
MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Isolation of Single Live FDB Muscle Fibers (Myofibers)
2.3. Confocal Imaging and Image Analysis
2.4. Imaging of T-Tubule Network Integrity and Mitochondrial Membrane Potential at NMJ
2.5. Evaluation of Mitochondrial ROS Level in Live FDB Myofibers
2.6. Evaluation of Membrane Repair Function in FDB Myofibers
2.7. Quantification of Membrane Integrity of Diaphragm Muscle
2.8. Immunohistochemistry of Mouse and Human Muscle
2.9. Immunoblotting Assay
2.10. Evaluation of Serum Creatine Kinase (CK) Activity
2.11. Quantification of Motor Neurons in Lumbar Spinal Cord with Nissl Staining
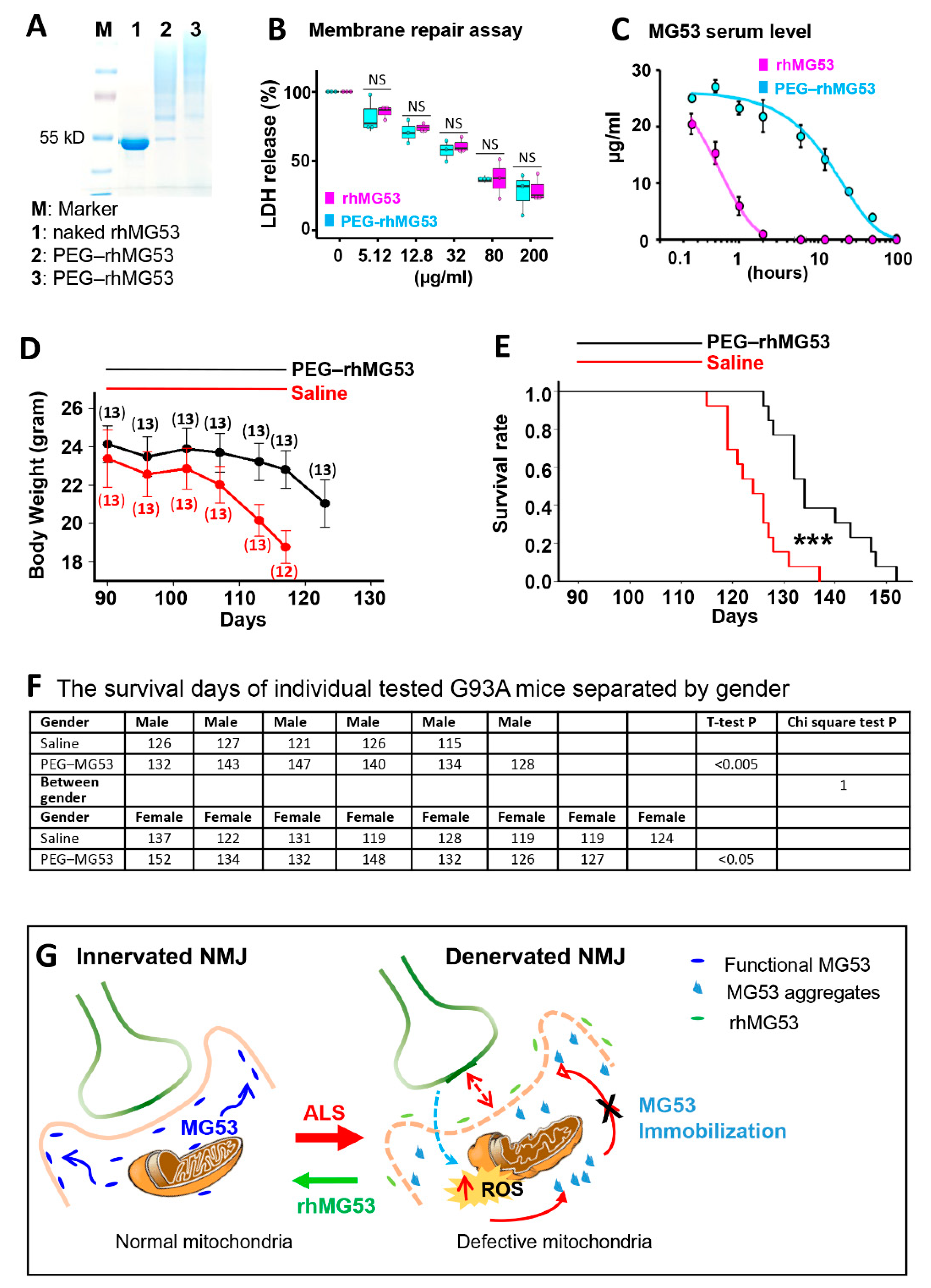
2.12. PEGylation of rhMG53
2.13. Pharmacokinetic Evaluation of PEG–rhMG53 in Rats
2.14. Intravenous Administration of rhMG53 and PEG–rhMG53
2.15. Numerical Data Presentation and Statistics
3. Results
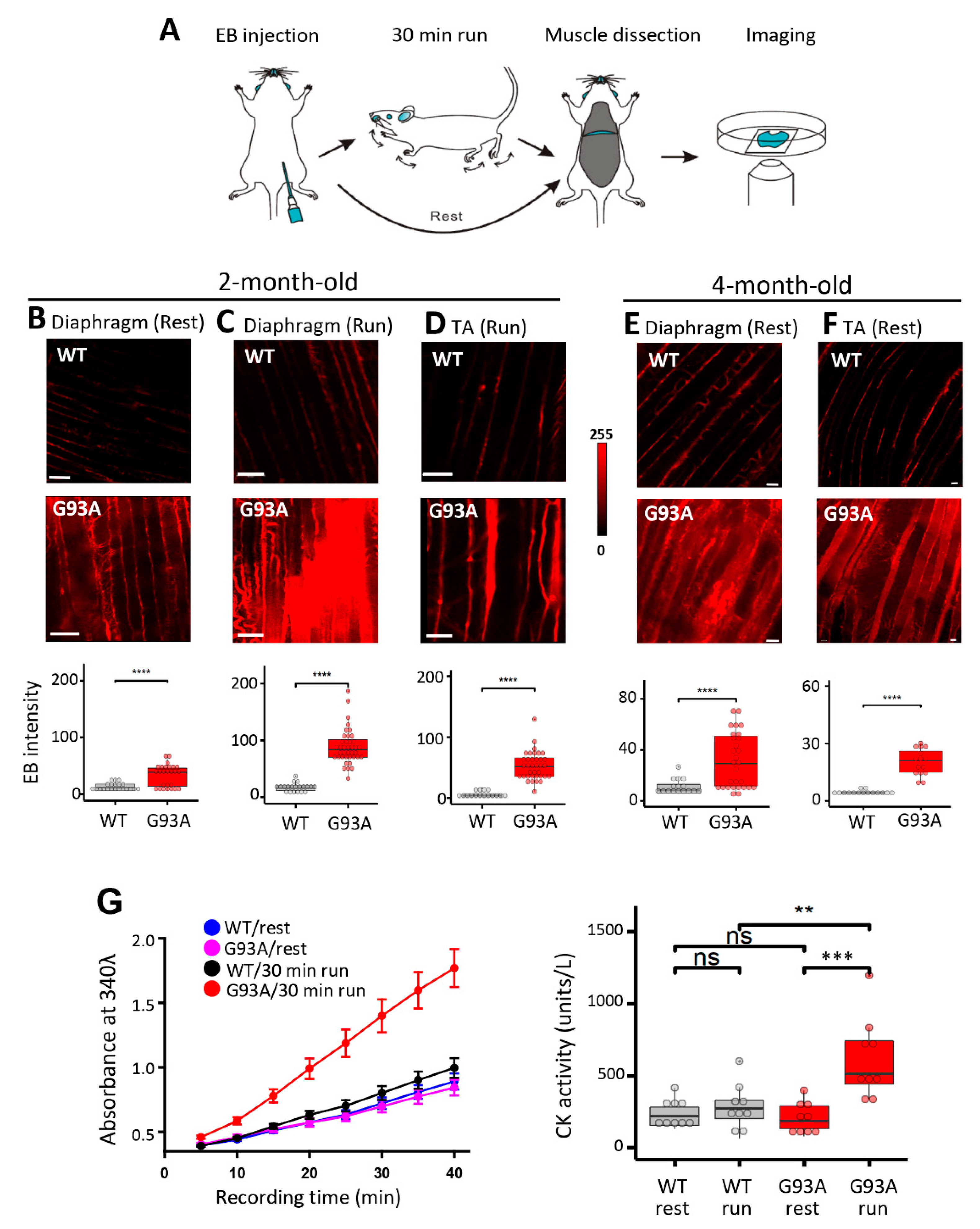
3.1. Increased Susceptibility to Diaphragm Injury Is an Early Pathology Event in SOD1(G93A) Mice
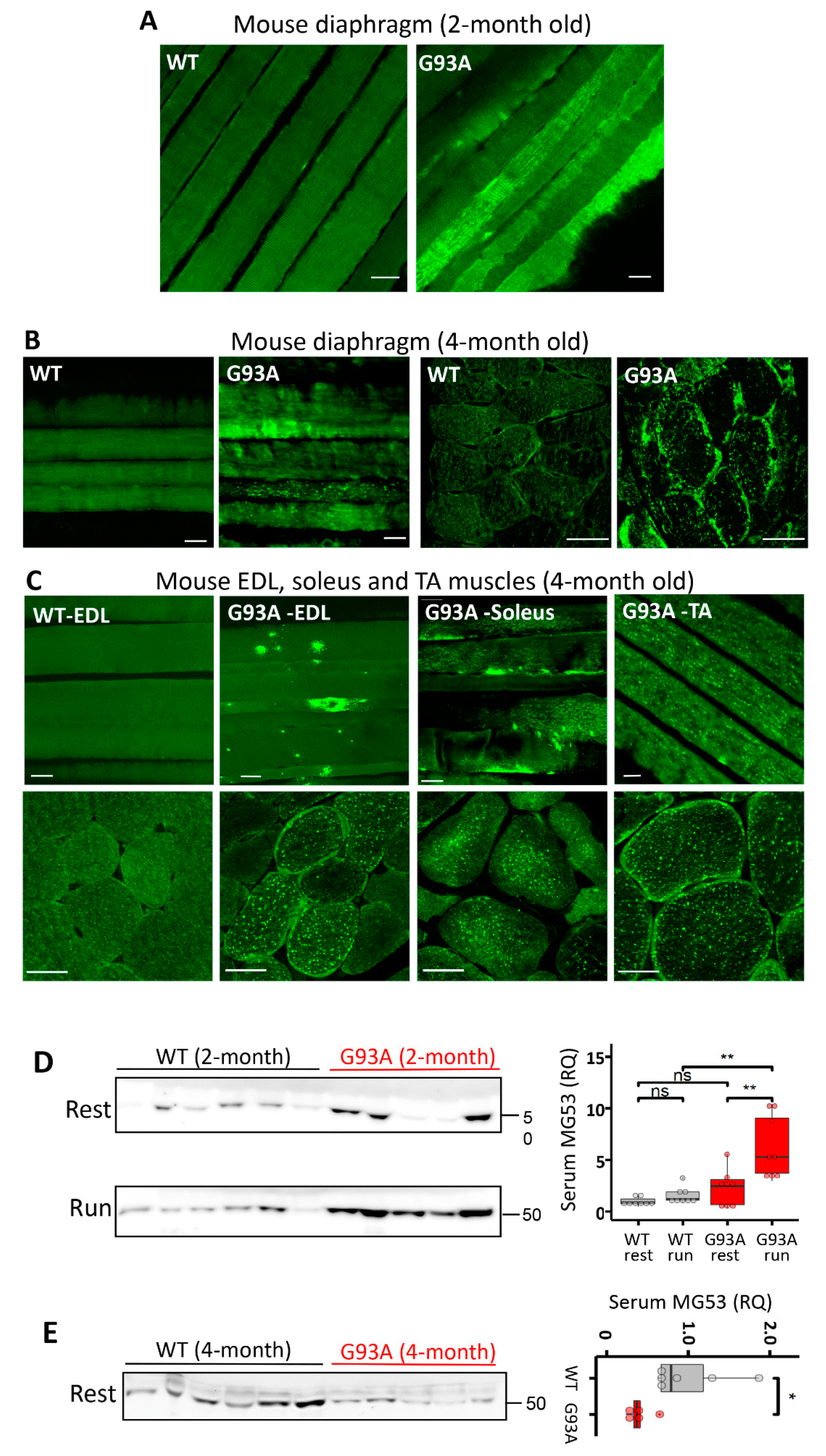
3.2. MG53 Is Implicated in the Membrane Fragility of G93A Diaphragm, Fast- and Slow-Type Muscles
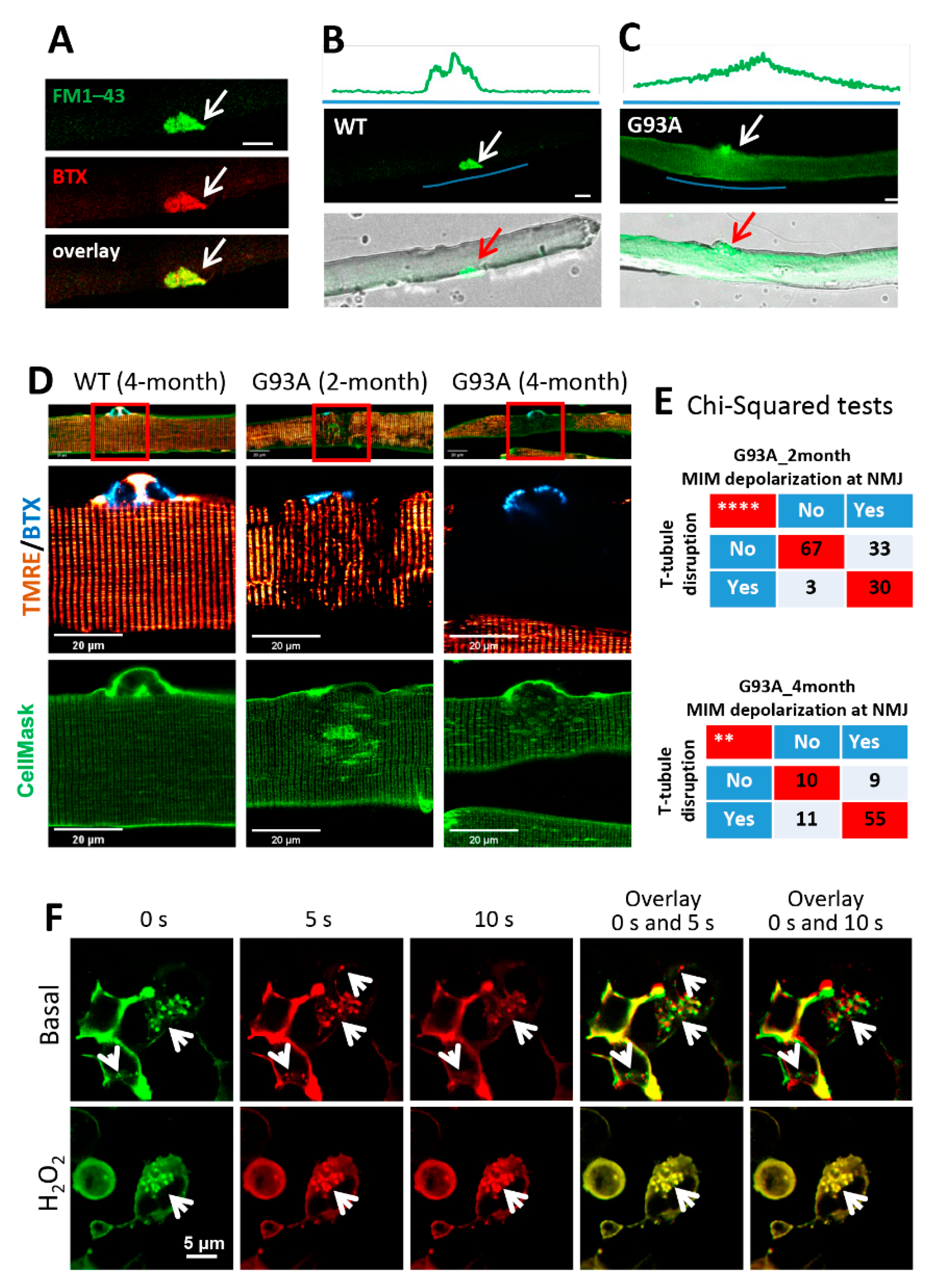
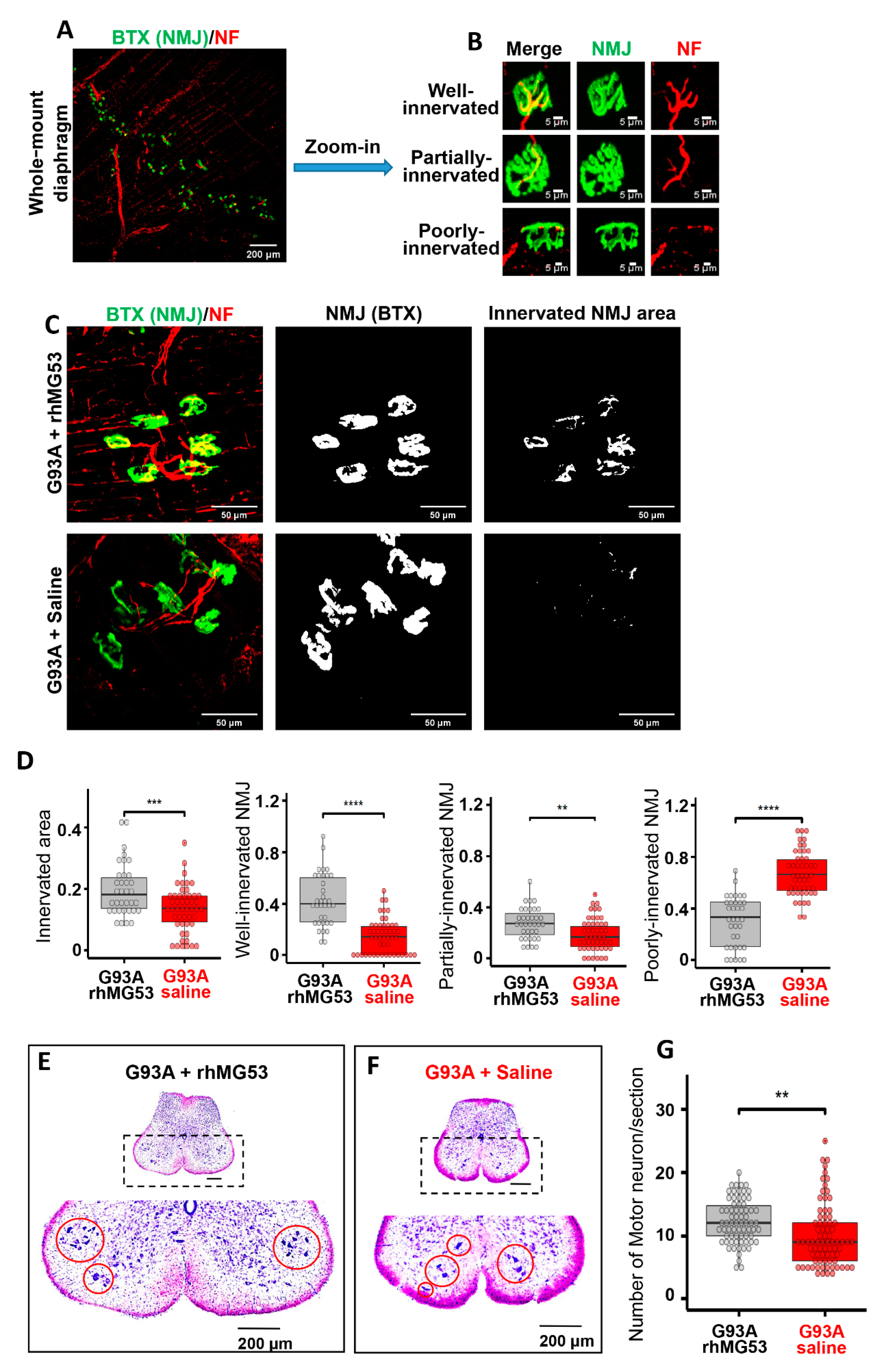
3.3. NMJ Is an Active Site of Injury Repair by MG53 that Is Lost in ALS
3.4. Mitochondrial Dysfunction Is Associated with the Disruption of Cell Membrane Integrity at NMJs of the ALS Muscle
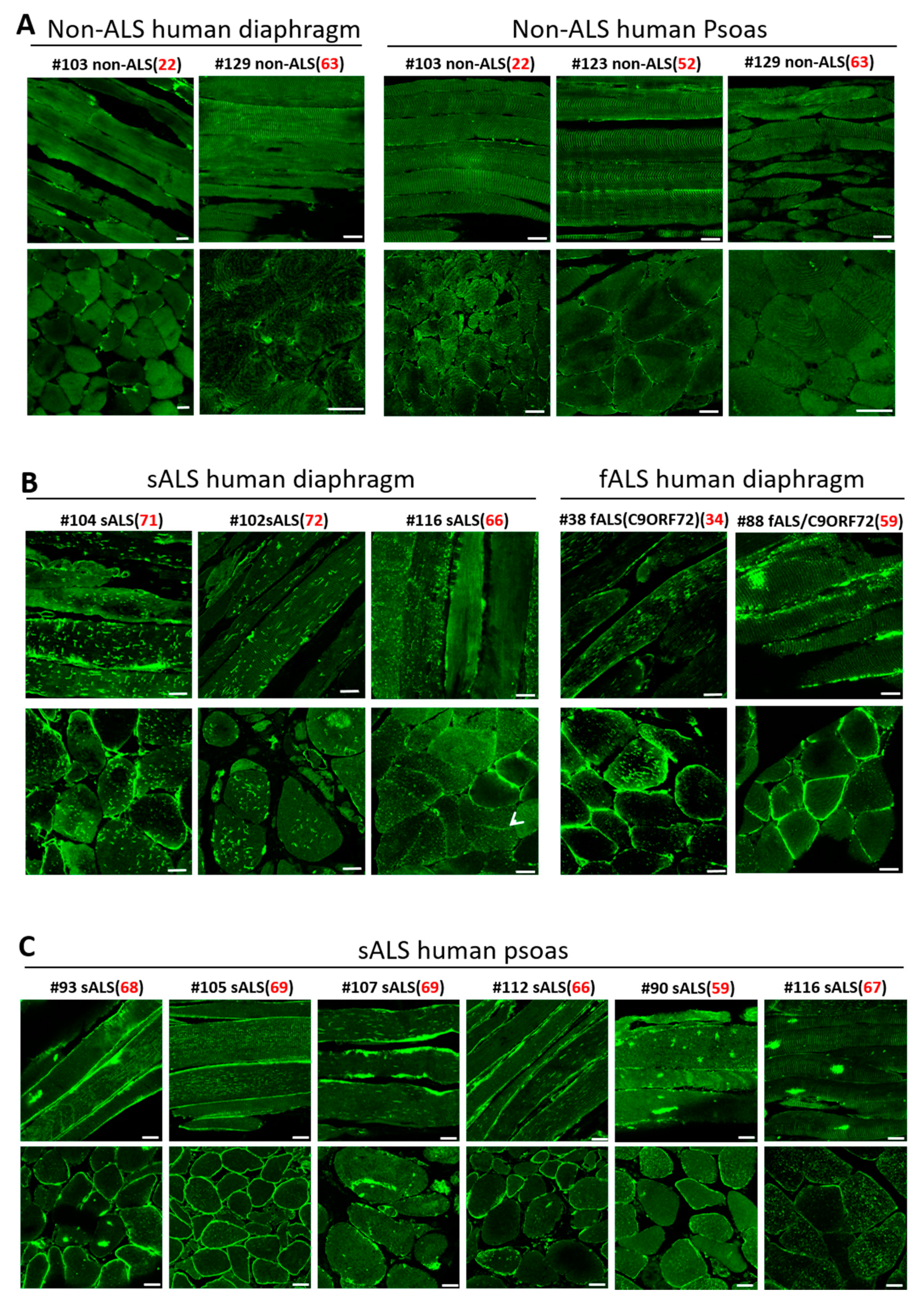
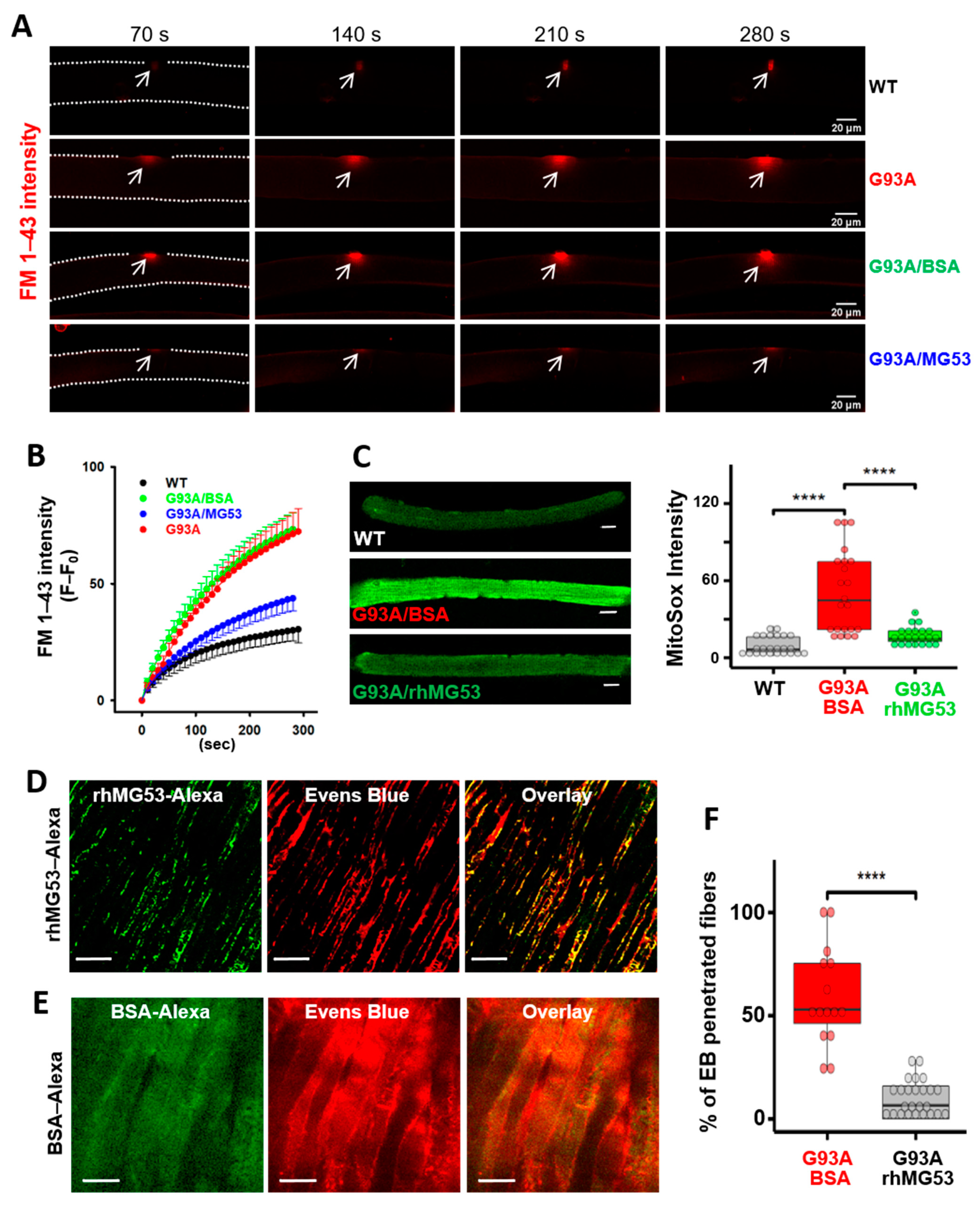
3.5. Impaired MG53 Membrane Repair Function Is a Common Pathological Feature of ALS Muscle
3.6. Recombinant Human MG53 Protein Preserves Membrane Integrity of Diaphragm in G93A Mice
3.7. Systemic Application of rhMG53 Preserves NMJ Integrity and Extends Life Span of G93A Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joyce, P.I.; Fratta, P.; Fisher, E.M.; Acevedo-Arozena, A. SOD1 and TDP-43 animal models of amyotrophic lateral sclerosis: Recent advances in understanding disease toward the development of clinical treatments. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2011, 22, 420–448. [Google Scholar] [CrossRef]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernan, M.A. Incidence and lifetime risk of motor neuron disease in the United Kingdom: A population-based study. Eur. J. Neurol. Off. J. Eur. Fed. Neurol. Soc. 2009, 16, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Swash, M.; Pinto, S. Diaphragmatic neurophysiology and respiratory markers in ALS. Front. Neurol. 2019, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [Green Version]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. MN 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef]
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musaro, A. Neuromuscular junction as an entity of nerve-muscle communication. Cells 2019, 8, 906. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A. Understanding ALS: New therapeutic approaches. FEBS J. 2013, 280, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.; Hruscha, A.; Hogl, S.; Banzhaf-Strathmann, J.; Strecker, K.; van der Zee, J.; Teucke, M.; Eimer, S.; Hegermann, J.; Kittelmann, M.; et al. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. USA 2013, 110, 4986–4991. [Google Scholar] [CrossRef] [Green Version]
- Cykowski, M.D.; Dickson, D.W.; Powell, S.Z.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Dipeptide repeat (DPR) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol. 2019, 138, 667–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef] [Green Version]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H.; et al. Absence of physiological Ca2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Rios, E.; Ma, J.; Zhou, J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lin, P.; De, G.; Choi, K.H.; Takeshima, H.; Weisleder, N.; Ma, J. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 2011, 286, 12820–12824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Zhang, Y.; Song, R.; Hwang, M.; Jin, L.; et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574. [Google Scholar] [CrossRef] [Green Version]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Chen, K.; Lin, P.; Lieber, G.; Nishi, M.; Yan, R.; Wang, Z.; Yao, Y.; Li, Y.; Whitson, B.A.; et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 2014, 5, 4387. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, H.; Zheng, Y.; Xu, Z.; Li, L.; Tan, T.; Park, K.H.; Hou, J.; Zhang, C.; Li, D.; et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell. Cardiol. 2015, 80, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra236. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Zhang, B.; Zhu, H.; Li, H.; Han, Y.; Chen, K.; Wang, Z.; Zeng, J.; Liu, Y.; Wang, X.; et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 2016, 7, 22474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S.; Royer, L.; Yi, J.; Brum, G.; Meissner, G.; Rios, E.; Zhou, J. Ca(2+) sparks operated by membrane depolarization require isoform 3 ryanodine receptor channels in skeletal muscle. Proc. Natl. Acad. Sci. USA 2007, 104, 5235–5240. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Wang, Q.; Zhou, X.; Tan, T.; Park, K.H.; Kramer, H.F.; McDougal, A.; Laping, N.J.; Kumar, S.; Adesanya, T.M.A.; et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat. Commun. 2019, 10, 4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, H.L.; Tan, T.; Yang, C.; Gemensky-Metzler, A.J.; Wehrman, R.F.; Jiang, Q.; Peterson, C.M.W.; Geng, B.; Zhou, X.; Wang, Q.; et al. MG53 promotes corneal wound healing and mitigates fibrotic remodeling in rodents. Commun. Biol. 2019, 2, 71. [Google Scholar] [CrossRef]
- Adesanya, T.M.A.; Russell, M.; Park, K.H.; Zhou, X.; Sermersheim, M.A.; Gumpper, K.; Koenig, S.N.; Tan, T.; Whitson, B.A.; Janssen, P.M.L.; et al. MG 53 protein protects aortic valve interstitial cells from membrane injury and fibrocalcific remodeling. J. Am. Heart Assoc. 2019, 8, e009960. [Google Scholar] [CrossRef] [Green Version]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N.; Stadler, J.; Tilbury, L.; Smith, D. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 9–16. [Google Scholar] [CrossRef] [Green Version]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Models Mech. 2019, 12, dmm037424. [Google Scholar] [CrossRef] [Green Version]
- McGoldrick, P.; Joyce, P.I.; Fisher, E.M.; Greensmith, L. Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2013, 1832, 1421–1436. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, R.; Nishikawa, A.; Tanaka, H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: Evidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 1995, 118, 959–964. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wang, L.; Yue, H.; Whitson, B.A.; Haggard, E.; Xu, X.; Ma, J. MG53, a tissue repair protein with broad applications in regenerative medicine. Cells 2021, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Miyake, K.; Vogel, S.S. The endomembrane requirement for cell surface repair. Proc. Natl. Acad. Sci. USA 2003, 100, 4592–4597. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, M.; Ko, J.K.; Weisleder, N.; Takeshima, H.; Ma, J. Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am. J. Physiol. Cell Physiol. 2011, 301, C106–C114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Liu, J.; Bian, Z.; Cui, Y.; Zhou, X.; Zhang, B.; Adesanya, T.M.; Yi, F.; Park, K.H.; Tan, T.; et al. Effect of metabolic syndrome on mitsugumin 53 expression and function. PLoS ONE 2015, 10, e0124128. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yi, J.; Li, X.; Xiao, Y.; Dhakal, K.; Zhou, J. ALS-associated mutation SOD1(G93A) leads to abnormal mitochondrial dynamics in osteocytes. Bone 2018, 106, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N’ Guessan, B.; Tranchant, C.; Loeffler, J.P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: A temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Napoli, L.; Crugnola, V.; Lamperti, C.; Silani, V.; Di Mauro, S.; Bresolin, N.; Moggio, M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1612–1613. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Siciliano, G.; Pastorini, E.; Pasquali, L.; Manca, M.L.; Iudice, A.; Murri, L. Impaired oxidative metabolism in exercising muscle from ALS patients. J. Neurol. Sci. 2001, 191, 61–65. [Google Scholar] [CrossRef]
- Soraru, G.; Vergani, L.; Fedrizzi, L.; D’ Ascenzo, C.; Polo, A.; Bernazzi, B.; Angelini, C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropathol. Appl. Neurobiol. 2007, 33, 204–211. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Campanari, M.L.; Bourefis, A.R.; Kabashi, E. Diagnostic challenge and neuromuscular junction contribution to ALS pathogenesis. Front. Neurol. 2019, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Cappello, V.; Francolini, M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph. Lateral Scler. 2010, 11, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Mei, L. Morphological analysis of neuromuscular junctions by immunofluorescent staining of whole-mount mouse diaphragms. Methods Mol. Biol. 2013, 1018, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Weisleder, N.; Takeshima, H.; Ma, J. Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Commun. Integr. Biol. 2009, 2, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, S.; Holtmann, B.; Sendtner, M.; Metzger, F. Therapeutic effects of PEGylated insulin-like growth factor I in the pmn mouse model of motoneuron disease. Exp. Neurol. 2011, 232, 261–269. [Google Scholar] [CrossRef]
- Basu, A.; Yang, K.; Wang, M.; Liu, S.; Chintala, R.; Palm, T.; Zhao, H.; Peng, P.; Wu, D.; Zhang, Z.; et al. Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug. Chem. 2006, 17, 618–630. [Google Scholar] [CrossRef]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Kang, J.S.; Deluca, P.P.; Lee, K.C. Emerging PEGylated drugs. Expert Opin. Emerg. Drugs 2009, 14, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Duann, P.; Lin, P.H.; Zhao, L.; Fan, Z.; Tan, T.; Zhou, X.; Sun, M.; Fu, M.; Orange, M.; et al. Modulation of wound healing and scar formation by MG53 protein-mediated cell membrane repair. J. Biol. Chem. 2015, 290, 24592–24603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.S.; Park, J.S.; Ham, Y.M.; Nguyen, N.; Lee, N.R.; Hong, J.; Kim, B.W.; Lee, H.; Lee, C.S.; Jeong, B.C.; et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat. Commun. 2013, 4, 2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzova-Davis, S.; Haller, E.; Saporta, S.; Kolomey, I.; Nicosia, S.V.; Sanberg, P.R. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 2007, 1157, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Shefner, J.M. Effects of strength training in amyotrophic lateral sclerosis: How much do we know? Muscle Nerve 2019, 59, 6–7. [Google Scholar] [CrossRef] [Green Version]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The role of exercise as a non-pharmacological therapeutic approach for amyotrophic lateral sclerosis: Beneficial or detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Di, P.W.C.; Di, P.S.G.C. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): A multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015, 14, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Bermejo, J.; Morelot-Panzini, C.; Tanguy, M.L.; Meininger, V.; Pradat, P.F.; Lenglet, T.; Bruneteau, G.; Forestier, N.L.; Couratier, P.; Guy, N.; et al. Early diaphragm pacing in patients with amyotrophic lateral sclerosis (RespiStimALS): A randomised controlled triple-blind trial. Lancet Neurol. 2016, 15, 1217–1227. [Google Scholar] [CrossRef]
- McDermott, C.J.; Bradburn, M.J.; Maguire, C.; Cooper, C.L.; Baird, W.O.; Baxter, S.K.; Cohen, J.; Cantrill, H.; Dixon, S.; Ackroyd, R.; et al. DiPALS: Diaphragm pacing in patients with amyotrophic lateral sclerosis—A randomised controlled trial. Health Technol. Assess. 2016, 20, 1–186. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.G.; Lewis, R.A. Diaphragm pacing in patients with amyotrophic lateral sclerosis. Lancet Neurol. 2016, 15, 542. [Google Scholar] [CrossRef] [Green Version]
- Wood, H. Motor neuron disease: Diaphragm pacing is associated with reduced survival in ALS patients with respiratory insufficiency. Nat. Rev. Neurol. 2015, 11, 484. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, J.; Li, A.; Li, X.; Park, K.; Zhou, X.; Yi, F.; Xiao, Y.; Yoon, D.; Tan, T.; Ostrow, L.W.; et al. MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression. Antioxidants 2021, 10, 1522. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10101522
Yi J, Li A, Li X, Park K, Zhou X, Yi F, Xiao Y, Yoon D, Tan T, Ostrow LW, et al. MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression. Antioxidants. 2021; 10(10):1522. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10101522
Chicago/Turabian StyleYi, Jianxun, Ang Li, Xuejun Li, Kiho Park, Xinyu Zhou, Frank Yi, Yajuan Xiao, Dosuk Yoon, Tao Tan, Lyle W. Ostrow, and et al. 2021. "MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression" Antioxidants 10, no. 10: 1522. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10101522