
Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases



Abstract
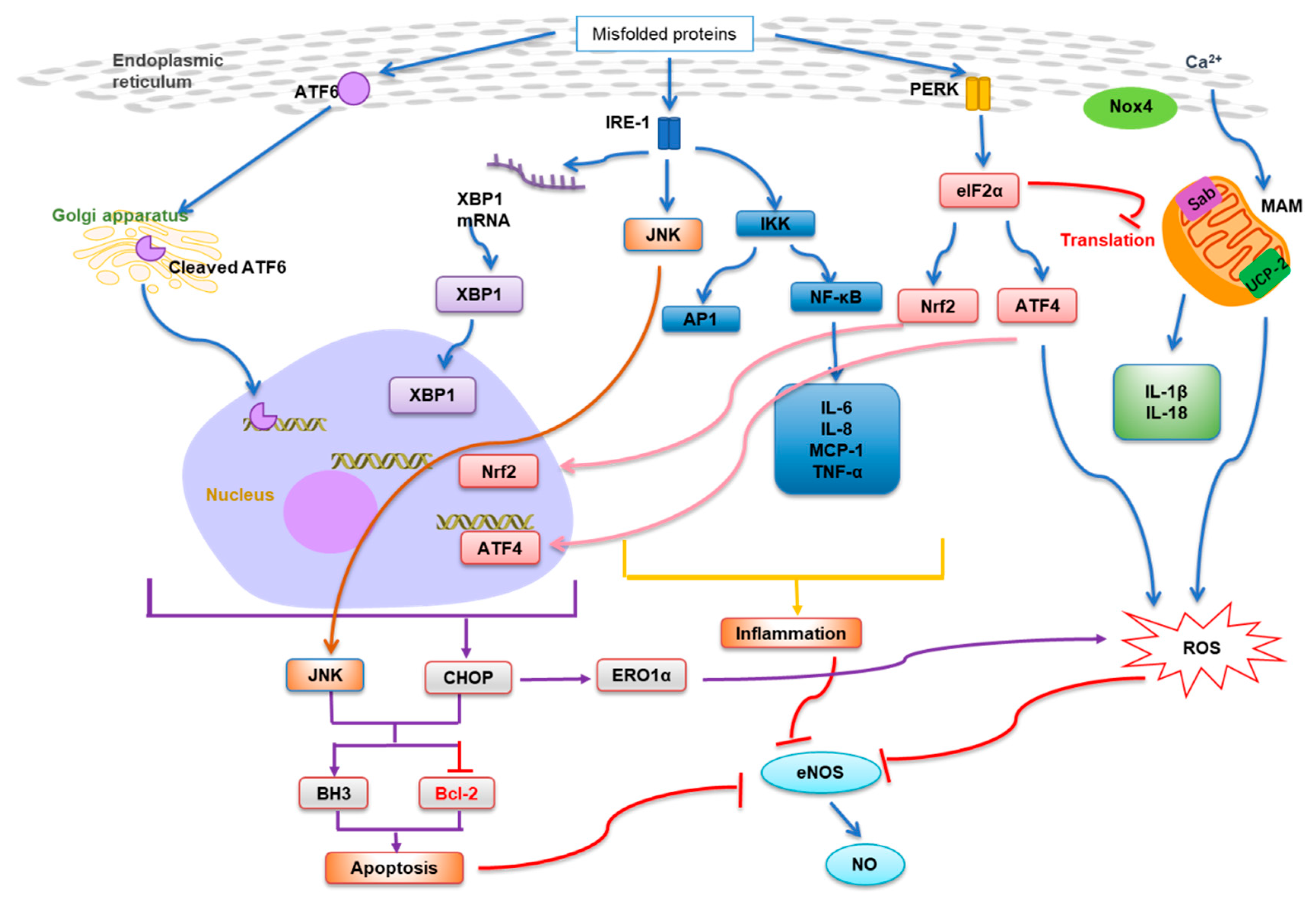
:1. Introduction
2. Regulation of ER Stress and Integration with Other Signaling Networks
3. ER Stress Linking to Cardiovascular Complications in Diabetes and Obesity
4. ER Stress in Atherosclerosis
5. Hypertension
6. Pulmonary Arterial Hypertension
7. Hyperhomocysteinemia
8. Myocardial Infarction
9. Stroke
10. ER Stress as Drug Target to Combat against CVD
11. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, I.J.; McMillan, D.R. Stress (heat shock) proteins: Molecular chaperones in cardiovascular biology and disease. Circ. Res. 1998, 83, 117–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Engelman, R.M.; Moraru, I.I.; Rousou, J.A.; Flack, J.E., III; Deaton, D.W.; Maulik, N.; Das, D.K. Heat shock. A new approach for myocardial preservation in cardiac surgery. Circulation 1992, 86, II358–II363. [Google Scholar] [PubMed]
- Quinones, Q.J.; de Ridder, G.G.; Pizzo, S.V. GRP78: A chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 2008, 23, 1409–1416. [Google Scholar]
- Lord, J.M.; Davey, J.; Frigerio, L.; Roberts, L.M. Endoplasmic reticulum-associated protein degradation. Semin. Cell Dev. Biol. 2000, 11, 159–164. [Google Scholar] [CrossRef]
- Demasi, M.; Laurindo, F.R. Physiological and pathological role of the ubiquitin-proteasome system in the vascular smooth muscle cell. Cardiovasc. Res. 2012, 95, 183–193. [Google Scholar] [CrossRef]
- Dhawan, V.; Bakshi, C.; Rather, R.A. Molecular Targets and Novel Therapeutics to Target Oxidative Stress in Cardiovascular Diseases. In Oxidative Stress in Heart Diseases; Chakraborti, S., Dhalla, N., Ganguly, N., Dikshit, M., Eds.; Springer: Singapore, 2019; pp. 59–82. [Google Scholar]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidant 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Leopold, J.A.; Loscalzo, J. Oxidative mechanisms and atherothrombotic cardiovascular disease. Drug Discov. Today Strat. 2008, 5, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachidanandam, K.; Fagan, S.C.; Ergul, A. Oxidative stress and cardiovascular disease: Antioxidants and unresolved issues. Cardiovasc. Drug Rev. 2005, 23, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaconstantinou, J. The Role of Signaling Pathways of Inflammation and Oxidative Stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells 2019, 8, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, N.; Zazueta, C.; Aguilera-Aguirre, L. Oxidative Stress and Inflammation in Cardiovascular Disease. Oxid. Med. Cell. Longev. 2017, 2017, 5853238. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Oxidative Stress in Cell Death and Cardiovascular Diseases. Oxid. Med. Cell. Longev 2019, 2019, 9030563. [Google Scholar] [CrossRef] [Green Version]
- Pickering, R.J. Oxidative Stress and Inflammation in Cardiovascular Diseases. Antioxidant 2021, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Patel, A.; Anderson, C.S.; Dong, J.; Ma, C. Epidemiology of Cardiovascular Disease in China and Opportunities for Improvement: JACC International. J. Am. Coll. Cardiol. 2019, 73, 3135–3147. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhang, L.; Lu, Y.; Su, M.; Li, X.; Liu, J.; Zhang, H.; Nasir, K.; Masoudi, F.A.; Krumholz, H.M.; et al. Secondary prevention of cardiovascular disease in China. Heart 2020, 106, 1349–1356. [Google Scholar] [CrossRef]
- Koopman, M.; Hetz, C.; Nollen, E.A.A. Saved by the Matrix: UPR Independent Survival under ER Stress. Cell 2019, 179, 1246–1248. [Google Scholar] [CrossRef]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Xu, W.; Wang, C.; Hua, J. X-box binding protein 1 (XBP1) function in diseases. Cell Biol. Int. 2021, 45, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Kubra, K.T.; Akhter, M.S.; Uddin, M.A.; Barabutis, N. Unfolded protein response in cardiovascular disease. Cell. Signal. 2020, 73, 109699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, X.; Gillette, T.G.; Deng, Y.; Wang, Z.V. Unfolded Protein Response as a Therapeutic Target in Cardiovascular Disease. Curr. Top. Med. Chem. 2019, 19, 1902–1917. [Google Scholar] [CrossRef]
- Zhu, Q. The ER stress-autophagy axis: Implications for cognitive dysfunction in diabetes mellitus. Clin. Sci. (Lond.) 2020, 134, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Shaygannia, E.; Rahmani, M.; Eskandari, A.; Golsefid, A.A.; Tavalaee, M.; Gharagozloo, P.; Drevet, J.R.; Nasr-Esfahani, M.H. Endoplasmic Reticulum Stress (ER Stress) and Unfolded Protein Response (UPR) Occur in a Rat Varicocele Testis Model. Oxid. Med. Cell. Longev. 2020, 2020, 5909306. [Google Scholar] [CrossRef] [PubMed]
- Danecka, M.D.; Jurczak, W.; Olszanecka, A.; Krawczyk, K.; Skotnicki, A.B. Primary cardioprotection in lymphoma patients with high risk of cardiovascular disease, treated with (R) CHOP regimen: A single center retrospective analysis. J. Clin. Oncol. 2015, 33, e19515. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Zhang, Y.; Ji, W.; Wang, L.; Lee, S.C. Periplogenin Activates ROS-ER Stress Pathway to Trigger Apoptosis via BIP-eIF2alpha- CHOP and IRE1alpha-ASK1-JNK Signaling Routes. Anticancer Agents Med. Chem. 2021, 21, 61–70. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Blanton, R.M.; Keaney, J.F.; Kant, S. JNK and cardiometabolic dysfunction. Biosci. Rep. 2019, 39, BSR20190267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Z.; Ling, X.; Cao, S.S.; Cheng, Q.Y. Discovery and characterization of a small chemical compound that shows exceptional antitumor activity and targets multiple antiapoptotic proteins in the inhibitor of apoptosis (IAP) and Bcl-2 families. Cancer Res. 2011, 71, 4525. [Google Scholar]
- Li, G.; Qi, W.Q.; Li, X.X.; Zhao, J.W.; Luo, M.H.; Chen, J.J. Recent Advances in c-Jun N-Terminal Kinase (JNK) Inhibitors. Curr. Med. Chem. 2021, 28, 607–627. [Google Scholar] [CrossRef]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. ER stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Bioenergetics 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoma, A.; Lyon, M.; Al-Shanti, N.; Nye, G.A.; Cooper, R.G.; Lightfoot, A.P. Eukarion-134 Attenuates Endoplasmic Reticulum Stress-Induced Mitochondrial Dysfunction in Human Skeletal Muscle Cells. Antioxidant 2020, 9, 710. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.Y.; Wong, W.T.; Xu, A.; Lu, Y.; Zhang, Y.; Wang, L.; Cheang, W.S.; Wang, Y.; Yao, X.; Huang, Y. Uncoupling protein-2 protects endothelial function in diet-induced obese mice. Circ. Res. 2012, 110, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Piao, J.H.; Nakajima, A.; Sakon-Komazawa, S.; Kojima, Y.; Mori, K.; Yagita, H.; Okumura, K.; Harding, H.; Nakano, H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 2005, 280, 33917–33925. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhu, J.; Wang, Y.H.; Hang, C.H. ROS-Mediated Mitochondrial Dysfunction and ER Stress Contribute to Compression-Induced Neuronal Injury. Neuroscience 2019, 416, 268–280. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, A. ROS-mediated PERK-eIF2alpha-ATF4 pathway plays an important role in arsenite-induced L-02 cells apoptosis via regulating CHOP-DR5 signaling. Environ. Toxicol. 2020, 35, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Digaleh, H.; Kiaei, M.; Khodagholi, F. Nrf2 and Nrf1 signaling and ER stress crosstalk: Implication for proteasomal degradation and autophagy. Cell. Mol. Life Sci. 2013, 70, 4681–4694. [Google Scholar] [CrossRef]
- Ooi, B.K.; Goh, B.H.; Yap, W.H. Oxidative Stress in Cardiovascular Diseases: Involvement of Nrf2 Antioxidant Redox Signaling in Macrophage Foam Cells Formation. Int. J. Mol. Sci. 2017, 18, 2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.M.; Camargo, L.L.; Filho, S.V.; Lopes, L.R. Endoplasmic reticulum stress and Nox-mediated reactive oxygen species signaling in the peripheral vasculature: Potential role in hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Asp. Med. 2018, 63, 18–29. [Google Scholar] [CrossRef]
- Wood, S.K. The role of inflammation and oxidative stress in depression and cardiovascular disease. In Cardiovascular Implications of Stress and Depression; Chantler, P.D., Larkin, K.T., Eds.; Academic Press: London, UK, 2020; pp. 175–209. [Google Scholar]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Kim, K.; Kim, J.H.; Park, Y. The Role of Endoplasmic Reticulum Stress in Cardiovascular Disease and Exercise. Int. J. Vasc. Med. 2017, 2017, 2049217. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Younce, C.W.; Wang, K.K.; Kolattukudy, P.E. Hyperglycaemia-induced cardiomyocyte death is mediated via MCP-1 production and induction of a novel zinc-finger protein MCPIP. Cardiovasc. Res. 2010, 87, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Zhang, M.; Wang, S.; Liang, B.; Zhao, Z.; Liu, C.; Wu, M.; Choi, H.C.; Lyons, T.J.; Zou, M.H. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010, 59, 1386–1396. [Google Scholar] [CrossRef] [Green Version]
- Burgos-Moron, E.; Abad-Jimenez, Z.; Maranon, A.M.; Iannantuoni, F.; Escribano-Lopez, I.; Lopez-Domenech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, R.; Kundur, A.R.; Singh, I. Oxidative stress biomarkers in type 2 diabetes mellitus for assessment of cardiovascular disease risk. Diabetes. Metab. Synd. 2018, 12, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhu, H.; He, C.; He, T.; Pan, S.; Zhao, N.; Zhu, L.; Guan, G.; Liu, P.; Zhang, Y.; et al. Nicorandil attenuates high glucose-induced insulin resistance by suppressing oxidative stress-mediated ER stress PERK signaling pathway. BMJ Open Diabetes Res. Care 2021, 9, 4525. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Kumar, S. Natural AMPK Activators: An Alternative Approach for the Treatment and Management of Metabolic Syndrome. Curr. Med. Chem. 2017, 24, 1007–1047. [Google Scholar] [CrossRef]
- Desjardins, E.M.; Steinberg, G.R. Emerging Role of AMPK in Brown and Beige Adipose Tissue (BAT): Implications for Obesity, Insulin Resistance, and Type 2 Diabetes. Curr. Diab. Rep. 2018, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, J.; Liang, H.; Yang, S.; Zhang, Y.; Han, W.; Chen, C.; Cao, N.; Liang, P.; Du, X.; et al. Kang Le Xin Reduces Blood Pressure Through Inducing Endothelial-Dependent Vasodilation by Activating the AMPK-eNOS Pathway. Front. Pharmacol. 2020, 10, 1548. [Google Scholar] [CrossRef]
- Hwang, H.J.; Jung, T.W.; Kim, J.W.; Kim, J.A.; Lee, Y.B.; Hong, S.H.; Roh, E.; Choi, K.M.; Baik, S.H.; Yoo, H.J. Protectin DX prevents H2O2-mediated oxidative stress in vascular endothelial cells via an AMPK-dependent mechanism. Cell. Signal. 2019, 53, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, H.; Shen, E.; Wan, L.; Arnold, J.M.; Peng, T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010, 59, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; An, E.J.; Kim, J.; Bae, Y.S. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol. Ther. (Seoul) 2020, 28, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Duraisamy, A.J.; Kowluru, A.; Kowluru, R.A. Functional Regulation of an Oxidative Stress Mediator, Rac1, in Diabetic Retinopathy. Mol. Neurobiol. 2019, 56, 8643–8655. [Google Scholar] [CrossRef]
- Ouerd, S.; Idris-Khodja, N.; Trindade, M.; Ferreira, N.S.; Berillo, O.; Coelho, S.C.; Neves, M.F.; Jandeleit-Dahm, K.A.; Paradis, P.; Schiffrin, E.L. Endothelium-restricted endothelin-1 overexpression in type 1 diabetes worsens atherosclerosis and immune cell infiltration via NOX1. Cardiovasc. Res. 2021, 117, 1144–1153. [Google Scholar] [CrossRef]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.H.; Juguilon, H.; et al. AMPK and PPAR delta Agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M.; Lee, S.S.; Li, W.; Ward, J.M.; Gavrilova, O.; Everett, C.; Reitman, M.L.; Hudson, L.D.; Gonzalez, F.J. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol. Cell. Biol. 2000, 20, 5119–5128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piqueras, L.; Reynolds, A.R.; Hodivala-Dilke, K.M.; Alfranca, A.; Redondo, J.M.; Hatae, T.; Tanabe, T.; Warner, T.D.; Bishop-Bailey, D. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Wong, W.T.; Wang, L.; Cheng, C.K.; Lau, C.W.; Ma, R.C.W.; Xu, A.; Wang, N.; Huang, Y.; Tian, X.Y. Resveratrol ameliorates endothelial dysfunction in diabetic and obese mice through sirtuin 1 and peroxisome proliferator-activated receptor delta. Pharmacol. Res. 2019, 139, 384–394. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of Lipid Peroxidation and PPAR-delta in Amplifying Glucose-Stimulated Insulin Secretion. Diabetes 2011, 60, 2830–2842. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPAR delta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M.; Curtiss, L.K. Transcriptional repression of atherogenic inflammation: Modulation by PPARdelta. Science 2003, 302, 453–457. [Google Scholar] [CrossRef]
- Tian, X.Y.; Wong, W.T.; Wang, N.P.; Lu, Y.; Cheang, W.S.; Liu, J.; Liu, L.M.; Liu, Y.H.; Lee, S.S.T.; Chen, Z.Y.; et al. PPAR delta Activation Protects Endothelial Function in Diabetic Mice. Diabetes 2012, 61, 3285–3293. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Fang, S.; Liu, X.; Li, J.; Wang, X.; Cui, J.; Chen, T.; Li, Z.; Yang, F.; Tian, J.; et al. Omentin-1 protects against high glucose-induced endothelial dysfunction via the AMPK/PPARdelta signaling pathway. Biochem. Pharmacol. 2020, 174, 113830. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhe, R.; Guo, X.; Chen, Y.; Zou, Y.; Zhou, L.; Wang, Z. The Role of PPARdelta Agosnist GW501516 in Rats with Gestational Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.J.; Verma, S.; Anderson, T.J. Improved endothelial function with metformin in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2001, 37, 1344–1350. [Google Scholar] [CrossRef] [Green Version]
- Tangney, N.; De Buitleir, C.; O’Brien, T.; Liew, A. Efficacy and safety of initial combined therapy with metformin plus a dipeptidyl peptidase-4 inhibitor versus metformin monotherapy in type 2 diabetes mellitus: A systematic review and meta-analysis of phase 3 randomised controlled trials. Ir. J. Med. Sci. 2019, 188, S248. [Google Scholar]
- Adler, A.I. Cardiovascular risk reduction in diabetes: Underemphasised and overdue. Messages from major trials. Clin. Med. 2001, 1, 472–477. [Google Scholar] [CrossRef]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.P.; Bhopale, K.K.; Caracheo, A.A.; Kaphalia, L.; Loganathan, G.; Balamurugan, A.N.; Rastellini, C.; Kaphalia, B.S. Differential cytotoxicity, ER/oxidative stress, dysregulated AMPKalpha signaling, and mitochondrial stress by ethanol and its metabolites in human pancreatic acinar cells. Alcohol. Clin. Exp. Res. 2021, 45, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Cui, W.; Chen, S.; Shao, Z.; Li, Y.; Wang, W.; Mao, L.; Li, J.; Mei, X. Metformin alleviates high glucose-induced ER stress and inflammation by inhibiting the interaction between caveolin1 and AMPKalpha in rat astrocytes. Biochem. Biophys. Res. Commun. 2021, 534, 908–913. [Google Scholar] [CrossRef]
- Chen, C.; Kassan, A.; Castaneda, D.; Gabani, M.; Choi, S.K.; Kassan, M. Metformin prevents vascular damage in hypertension through the AMPK/ER stress pathway. Hypertens. Res. 2019, 42, 960–969. [Google Scholar] [CrossRef]
- Cheang, W.S.; Wong, W.T.; Zhao, L.; Xu, J.; Wang, L.; Lau, C.W.; Chen, Z.Y.; Ma, R.C.; Xu, A.; Wang, N.; et al. PPARdelta Is Required for Exercise to Attenuate Endoplasmic Reticulum Stress and Endothelial Dysfunction in Diabetic Mice. Diabetes 2017, 66, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Li, J.M.; Wang, J.J.; Yu, Q.; Wang, M.; Zhang, S.X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009, 583, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.H.; Tong, P.; Gu, L.M.; Li, W.J. Astragalus polysaccharide attenuates metabolic memory-triggered ER stress and apoptosis via regulation of miR-204/SIRT1 axis in retinal pigment epithelial cells. Biosci. Rep. 2020, 40, BSR20192121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.J.; Li, J.; Hosoya, K.I.; Ratan, R.; Townes, T.; Zhang, S.X. Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia 2012, 55, 2533–2545. [Google Scholar] [CrossRef] [Green Version]
- Hopfgarten, J.; Stenwall, P.A.; Wiberg, A.; Anagandula, M.; Ingvast, S.; Rosenling, T.; Korsgren, O.; Skog, O. Gene expression analysis of human islets in a subject at onset of type 1 diabetes. Acta Diabetol. 2014, 51, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, M.; Kassan, M.; Choi, S.K.; Partyka, M.; Trebak, M.; Henrion, D.; Matrougui, K. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension 2012, 60, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.J.; Zou, C.; Zhao, M.J.; Zheng, Z. ASK1 induces retinal microvascular endothelial cell apoptosis through ER stress-associated pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 1324–1332. [Google Scholar] [PubMed]
- Ju, X.; Yang, X.; Yan, T.; Chen, H.; Song, Z.; Zhang, Z.; Wu, W.; Wang, Y. EGFR inhibitor, AG1478, inhibits inflammatory infiltration and angiogenesis in mice with diabetic retinopathy. Clin. Exp. Pharmacol. Physiol. 2019, 46, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, A.; Choi, S.K.; Galan, M.; Kassan, M.; Partyka, M.; Kadowitz, P.; Henrion, D.; Trebak, M.; Belmadani, S.; Matrougui, K. Chronic inhibition of endoplasmic reticulum stress and inflammation prevents ischaemia-induced vascular pathology in type II diabetic mice. J. Pathol. 2012, 227, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Li, J.; Hou, F.; Wang, X.; Liu, B. Mangiferin inhibits endoplasmic reticulum stress-associated thioredoxin-interacting protein/NLRP3 inflammasome activation with regulation of AMPK in endothelial cells. Metabolism 2015, 64, 428–437. [Google Scholar] [CrossRef]
- Aswal, S.; Kumar, A.; Chauhan, A.; Semwal, R.B.; Kumar, A.; Semwal, D.K. A Molecular Approach on the Protective Effects of Mangiferin Against Diabetes and Diabetes-related Complications. Curr. Diabetes Rev. 2020, 16, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Leng, B.; Li, C.; Sun, Y.; Zhao, K.; Zhang, L.; Lu, M.L.; Wang, H.X. Protective Effect of Astragaloside IV on High Glucose-Induced Endothelial Dysfunction via Inhibition of P2X7R Dependent P38 MAPK Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 5070415. [Google Scholar] [CrossRef]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.H. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010, 121, 792–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Salvayre, R.; Negre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011, 18, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taborda, N.A.; Blanquiceth, Y.; Urcuqui-Inchima, S.; Latz, E.; Hernandez, J.C. High-Density Lipoproteins Decrease Proinflammatory Activity and Modulate the Innate Immune Response. J. Interferon Cytokine Res. 2019, 39, 760–770. [Google Scholar] [CrossRef]
- Sulliman, N.C.; Ghaddar, B.; Gence, L.; Patche, J.; Rastegar, S.; Meilhac, O.; Diotel, N. HDL biodistribution and brain receptors in zebrafish, using HDLs as vectors for targeting endothelial cells and neural progenitors. Sci. Rep. UK 2021, 11, 6439. [Google Scholar] [CrossRef]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, A.; Ravanan, P. Kaempferol mitigates Endoplasmic Reticulum Stress Induced Cell Death by targeting caspase 3/7. Sci. Rep. 2018, 8, 2189. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.Q.; Fei, M.Z.; Wu, C.T.; Wang, W.; Luo, R.; Shen, L.P.; Zhang, Z. Atorvastatin inhibits endoplasmic reticulum stress through AMPK signaling pathway in atherosclerosis in mice. Exp. Ther. Med. 2020, 19, 2266–2272. [Google Scholar] [CrossRef]
- Hong, J.; Kim, K.; Park, E.; Lee, J.; Markofski, M.M.; Marrelli, S.P.; Park, Y. Exercise ameliorates endoplasmic reticulum stress-mediated vascular dysfunction in mesenteric arteries in atherosclerosis. Sci. Rep. 2018, 8, 7938. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pan, W.; Wang, C.; Dong, H.; Xing, L.; Hou, J.; Fang, S.; Li, H.; Yang, F.; Yu, B. H2S attenuates endoplasmic reticulum stress in hypoxia-induced pulmonary artery hypertension. Biosci. Rep. 2019, 39, BSR20190304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.N.; Cao, X.; Guruju, M.R.; Pierce, J.P.; Morgan, D.A.; Wang, G.; Iadecola, C.; Mark, A.L.; Davisson, R.L. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J. Clin. Investig. 2012, 122, 3960–3964. [Google Scholar] [CrossRef]
- Horwath, J.A.; Hurr, C.; Butler, S.D.; Guruju, M.; Cassell, M.D.; Mark, A.L.; Davisson, R.L.; Young, C.N. Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI Insight 2017, 2, e90170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassan, M.; Galan, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic reticulum stress is involved in cardiac damage and vascular endothelial dysfunction in hypertensive mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitler, K.M.; Matsumoto, T.; Webb, R.C. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H344–H353. [Google Scholar] [CrossRef] [Green Version]
- Cheang, W.S.; Ngai, C.Y.; Tam, Y.Y.; Tian, X.Y.; Wong, W.T.; Zhang, Y.; Lau, C.W.; Chen, Z.Y.; Bian, Z.X.; Huang, Y.; et al. Black tea protects against hypertension-associated endothelial dysfunction through alleviation of endoplasmic reticulum stress. Sci. Rep. UK 2015, 5, 10340. [Google Scholar] [CrossRef] [PubMed]
- Murugan, D.; Lau, Y.S.; Lau, C.W.; Mustafa, M.R.; Huang, Y. Angiotensin 1-7 Protects against Angiotensin II-Induced Endoplasmic Reticulum Stress and Endothelial Dysfunction via Mas Receptor. PLoS ONE 2015, 10, e0145413. [Google Scholar]
- Spitler, K.M.; Webb, R.C. Endoplasmic reticulum stress contributes to aortic stiffening via proapoptotic and fibrotic signaling mechanisms. Hypertension 2014, 63, e40–e45. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, T.; Kawai, T.; Forrester, S.J.; Obama, T.; Tsuji, T.; Fukuda, Y.; Elliott, K.J.; Tilley, D.G.; Davisson, R.L.; Park, J.Y.; et al. Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 2015, 65, 1349–1355. [Google Scholar] [CrossRef]
- Obama, T.; Takayanagi, T.; Kobayashi, T.; Bourne, A.M.; Elliott, K.J.; Charbonneau, M.; Dubois, C.M.; Eguchi, S. Vascular induction of a disintegrin and metalloprotease 17 by angiotensin II through hypoxia inducible factor 1alpha. Am. J. Hypertens. 2015, 28, 10–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Zhang, L.; Miao, K.; Wang, Y. A crucial role of endoplasmic reticulum stress in cellular responses during pulmonary arterial hypertension. Am. J. Transl. Res. 2020, 12, 1481–1490. [Google Scholar] [PubMed]
- Yang, J.; Griffiths, M.; Nies, M.K.; Brandal, S.; Damico, R.; Vaidya, D.; Tao, X.T.; Simpson, C.E.; Kolb, T.M.; Mathai, S.C.; et al. Insulin-like growth factor binding protein-2: A new circulating indicator of pulmonary arterial hypertension severity and survival. BMC Med. 2020, 18, 268. [Google Scholar] [CrossRef] [PubMed]
- Zhuan, B.; Wang, X.; Wang, M.D.; Li, Z.C.; Yuan, Q.; Xie, J.; Yang, Z. Hypoxia induces pulmonary artery smooth muscle dysfunction through mitochondrial fragmentation-mediated endoplasmic reticulum stress. Aging US 2020, 12, 23684–23697. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Sutendra, G.; Michelakis, E.D. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 2013, 127, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, M.; Furuhashi, M.; Ishimura, S.; Mita, T.; Fuseya, T.; Okazaki, Y.; Yoshida, H.; Tsuchihashi, K.; Miura, T. Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1314–H1323. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Adi, D.; Long, M.; Wang, J.; Liu, F.; Gai, M.T.; Aierken, A.; Li, M.Y.; Li, Q.; Wu, L.Q.; et al. 4-Phenylbutyric Acid Induces Protection against Pulmonary Arterial Hypertension in Rats. PLoS ONE 2016, 11, e0157538. [Google Scholar] [CrossRef]
- Mao, S.Z.; Fan, X.F.; Xue, F.; Chen, R.; Chen, X.Y.; Yuan, G.S.; Hu, L.G.; Liu, S.F.; Gong, Y.S. Intermedin modulates hypoxic pulmonary vascular remodeling by inhibiting pulmonary artery smooth muscle cell proliferation. Pulm. Pharmacol. Ther. 2014, 27, 1–9. [Google Scholar] [CrossRef]
- Cao, X.; He, Y.; Li, X.; Xu, Y.; Liu, X. The IRE1alpha-XBP1 pathway function in hypoxia-induced pulmonary vascular remodeling, is upregulated by quercetin, inhibits apoptosis and partially reverses the effect of quercetin in PASMCs. Am. J. Transl. Res. 2019, 11, 641–654. [Google Scholar]
- Sutendra, G.; Dromparis, P.; Wright, P.; Bonnet, S.; Haromy, A.; Hao, Z.; McMurtry, M.S.; Michalak, M.; Vance, J.E.; Sessa, W.C.; et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci. Transl. Med. 2011, 3, 88ra55. [Google Scholar] [CrossRef] [Green Version]
- Sutendra, G.; Dromparis, P.; Bonnet, S.; Haromy, A.; McMurtry, M.S.; Bleackley, R.C.; Michelakis, E.D. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFalpha contributes to the pathogenesis of pulmonary arterial hypertension. J. Mol. Med. 2011, 89, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Ghatnekar, A.; Chrobak, I.; Reese, C.; Stawski, L.; Seta, F.; Wirrig, E.; Paez-Cortez, J.; Markiewicz, M.; Asano, Y.; Harley, R.; et al. Endothelial GATA-6 Deficiency Promotes Pulmonary Arterial Hypertension. Am. J. Pathol. 2013, 182, 2391–2406. [Google Scholar] [CrossRef] [Green Version]
- Ichihara, A.; Toyama, T.; Kudryashova, T.; Lenna, S.; Looney, A.; Shen, Y.; Avolio, T.; Goncharov, D.; Lafyatis, R.A.; Seta, F.; et al. Endothelial GATA6 Coordinates Cross-Talk Between BMP and Oxidative Stress Axis in Pulmonary Arterial Hypertension. Am. J. Respir Crit. Care 2020, 201, A6363. [Google Scholar]
- Lenna, S.; Farina, A.G.; Martyanov, V.; Christmann, R.B.; Wood, T.A.; Farber, H.W.; Scorza, R.; Whitfield, M.L.; Lafyatis, R.; Trojanowska, M. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum. 2013, 65, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Outinen, P.A.; Sood, S.K.; Pfeifer, S.I.; Pamidi, S.; Podor, T.J.; Li, J.; Weitz, J.I.; Austin, R.C. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 1999, 94, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Ruiz, N.; Murillo-Gonzalez, F.E.; Rojas-Garcia, A.E.; Barron-Vivanco, B.S.; Bernal-Hernandez, Y.Y.; Gonzalez-Arias, C.A.; Ortega-Cervantes, L.; Ponce-Gallegos, J.; Lopez-Guarnido, O.; Medina-Diaz, I.M. PON1 status and homocysteine levels as potential biomarkers for cardiovascular disease. Exp. Gerontol. 2020, 140, 111062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–35874. [Google Scholar] [CrossRef] [Green Version]
- Hossain, G.S.; van Thienen, J.V.; Werstuck, G.H.; Zhou, J.; Sood, S.K.; Dickhout, J.G.; de Koning, A.B.L.; Tang, D.; Wu, D.C.; Falk, E.; et al. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the development of atherosclerosis in hyperhomocysteinemia. J. Biol. Chem. 2003, 278, 30317–30327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.C.; Sun, W.T.; Yu, C.M.; Pun, S.H.; Underwood, M.J.; He, G.W.; Yang, Q. ER stress mediates homocysteine-induced endothelial dysfunction: Modulation of IKCa and SKCa channels. Atherosclerosis 2015, 242, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qin, Z.X.; Liu, C.; Song, M.B.; Luo, X.L.; Zhao, H.Q.; Qian, D.H.; Chen, J.F.; Huang, L. Nox4 and soluble epoxide hydrolase synergistically mediate homocysteine-induced inflammation in vascular smooth muscle cells. Vasc. Pharmacol. 2019, 120, 106544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xie, X.; Chen, Y.; Hammock, B.D.; Kong, W.; Zhu, Y. Homocysteine upregulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Circ. Res. 2012, 110, 808–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadir, A.; Kavalakatt, S.; Madhu, D.; Cherian, P.; Al-Mulla, F.; Abubaker, J.; Tiss, A. Soluble Epoxide Hydrolase 2 Expression Is Elevated in Obese Humans and Decreased by Physical Activity. Int. J. Mol. Sci. 2020, 21, 2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Gao, X.; Yang, S.; Meng, M.; Yang, X.; Ge, B. The role of endoplasmic reticulum stress in endothelial dysfunction induced by homocysteine thiolactone. Fundam. Clin. Pharmacol. 2015, 29, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, L.; Zhang, Z.; Lu, X. Protective Effect of Enalapril against Methionine-Enriched Diet-Induced Hypertension: Role of Endoplasmic Reticulum and Oxidative Stress. Biomed. Res. Int. 2015, 2015, 724876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kil, J.S.; Jeong, S.O.; Chung, H.T.; Pae, H.O. Piceatannol attenuates homocysteine-induced endoplasmic reticulum stress and endothelial cell damage via heme oxygenase-1 expression. Amino Acids 2017, 49, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wu, C.; Chen, Z.; Lu, G.; Sun, J. Atorvastatin attenuates atherosclerotic plaque destabilization by inhibiting endoplasmic reticulum stress in hyperhomocysteinemic mice. Mol. Med. Rep. 2016, 13, 3574–3580. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Wu, C.F.; Chen, Z.Y.; Lu, G.P. Atorvastatin Inhibits Homocysteine-Induced Endoplasmic Reticulum Stress through Activation of AMP-Activated Protein Kinase. Cardiovasc. Ther. 2012, 30, 317–325. [Google Scholar] [CrossRef]
- Zhu, L.; Jia, F.; Wei, J.; Yu, Y.; Yu, T.; Wang, Y.; Sun, J.; Luo, G. Salidroside protects against homocysteine-induced injury in human umbilical vein endothelial cells via the regulation of endoplasmic reticulum stress. Cardiovasc. Ther. 2017, 35, 33–39. [Google Scholar] [CrossRef]
- Hu, H.; Wang, C.; Jin, Y.; Meng, Q.; Liu, Q.; Liu, K.; Sun, H. Alpha-lipoic acid defends homocysteine-induced endoplasmic reticulum and oxidative stress in HAECs. Biomed. Pharmacother. 2016, 80, 63–72. [Google Scholar] [CrossRef]
- Cheng, C.K.; Luo, J.Y.; Lau, C.W.; Cho, W.C.; Ng, C.F.; Ma, R.C.W.; Tian, X.Y.; Huang, Y. A GLP-1 analog lowers ER stress and enhances protein folding to ameliorate homocysteine-induced endothelial dysfunction. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef] [PubMed]
- Li, W.G.; Zaheer, A.; Coppey, L.; Oskarsson, H.J. Activation of JNK in the remote myocardium after large myocardial infarction in rats. Biochem. Biophys. Res. Commun. 1998, 246, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009, 81, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, O.; Higuchi, Y.; Hirotani, S.; Kashiwase, K.; Nakayama, H.; Hikoso, S.; Takeda, T.; Watanabe, T.; Asahi, M.; Taniike, M.; et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 15883–15888. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.F.; Chen, G.; Li, H.Z.; Zhang, Y.; Liu, Y.M.; He, H.Q.; Liu, C.Y.; Xie, Z.C.; Zhang, Z.P.; Wang, J. Panax Notoginseng Saponins Inhibits Ventricular Remodeling after Myocardial Infarction in Rats Through Regulating ATF3/MAP2K3/p38 MAPK and NF kappa B Pathway. Chin. J. Integr. Med. 2020, 26, 897–904. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.Z.; Guo, Y.M.; Zhao, Z.; Zhang, L.; Yang, D.W. Association of TNF-alpha with ventricular arrhythmias during early acute myocardial infraction. Int. J. Clin. Exp. Pathol. 2016, 9, 6136–6144. [Google Scholar]
- Li, F.F.; Yang, Y.M.; Xue, C.Y.; Tan, M.T.; Xu, L.; Gao, J.B.; Xu, L.H.; Zong, J.; Qian, W.H. Zinc Finger Protein ZBTB20 protects against cardiac remodelling post-myocardial infarction via ROS-TNF alpha/ASK1/JNK pathway regulation. J. Cell. Mol. Med. 2020, 24, 13383–13396. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Kataoka, K.; Shintaku, H.; Yamashita, T.; Tokutomi, Y.; Dong, Y.F.; Matsuba, S.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Novel mechanism and role of angiotensin II-Induced vascular endothelial injury in hypertensive diastolic heart failure. Arterioscler. Thromb. Vasc. 2007, 27, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, Z.C.; Chen, P.P.; Xie, R.F. Primary Mechanism Study of Panax notoginseng Flower (Herb) on Myocardial Infarction in Rats. Cardiol. Res. Pract. 2019, 2019, 8723076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russomanno, G.; Corbi, G.; Manzo, V.; Ferrara, N.; Rengo, G.; Puca, A.A.; Latte, S.; Carrizzo, A.; Calabrese, M.C.; Andriantsitohaina, R.; et al. The anti-ageing molecule sirt1 mediates beneficial effects of cardiac rehabilitation. Immun. Ageing 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, S.; Oyadomari, S.; Yano, S.; Morioka, M.; Gotoh, T.; Hamada, J.I.; Ushio, Y.; Mori, M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, L.; Zhang, X.; Xu, L.; Xie, B.; Shi, H.; Duan, Z.; Zhang, H.; Ren, F. Kaempferol protects mice from d-GalN/LPS-induced acute liver failure by regulating the ER stress-Grp78-CHOP signaling pathway. Biomed. Pharmacother. 2019, 111, 468–475. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Daidone, M.; Pinto, A. Endothelial Dysfunction and Inflammation in Ischemic Stroke Pathogenesis. Curr. Pharm. Des. 2020, 26, 4209–4219. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Fu, H.; Huang, H.; Lu, Q.; Qin, H.; Wu, Y.; Huang, H.; Mao, G.; Wei, Z.; et al. Hes1 Knockdown Exacerbates Ischemic Stroke Following tMCAO by Increasing ER Stress-Dependent Apoptosis via the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway. Neurosci. Bull. 2020, 36, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Gharibani, P.M.; Ma, Z.; Alexandrescu, A.; Genova, R.; Chen, P.C.; Modi, J.; Menzie, J.; Pan, C.; Tao, R.; et al. Neuroprotective Functions Through Inhibition of ER Stress by Taurine or Taurine Combination Treatments in a Rat Stroke Model. Adv. Exp. Med. Biol. 2017, 975 Pt 1, 193–205. [Google Scholar]
- Qiu, J.; Wang, X.; Wu, F.; Wan, L.; Cheng, B.; Wu, Y.; Bai, B. Low Dose of Apelin-36 Attenuates ER Stress-Associated Apoptosis in Rats with Ischemic Stroke. Front. Neurol. 2017, 8, 556. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Xu, X.; Shi, H.; Yu, X.; Wang, X.; Yan, Y.; Fu, X.; Hu, H.; Li, X.; et al. bFGF inhibits ER stress induced by ischemic oxidative injury via activation of the PI3K/Akt and ERK1/2 pathways. Toxicol. Lett. 2012, 212, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Oxidative stress in cardiovascular disease: A new avenue toward future therapeutic approaches. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic Reticulum Stress As a Therapeutic Target in Cardiovascular Disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Goldberg, A.L. Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid May Improve Liver and Muscle but Not Adipose Tissue Insulin Sensitivity in Obese Men and Women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.T.; Giacca, A.; Lewis, G.F. Sodium Phenylbutyrate, a Drug With Known Capacity to Reduce Endoplasmic Reticulum Stress, Partially Alleviates Lipid-Induced Insulin Resistance and beta-Cell Dysfunction in Humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentino, T.V.; Procopio, T.; Mancuso, E.; Arcidiacono, G.P.; Andreozzi, F.; Arturi, F.; Sciacqua, A.; Perticone, F.; Hribal, M.L.; Sesti, G. SRT1720 counteracts glucosamine-induced endoplasmic reticulum stress and endothelial dysfunction. Cardiovasc. Res. 2015, 107, 295–306. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drugs or Herbs | Action Mechanism | Cardiovascular Disease | References |
|---|---|---|---|
| 4-phenyl butyric acid (PBA) | ↑ insulin sensitivity; ↓ blood pressure, fatty liver disease, cardiomyocyte UPR activity, cardiac fibrosis, TGF-β1, cPLA2/COX, arterial remodeling, right ventricular hypertrophy. | Diabetes and obesity, hypertension, pulmonary arterial hypertension | [61,118,119,128,129,130] |
| taurine-conjugated ursodeoxycholic acid (TUDCA) | ↑ insulin sensitivity, endothelium-dependent relaxation; ↓ blood pressure, fatty liver disease, cardiomyocyte UPR activity, cardiac fibrosis, TGF-β1, cPLA2/COX, Nox2, Nox4, body weight. | Diabetic vasculopathy, diabetic retinopathy, hypertension | [61,118,119,128,129,130] |
| Metformin | ↑ AMPK, PPARδ, eNOS activity, NO production; ↓ ER stress, oxidative stress. | Diabetic vasculopathy | [91,92,93] |
| AG1478 | ↑ endothelium-dependent relaxation; ↓ Nox2, Nox4. | Diabetic retinopathy | [101] |
| Mangiferin | ↑ TXNIP, NLRP3 inflammasome, IL-1β, IL-6, NO. | Diabetic vasculopathy | [103] |
| nicorandil | ↑ insulin resistance; ↓ PERK inhibition. | Diabetes | [62] |
| Kaempferol | ↑ caspase-3, caspase-7; ↓ GRP78, CHOP. | Atherosclerosis | [112] |
| Atrovastatin | ↑ AMPK. | Atherosclerosis | [113] |
| hydrogen sulfide | chemical chaperones. | Pulmonary arterial hypertension | [115] |
| intermedin | chemical chaperones. | Pulmonary arterial hypertension | [131] |
| 4u8c | chemical chaperones;↓ IRE1α/XBP1 inhibition. | Pulmonary arterial hypertension | [132] |
| black tea | ↓ ER stress, oxidative stress, blood pressure, endothelial dysfunction. | Hyperhomocysteinemia | [120] |
| Enalapril | ↓ ER stress, blood pressure. | Hyperhomocysteinemia | [148] |
| Piceatannol | ↓ apoptosis, oxidative stress, ER stress. | Hyperhomocysteinemia | [149] |
| Atorvastatin | ↑ AMPK; ↓ ER stress. | Hyperhomocysteinemia | [150,151] |
| salidroside | ↓ Bip, CHOP, PERK phosphorylation, IRE1α phosphorylation. | Hyperhomocysteinemia | [152] |
| Alpha-lipoic acid | ↓ ER stress, oxidative stress, apoptosis, inflammation. | Hyperhomocysteinemia | [153] |
| Exendin-4 | ↑ AMPK; ↓ ER stress, superoxide anion production. | Hyperhomocysteinemia | [154]. |
| Zinc finger protein ZBTB20 | ↓ TNFα, ASK1, JNK, NADPH oxidase. | Myocardial infarction | [161] |
| Valsartan | ↓ ASK1. | Myocardial infarction | [162] |
| Panax notoginseng flower | ↑ HIF-1, VEGFA, KDR, Bcl-2, Bax. | Myocardial infarction | [163] |
| Taurine | ↓ TF-6 and IRE-1. | Stroke | [169] |
| Apelin-36 | ↓ CHOP/GRP78. | Stroke | [170] |
| Basic fibroblast growth factor | ↑ PI3K/Akt, ERK1/2; ↓ CHOP, ATF6. | Stroke | [171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Murugan, D.D.; Khan, H.; Huang, Y.; Cheang, W.S. Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases. Antioxidants 2021, 10, 1167. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081167
Zhou Y, Murugan DD, Khan H, Huang Y, Cheang WS. Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases. Antioxidants. 2021; 10(8):1167. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081167
Chicago/Turabian StyleZhou, Yan, Dharmani Devi Murugan, Haroon Khan, Yu Huang, and Wai San Cheang. 2021. "Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases" Antioxidants 10, no. 8: 1167. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081167