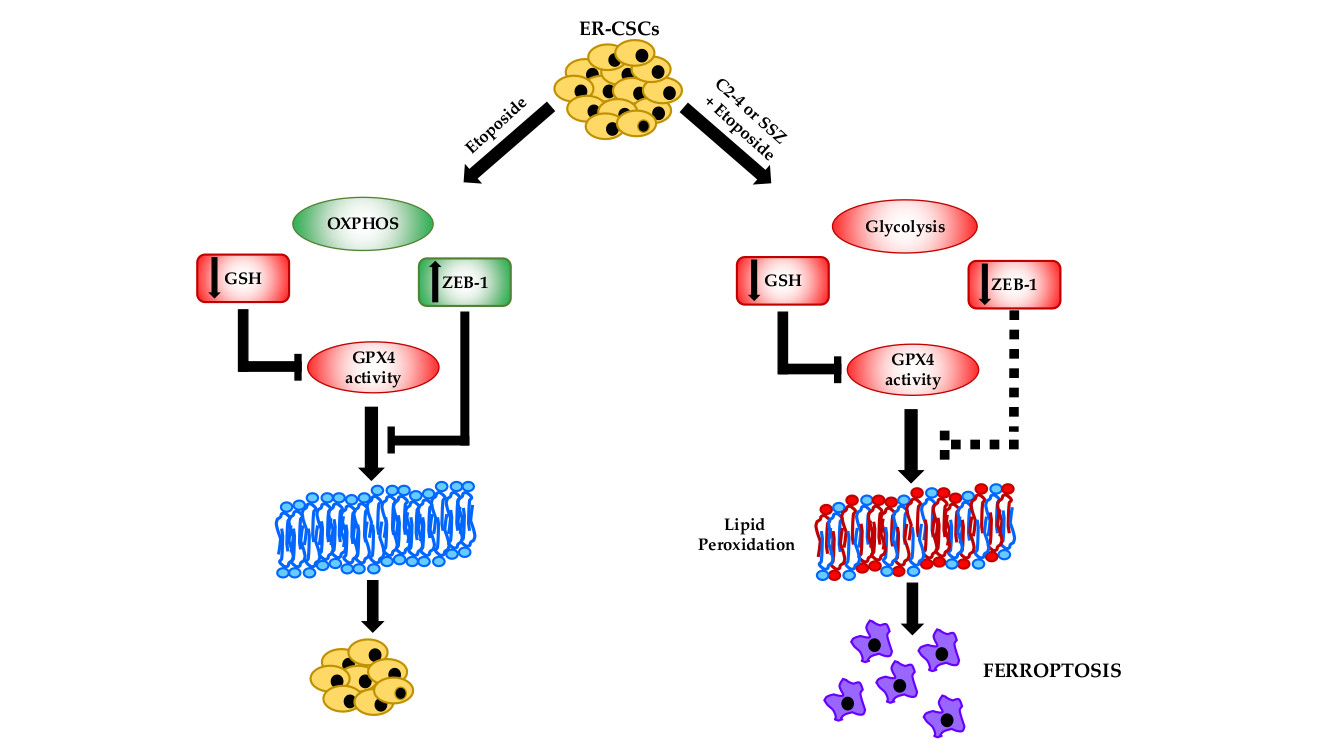
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
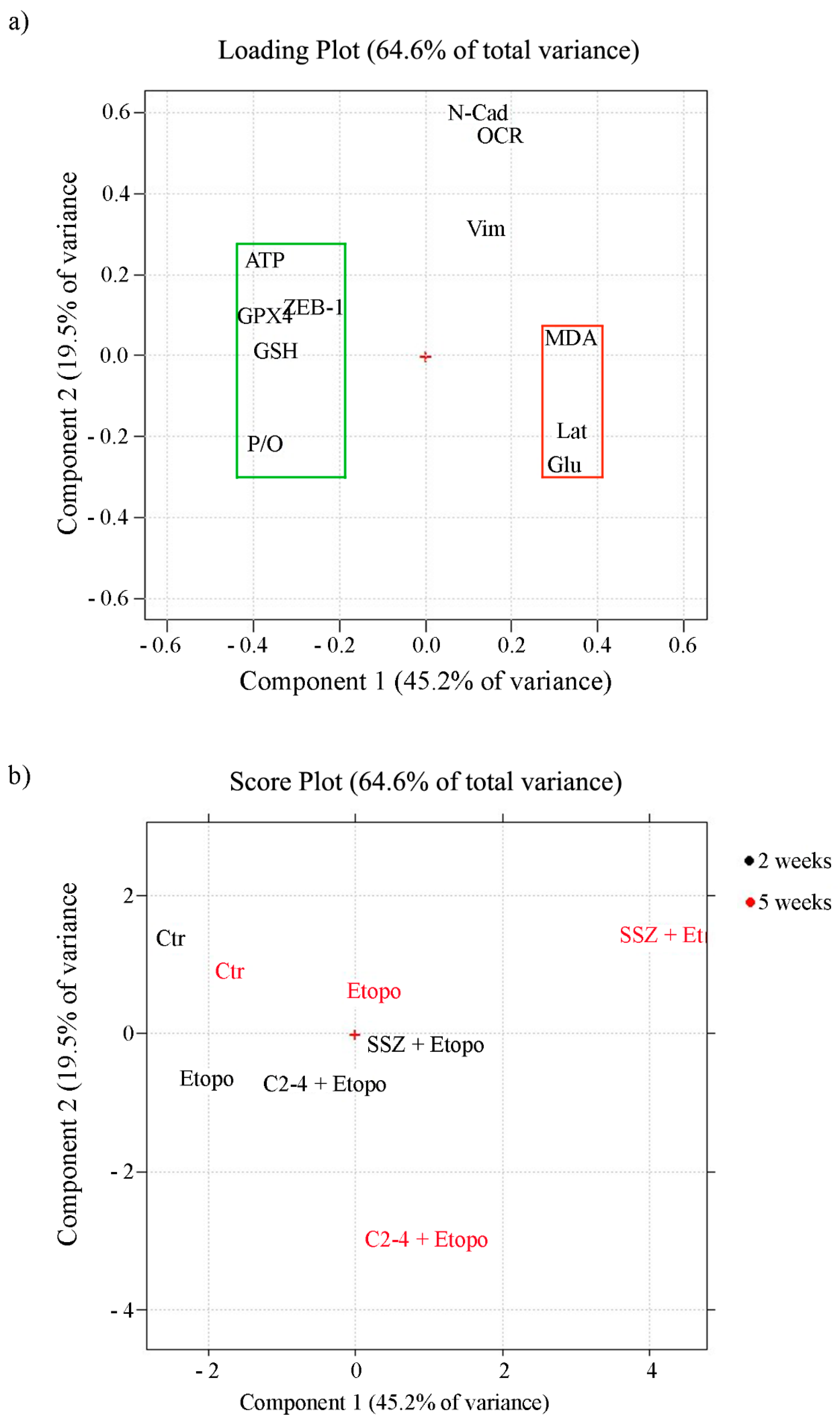{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cancer Stem Cell (Csc) Generation
2.3. Treatments
2.4. Immunofluorescence and Cytofluorimetric Analysis
2.5. HPLC Analysis of Intracellular Glutathione Levels
2.6. ATP Synthesis
2.7. Oxygen Consumption Rate (OCR)
2.8. Glucose Consumption
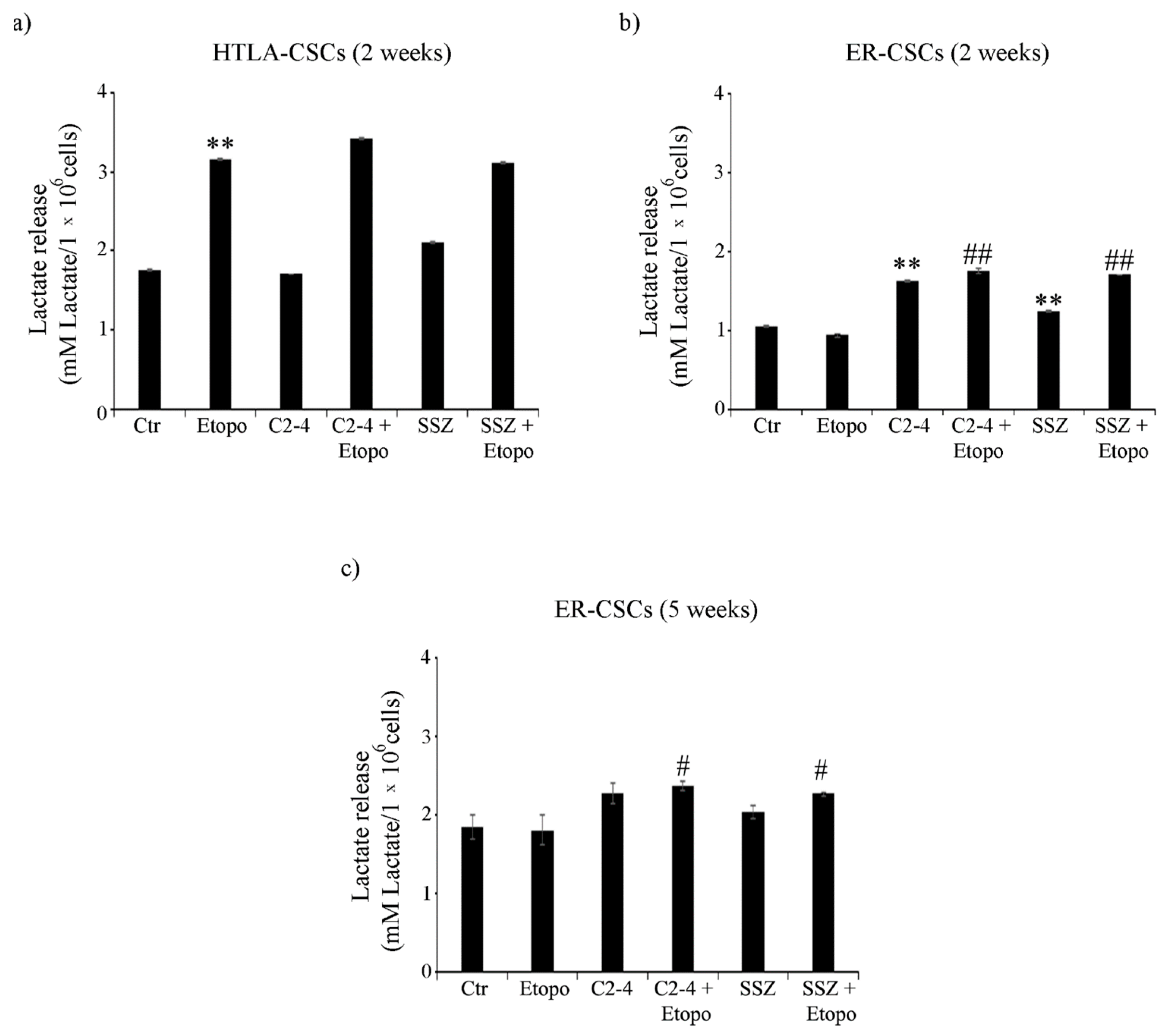
2.9. Lactate Release
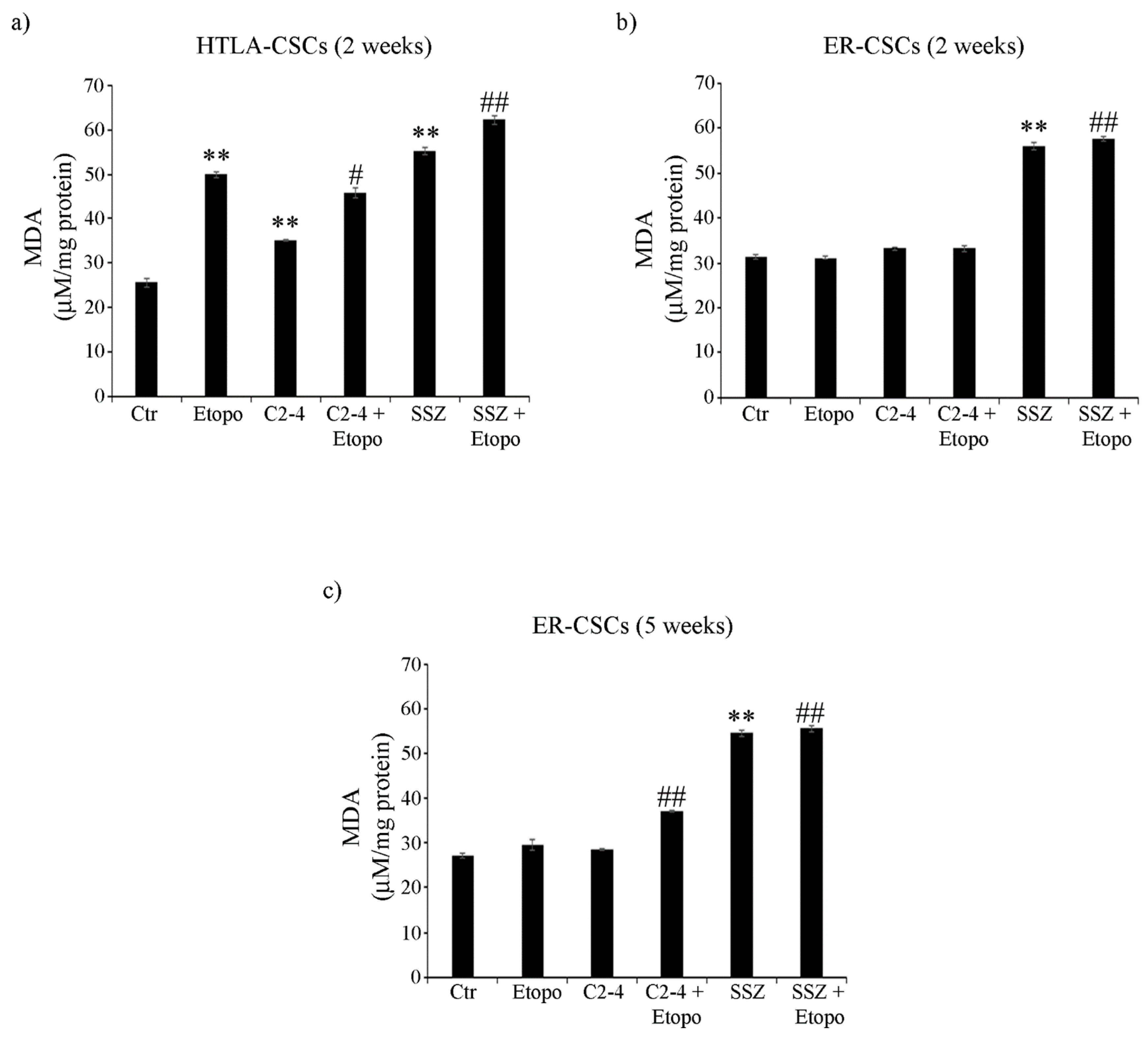
2.10. MDA Production
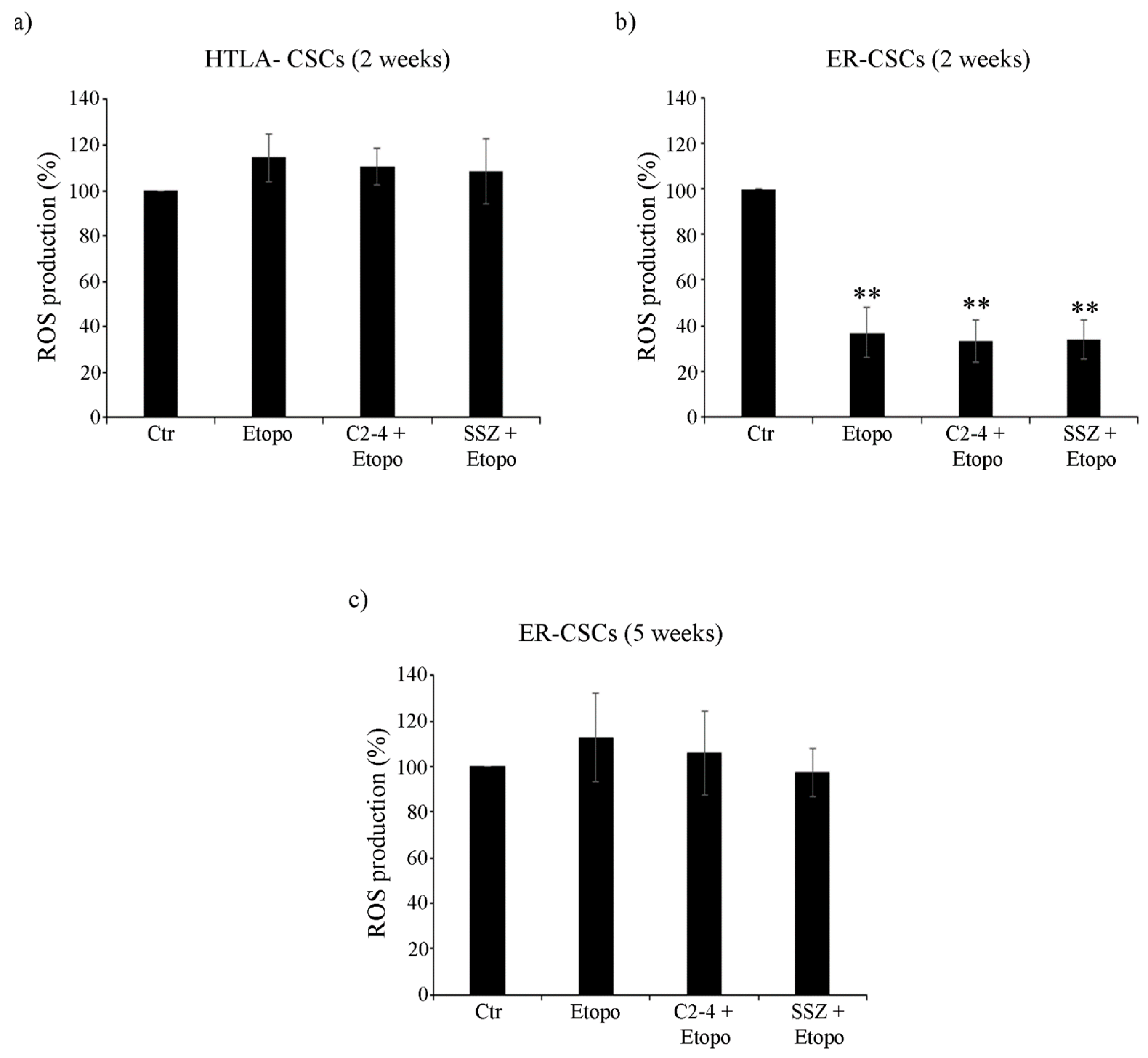
2.11. ROS Production
2.12. Western Blot Analyses
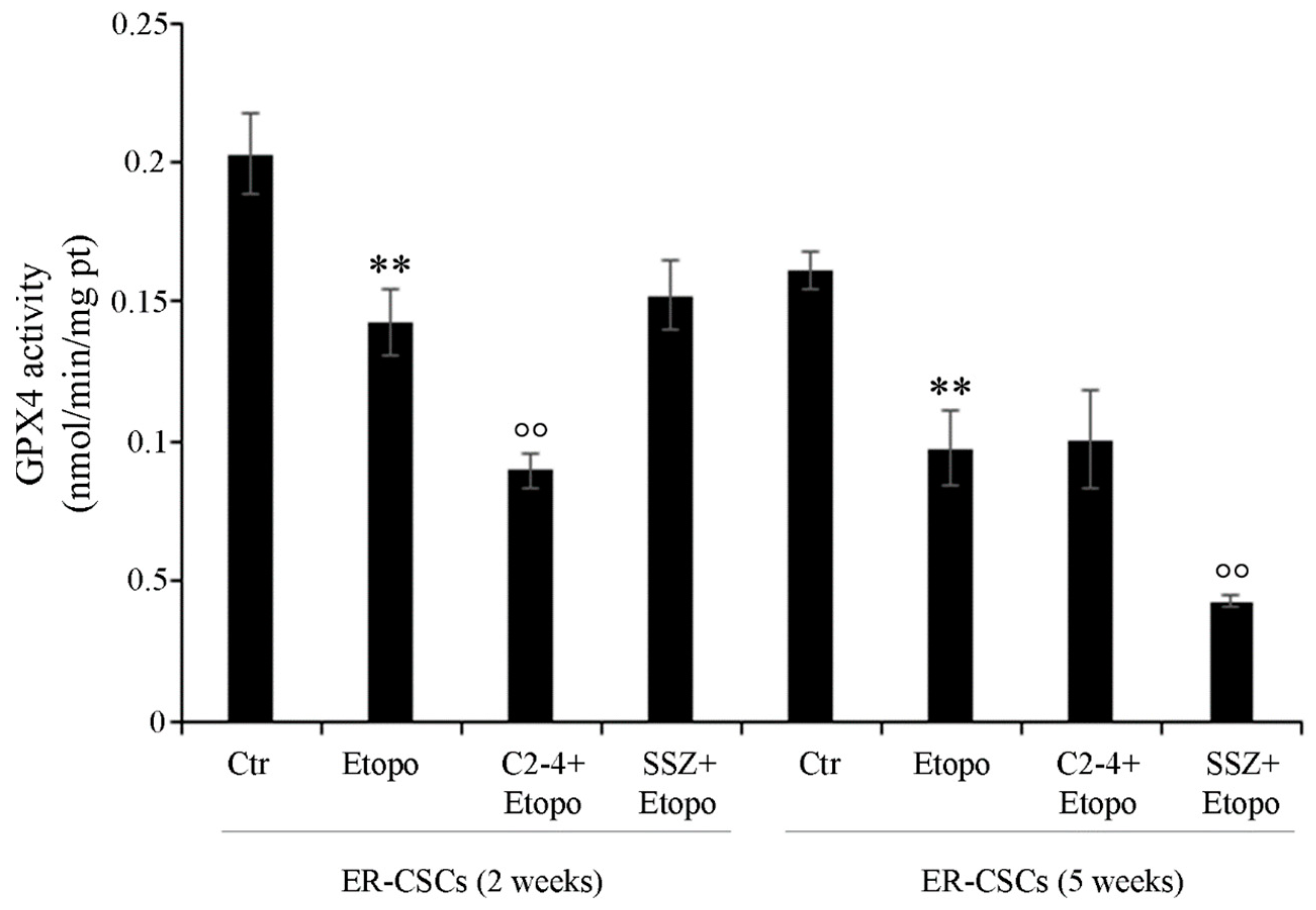
2.13. GPX4 Activity
2.14. Principal Component Analyses (PCA)
2.15. Statistical Analysis
3. Results and Discussion
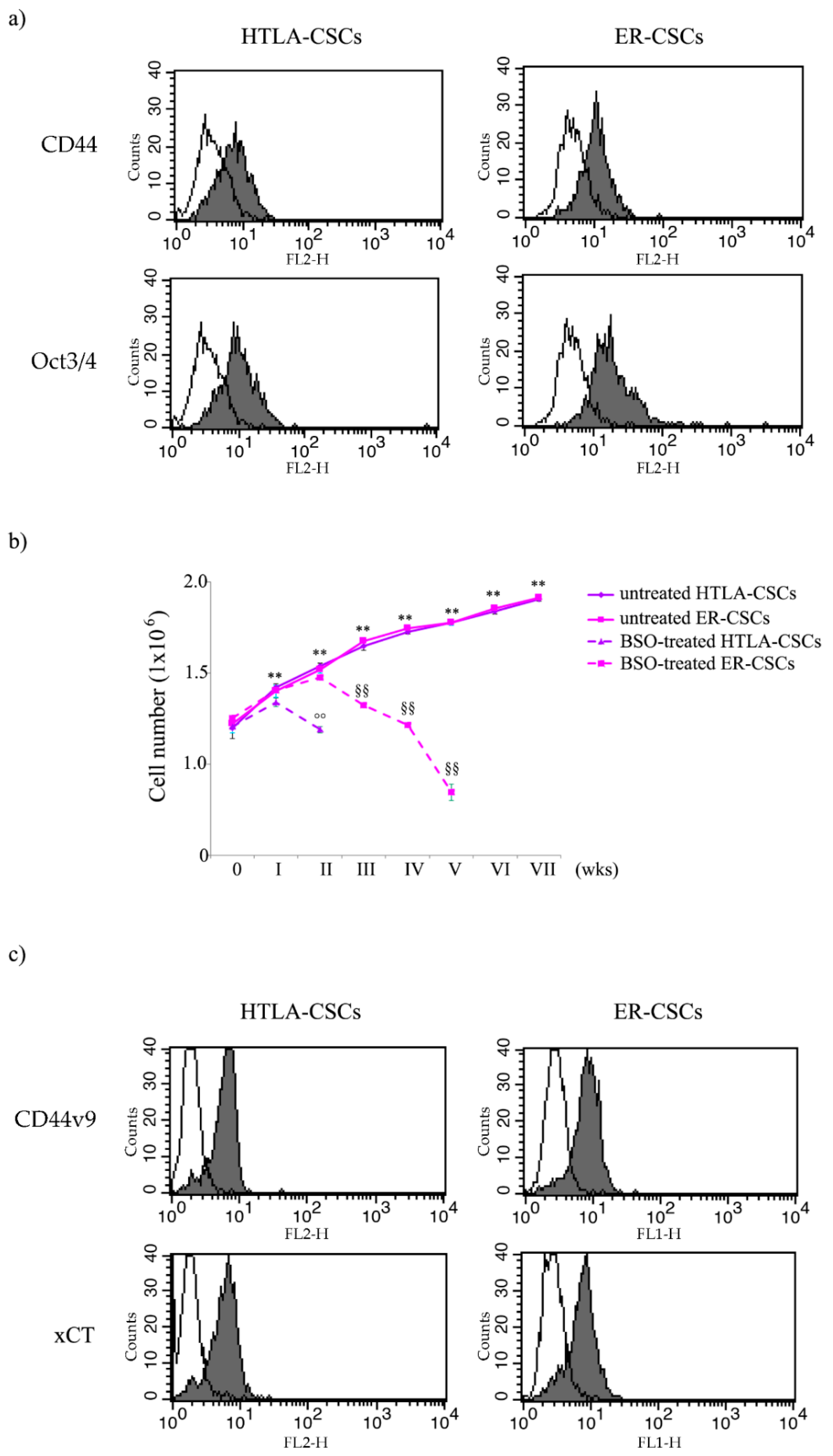
3.1. Neurospheres, Isolated from Parental HTLA-230 and HTLA-ER Cells, Are Cancer Stem Cells (CSCs) and GSH Plays a Crucial Role in Their Generation and Propagation
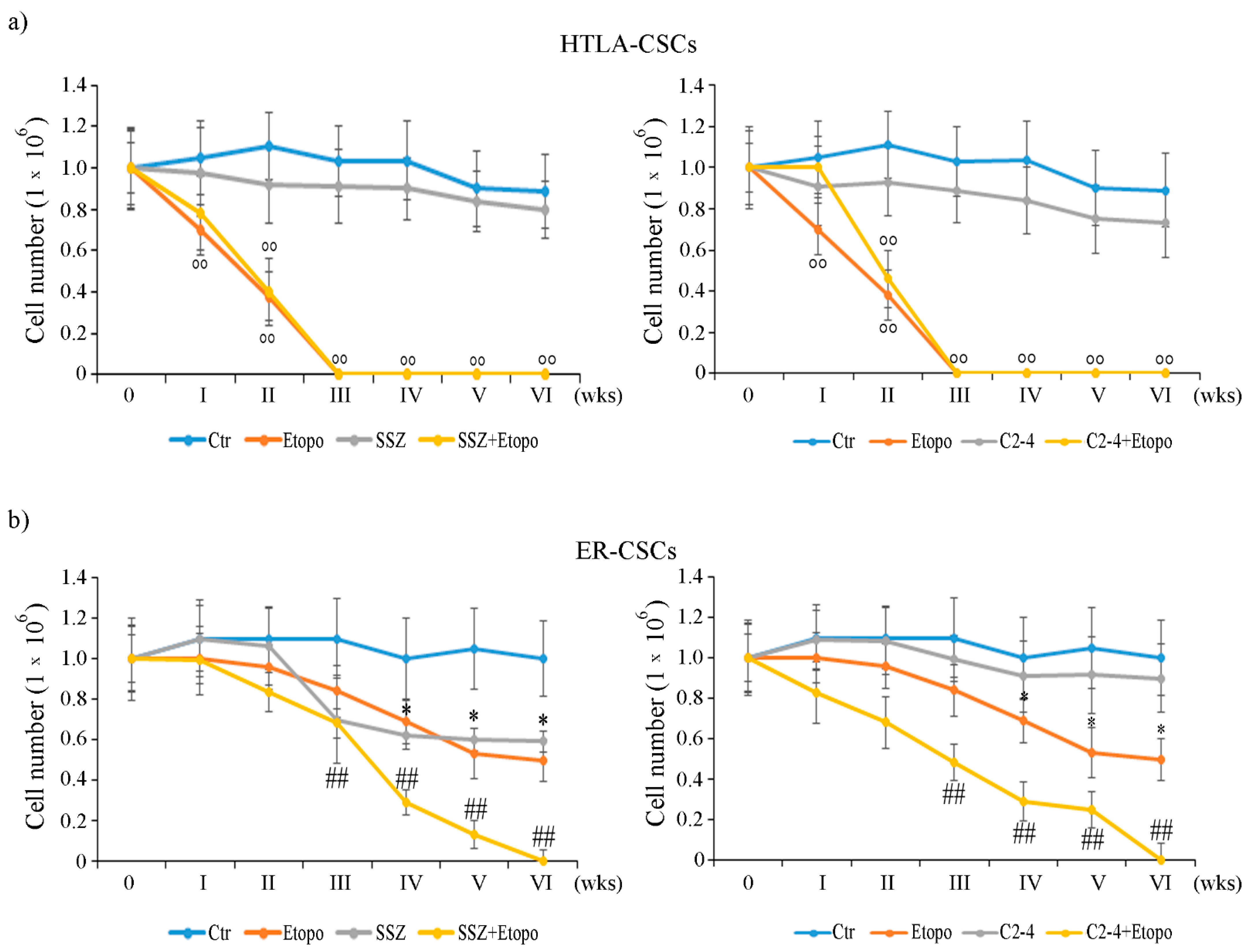
3.2. Etoposide Prevents the Formation of HTLA-CSCs after 3 Weeks of Treatment While It Completely Counteracts the Formation of ER-CSCs when It Is Combined with SSZ or C2-4 for 6 Weeks
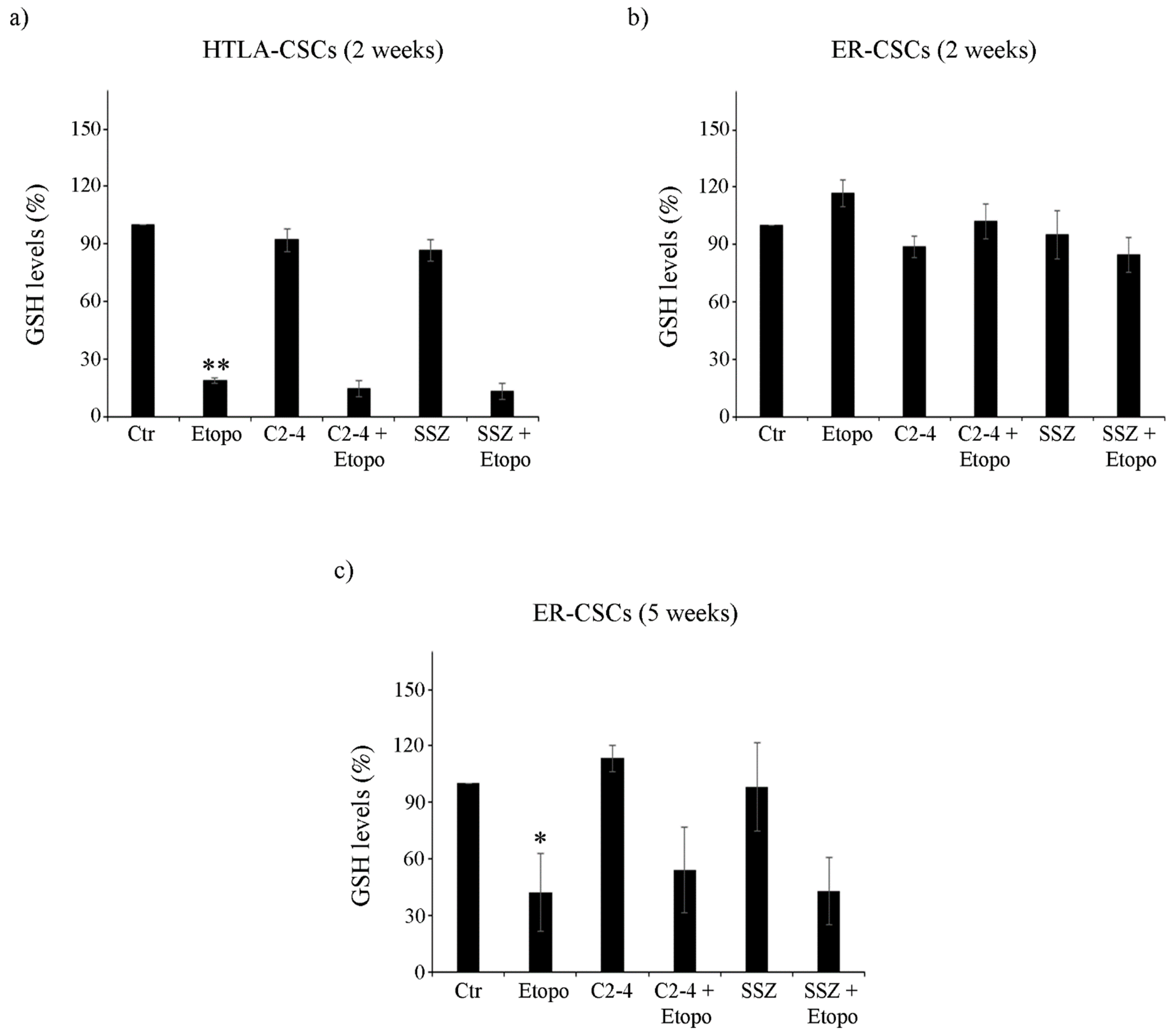
3.3. Etoposide Induces a Marked Depletion of GSH in HTLA-CSCs after Two Weeks While the Same Effect Is Not Observed in ER-CSCs until Five Weeks of Treatment
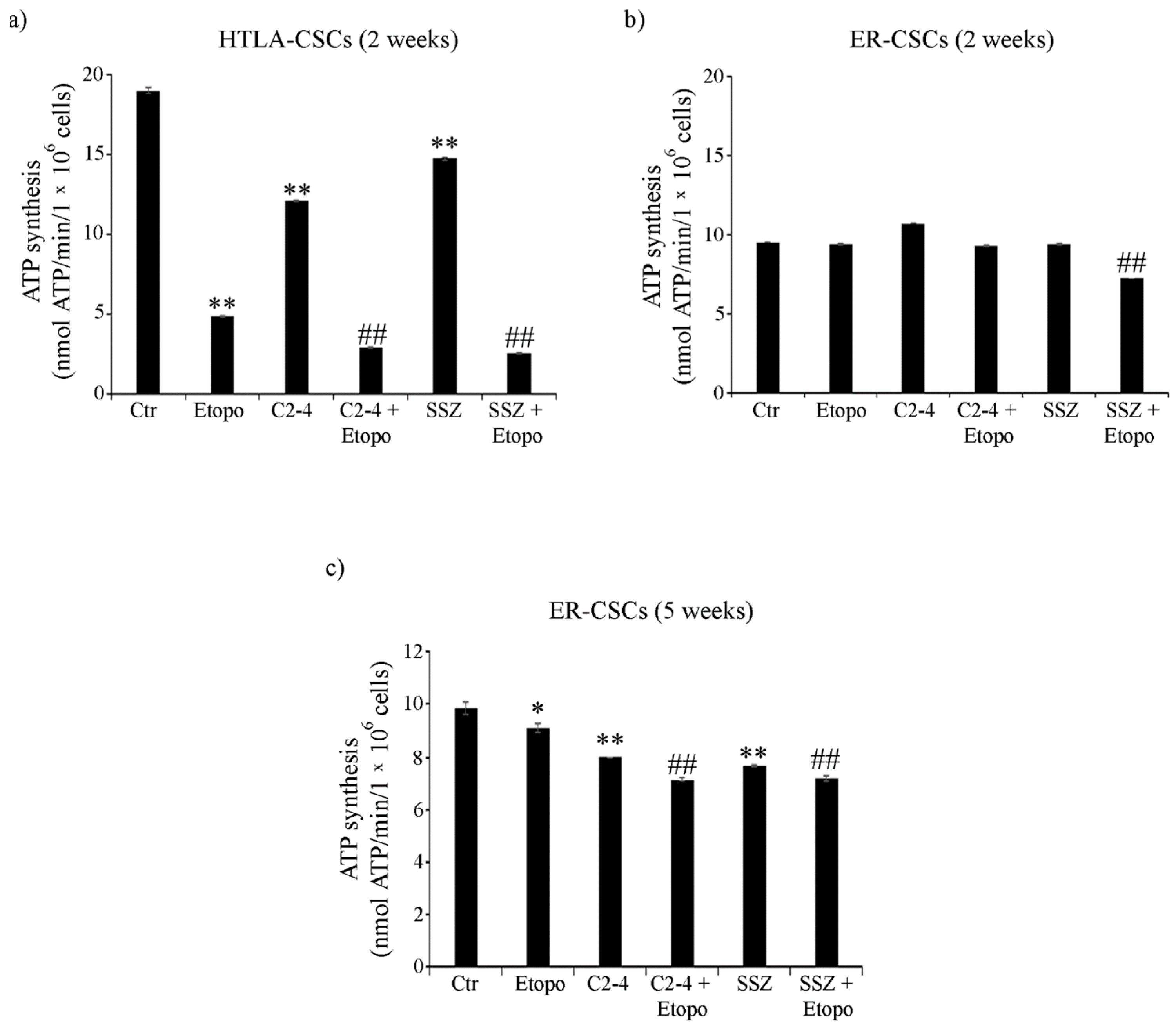
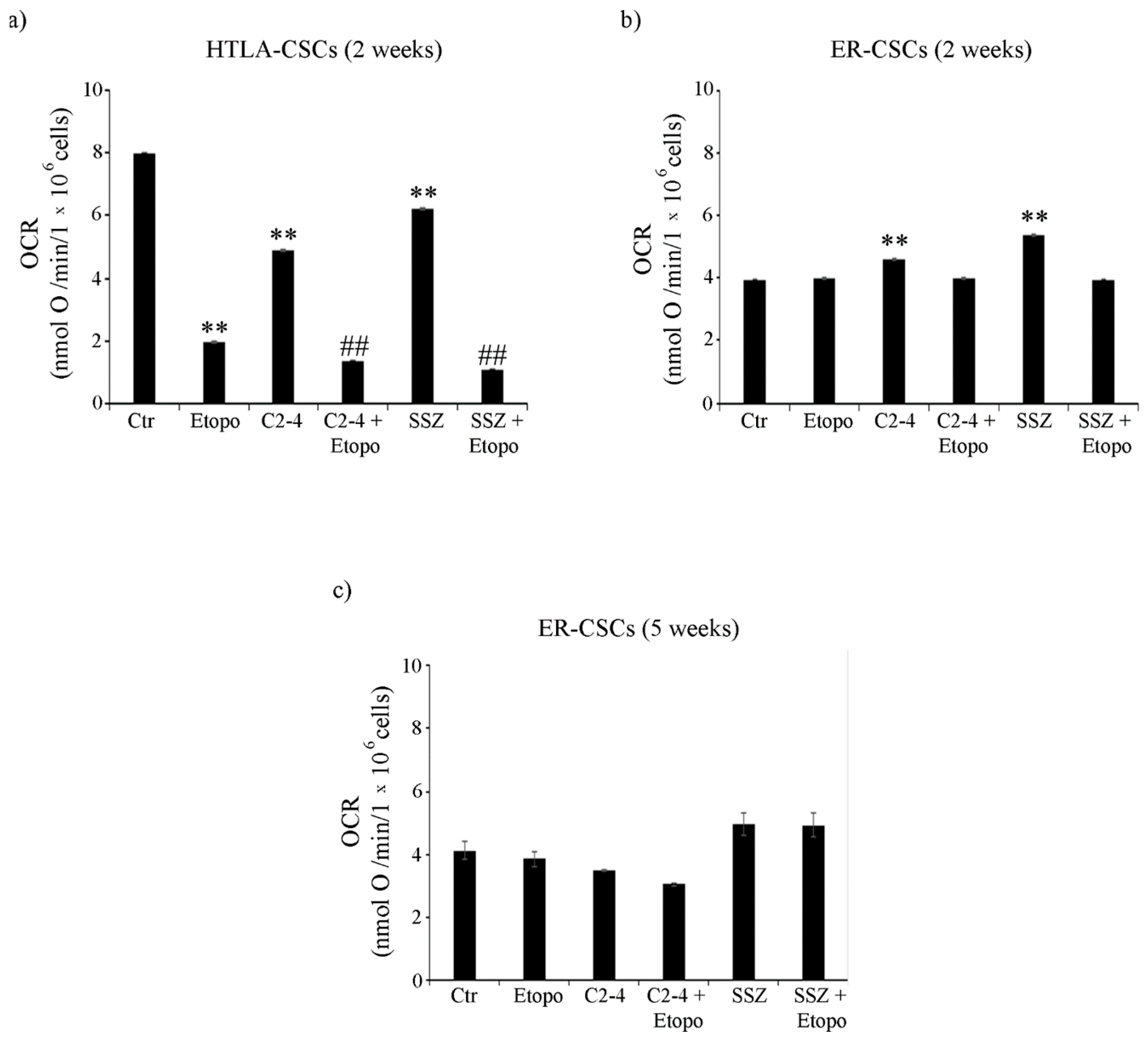
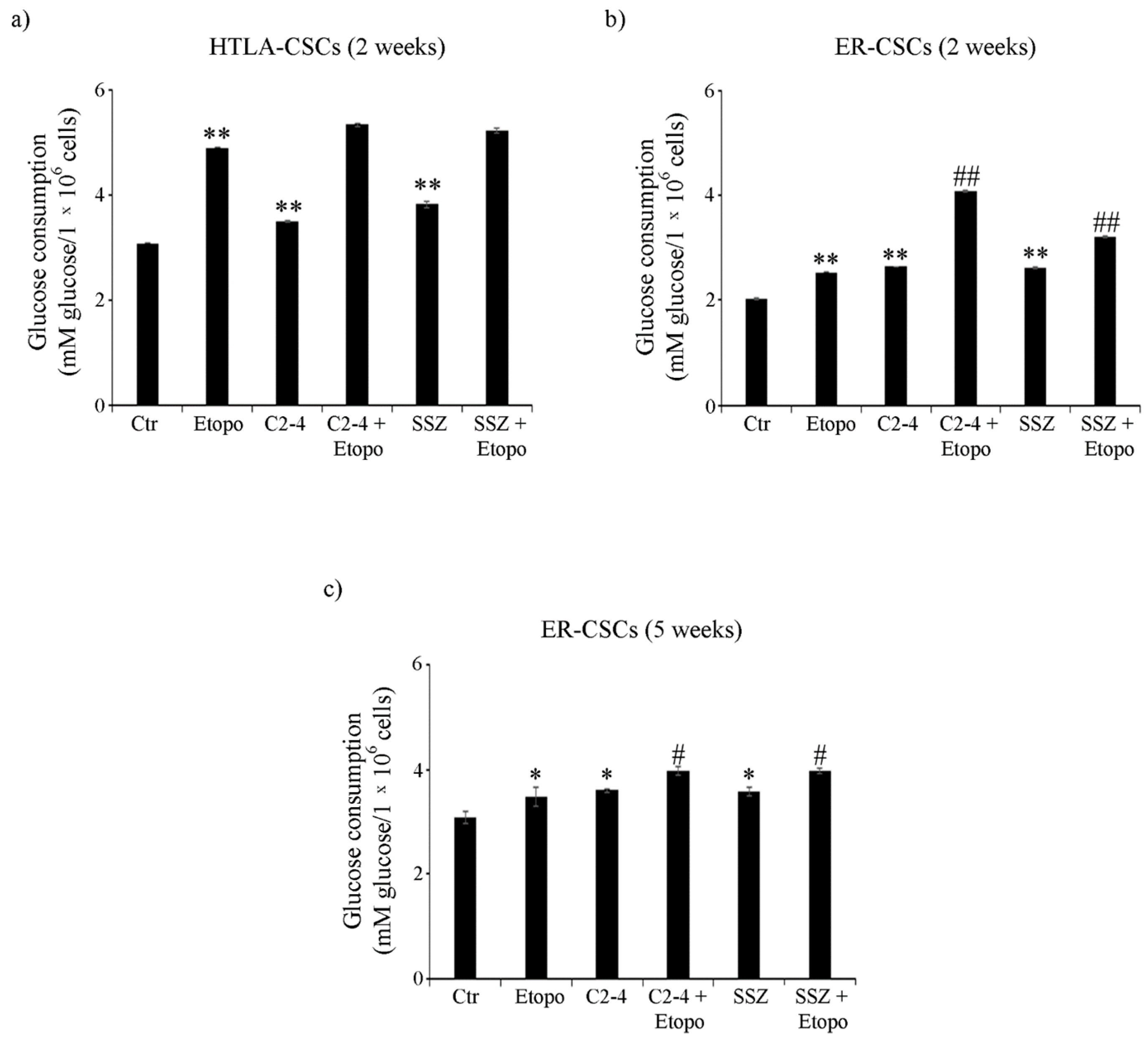
3.4. ER-CSCs Maintain an Efficient OXPHOS Metabolism Which Is Impaired Only by Co-Treatments
3.5. Co-Treatments Are Able to Induce Lipid Peroxidation in ER-CSCs
3.6. Co-Treatments Are Able to Induce Down-Regulation of GPX4 Activity and of ZEB-1 Expression
3.7. Chemoresistance of ER-CSCs Is Characterized by An Efficient OXPHOS Metabolism, High GSH Levels, ZEB-1 Up-Regulation and GPX4 Activation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- L’Abbate, A.; Macchia, G.; D’Addabbo, P.; Lonoce, A.; Tolomeo, D.; Trombetta, D.; Kok, K.; Bartenhagen, C.; Whelan, C.W.; Palumbo, O.; et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res. 2014, 42, 9131–9145. [Google Scholar] [CrossRef] [Green Version]
- Strobl-Mazzulla, P.H.; Bronner, M.E. Epithelial to mesenchymal transition: New and old insights from the classical neural crest model. Semin. Cancer Biol. 2012, 22, 411–416. [Google Scholar] [CrossRef] [Green Version]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.D.; Kattan, D.R.; Thomas, S.K.; Spengler, B.A.; Guo, H.F.; Biedler, J.L.; Cheung, N.K.; Ross, R.A. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004, 6, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Nagano, O.; Okazaki, S.; Saya, H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene 2013, 32, 5191–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, O.; Murakami, D.; Hartmann, D.; De Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Saya, H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004, 95, 930–935. [Google Scholar] [CrossRef]
- Domenicotti, C.; Marengo, B.; Verzola, D.; Garibotto, G.; Traverso, N.; Patriarca, S.; Maloberti, G.; Cottalasso, D.; Poli, G.; Passalacqua, M.; et al. Role of PKC-delta activity in glutathione-depleted neuroblastoma cells. Free Radic Biol. Med. 2003, 35, 504–516. [Google Scholar] [CrossRef]
- Marengo, B.; Raffaghello, L.; Pistoia, V.; Cottalasso, D.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Reactive oxygen species: Biological stimuli of neuroblastoma cell response. Cancer Lett. 2005, 228, 111–116. [Google Scholar] [CrossRef]
- Marengo, B.; De Ciucis, C.; Verzola, D.; Pistoia, V.; Raffaghello, L.; Patriarca, S.; Balbis, E.; Traverso, N.; Cottalasso, D.; Pronzato, M.A.; et al. Mechanisms of BSO (L-buthionine-S,R-sulfoximine)-induced cytotoxic effects in neuroblastoma. Free Radic Biol. Med. 2008, 44, 474–482. [Google Scholar] [CrossRef]
- Marengo, B.; De Ciucis, C.; Ricciarelli, R.; Passalacqua, M.; Nitti, M.; Zingg, J.M.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. PKCδ sensitizes neuroblastoma cells to L-buthionine-sulfoximine and etoposide inducing reactive oxygen species overproduction and DNA damage. PLoS ONE 2011, 6, e14661. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, D.; Wu, J.H.; Batist, G. Structure-based identification of novel human gamma-glutamylcysteine synthetase inhibitors. Mol. Pharmacol. 2007, 71, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef] [Green Version]
- Toma, J.G.; McKenzie, I.A.; Bagli, D.; Miller, F.D. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells 2005, 23, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Fariss, M.W.; Reed, D.J. High-performance liquid chromatography of thiols and disulfides: Dinitrophenol derivatives. Methods Enzymol. 1987, 143, 101–109. [Google Scholar] [CrossRef]
- Asensi, M.; Sastre, J.; Pallardo, F.V.; Lloret, A.; Lehner, M.; Garcia-de-la Asuncion, J.; Viña, J. Ratio of reduced to oxidized glutathione as indicator of oxidative stress status and DNA damage. Methods Enzymol. 1999, 299, 267–276. [Google Scholar] [CrossRef]
- Ravera, S.; Bartolucci, M.; Cuccarolo, P.; Litamè, E.; Illarcio, M.; Calzia, D.; Degan, P.; Morelli, A.; Panfoli, I. Oxidative stress in myelin sheath: The other face of the extramitochondrial oxidative phosphorylation ability. Free Radic Res. 2015, 49, 1156–1164. [Google Scholar] [CrossRef]
- Marengo, B.; De Ciucis, C.G.; Ricciarelli, R.; Furfaro, A.L.; Colla, R.; Canepa, E.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. p38MAPK inhibition: A new combined approach to reduce neuroblastoma resistance under etoposide treatment. Cell Death Dis. 2013, 4, e589. [Google Scholar] [CrossRef] [PubMed]
- Roveri, A.; Maiorino, M.; Ursini, F. Enzymatic and immunological measurements of soluble and membrane-bound phospholipid-hydroperoxide glutathione peroxidase. Methods Enzymol. 1994, 233, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Leardi, R. Chemometric methods in food authentication. In Modern Techniques for Food Authentication, 2nd ed.; Sun, D.W., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 17; pp. 687–729. [Google Scholar]
- Leardi, R.; Melzi, C.; Polotti, G. CAT (Chemometric Agile Software). Available online: http://gruppochemiometria.it/index.php/software (accessed on 27 April 2021).
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, A.; Hanot, M.; Malesys, C.; Maalouf, M.; Rousson, R.; Rodriguez-Lafrasse, C.; Ardail, D. Transient alteration of cellular redox buffering before irradiation triggers apoptosis in head and neck carcinoma stem and non-stem cells. PLoS ONE 2011, 6, e14558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemann, D.W.; Beyers, K.L. In vivo therapeutic potential of combination thiol depletion and alkylating chemotherapy. Br. J. Cancer 1993, 68, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villablanca, J.G.; Volchenboum, S.L.; Cho, H.; Kang, M.; Cohn, S.L.; Anderson, C.P.; Marachelian, A.; Groshen, S.; Tsao-Wei, D.; Matthay, K.K.; et al. A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatr. Blood Cancer 2016, 63, 1349–1356. [Google Scholar] [CrossRef]
- Okamoto, I.; Kawano, Y.; Matsumoto, M.; Suga, M.; Kaibuchi, K.; Ando, M.; Saya, H. Regulated CD44 cleavage under the control of protein kinase C, calcium influx, and the Rho family of small G proteins. J. Biol. Chem. 1999, 274, 25525–25534. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Luo, G.; Shi, X.; Long, Y.; Shen, W.; Li, Z.; Zhang, X. The Xc- inhibitor sulfasalazine improves the anti-cancer effect of pharmacological vitamin C in prostate cancer cells via a glutathione-dependent mechanism. Cell Oncol. 2020, 43, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.W.; Yu, Y.L.; Lu, J.Y.; Hung, Y.T.; Liu, H.C.; Yiang, G.T. Anti-Cancer Effects of Sulfasalazine and Vitamin E Succinate in MDA-MB 231 Triple-Negative Breast Cancer Cells. Int. J. Med. Sci. 2019, 16, 494–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogihara, K.; Kikuchi, E.; Okazaki, S.; Hagiwara, M.; Takeda, T.; Matsumoto, K.; Kosaka, T.; Mikami, S.; Saya, H.; Oya, M. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 2019, 110, 1431–1441. [Google Scholar] [CrossRef]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef]
- Wada, F.; Koga, H.; Akiba, J.; Niizeki, T.; Iwamoto, H.; Ikezono, Y.; Nakamura, T.; Abe, M.; Masuda, A.; Sakaue, T.; et al. High expression of CD44v9 and xCT in chemoresistant hepatocellular carcinoma: Potential targets by sulfasalazine. Cancer Sci. 2018, 109, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Balza, E.; Castellani, P.; Moreno, P.S.; Piccioli, P.; Medraño-Fernandez, I.; Semino, C.; Rubartelli, A. Restoring microenvironmental redox and pH homeostasis inhibits neoplastic cell growth and migration: Therapeutic efficacy of esomeprazole plus sulfasalazine on 3-MCA-induced sarcoma. Oncotarget 2017, 8, 67482–67496. [Google Scholar] [CrossRef] [Green Version]
- Mooney, M.R.; Geerts, D.; Kort, E.J.; Bachmann, A.S. Anti-tumor effect of sulfasalazine in neuroblastoma. Biochem. Pharmacol. 2019, 162, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Li, Q.; Gao, Y.; Zhao, L.; Liu, Y. Increased PKCα activity by Rack1 overexpression is responsible for chemotherapy resistance in T-cell acute lymphoblastic leukemia-derived cell line. Sci. Rep. 2016, 6, 33717. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Asai, D.; Kang, J.; Kishimura, A.; Mori, T.; Katayama, Y. Reversal of efflux of an anticancer drug in human drug-resistant breast cancer cells by inhibition of protein kinase Cα (PKCα) activity. Tumour. Biol. 2016, 37, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Abera, M.B.; Kazanietz, M.G. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol. Pharmacol. 2015, 87, 832–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, P.P.; Zhou, L.; Watt, J.C.; Ruvolo, V.R.; Burks, J.K.; Jiffar, T.; Kornblau, S.; Konopleva, M.; Andreeff, M. Targeting PKC-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells. J. Cell Biochem. 2011, 112, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.K.; Cheong, J.H. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch. Pharm. Res. 2019, 42, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Takahashi, J.; et al. Spheroid cancer stem cells display reprogrammed metabolism and obtain energy by actively running the tricarboxylic acid (TCA) cycle. Oncotarget 2016, 7, 33297–33305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Samuel, S.M.; Varghese, E.; Koklesová, L.; Líšková, A.; Kubatka, P.; Büsselberg, D. Counteracting Chemoresistance with Metformin in Breast Cancers: Targeting Cancer Stem Cells. Cancers 2020, 12, 2482. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Čipak Gašparović, A.; Milković, L.; Dandachi, N.; Stanzer, S.; Pezdirc, I.; Vrančić, J.; Šitić, S.; Suppan, C.; Balic, M. Chronic Oxidative Stress Promotes Molecular Changes Associated with Epithelial Mesenchymal Transition, NRF2, and Breast Cancer Stem Cell Phenotype. Antioxidants 2019, 8, 633. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Jolly, M.K. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a036905. [Google Scholar] [CrossRef]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Aznar, E.; Wiesmüller, L.; Sainz, B., Jr.; Hermann, P.C. EMT and Stemness-Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; AlQudsy, L.H.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, R.; Emaduddin, M.; Al-Saihati, H.; Moutasim, K.; Chan, J.; Spampinato, M.; Bhome, R.; Yuen, H.M.; Mescoli, C.; Vitale, A.; et al. Protein kinase C inhibitors override ZEB1-induced chemoresistance in HCC. Cell Death Dis. 2019, 10, 703. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants 2021, 10, 691. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050691
Monteleone L, Speciale A, Valenti GE, Traverso N, Ravera S, Garbarino O, Leardi R, Farinini E, Roveri A, Ursini F, et al. PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants. 2021; 10(5):691. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050691
Chicago/Turabian StyleMonteleone, Lorenzo, Andrea Speciale, Giulia Elda Valenti, Nicola Traverso, Silvia Ravera, Ombretta Garbarino, Riccardo Leardi, Emanuele Farinini, Antonella Roveri, Fulvio Ursini, and et al. 2021. "PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis" Antioxidants 10, no. 5: 691. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050691