
Exploring the Excited-State Nonadiabatic Effects in the Semisaturated Planar Tetracoordinated Carbon Molecule C7H4

 , and
, and
Abstract
:1. Introduction
2. Theoretical Framework
2.1. Electronic Structure Calculations
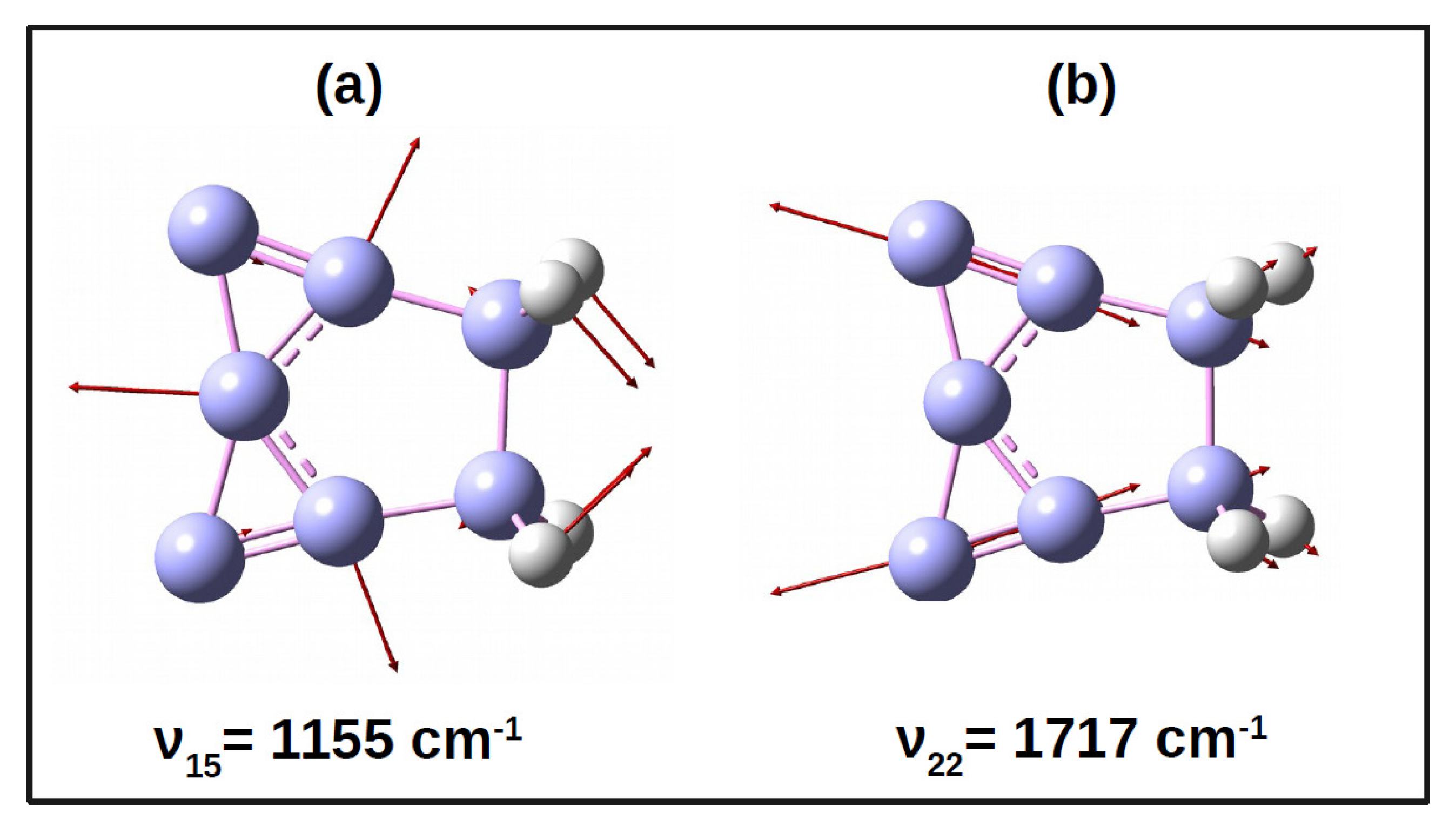
2.2. Vibronic Hamiltonian
2.3. Dynamics Simulations
3. Results and Discussion
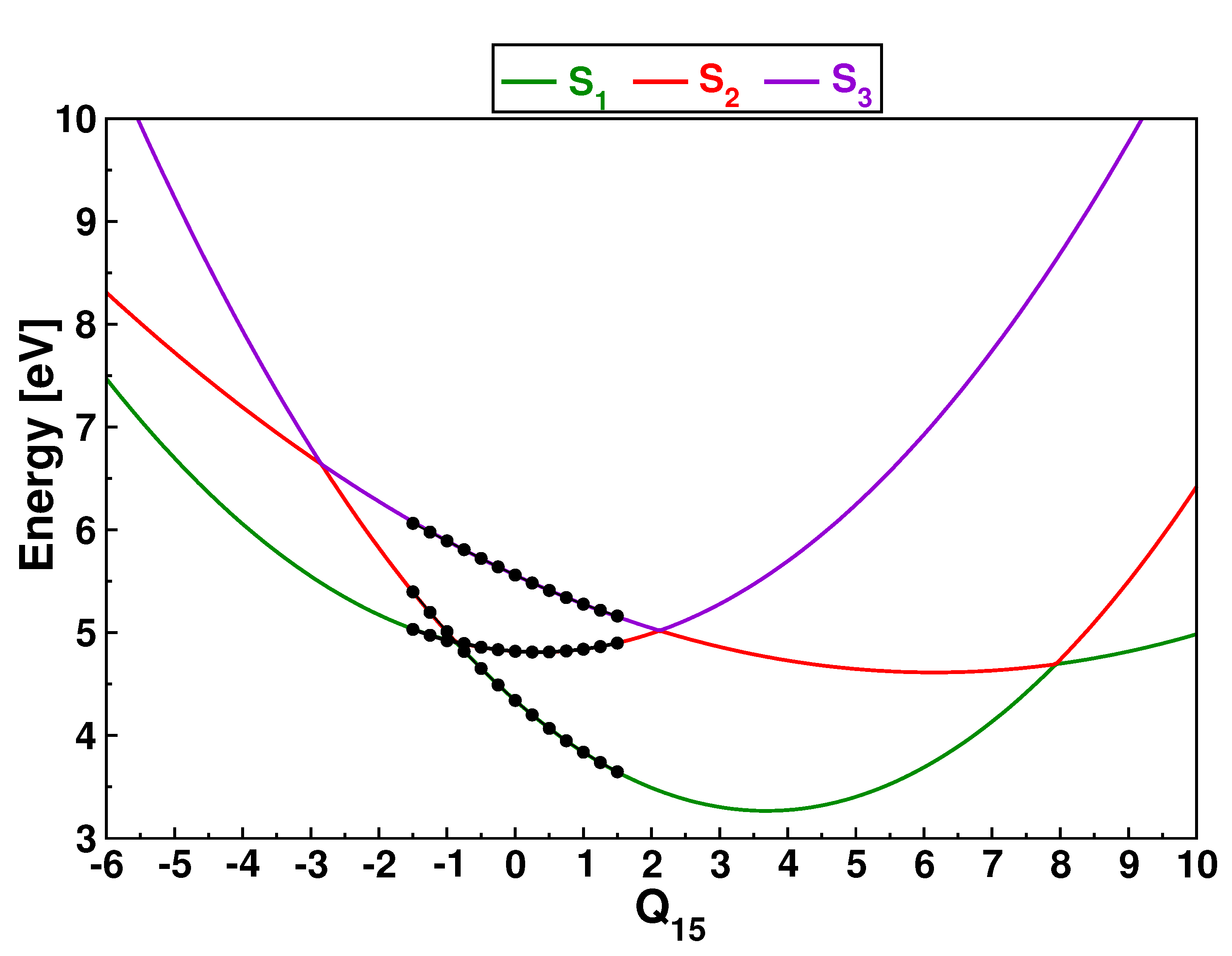
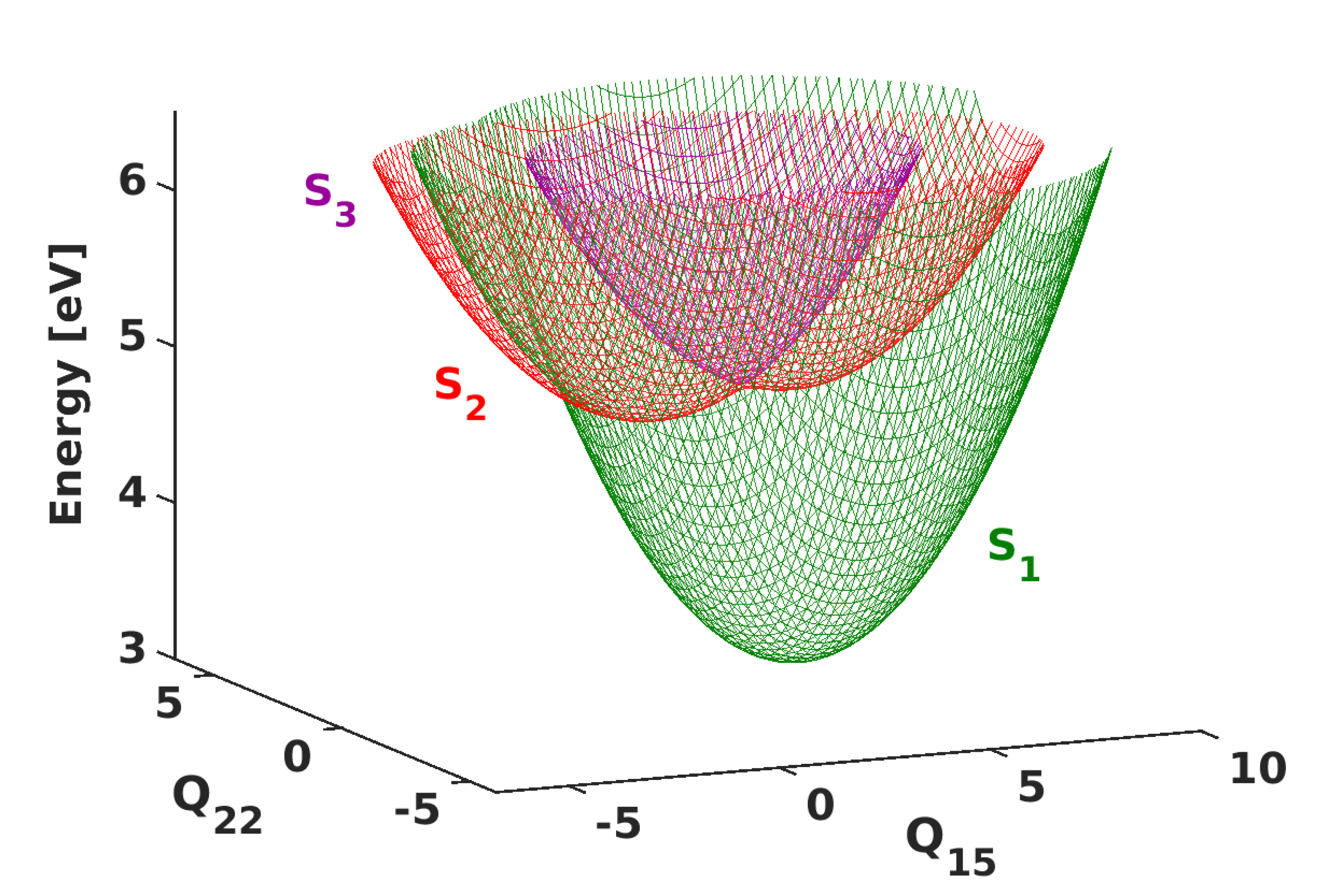
3.1. PESs and Conical Intersections
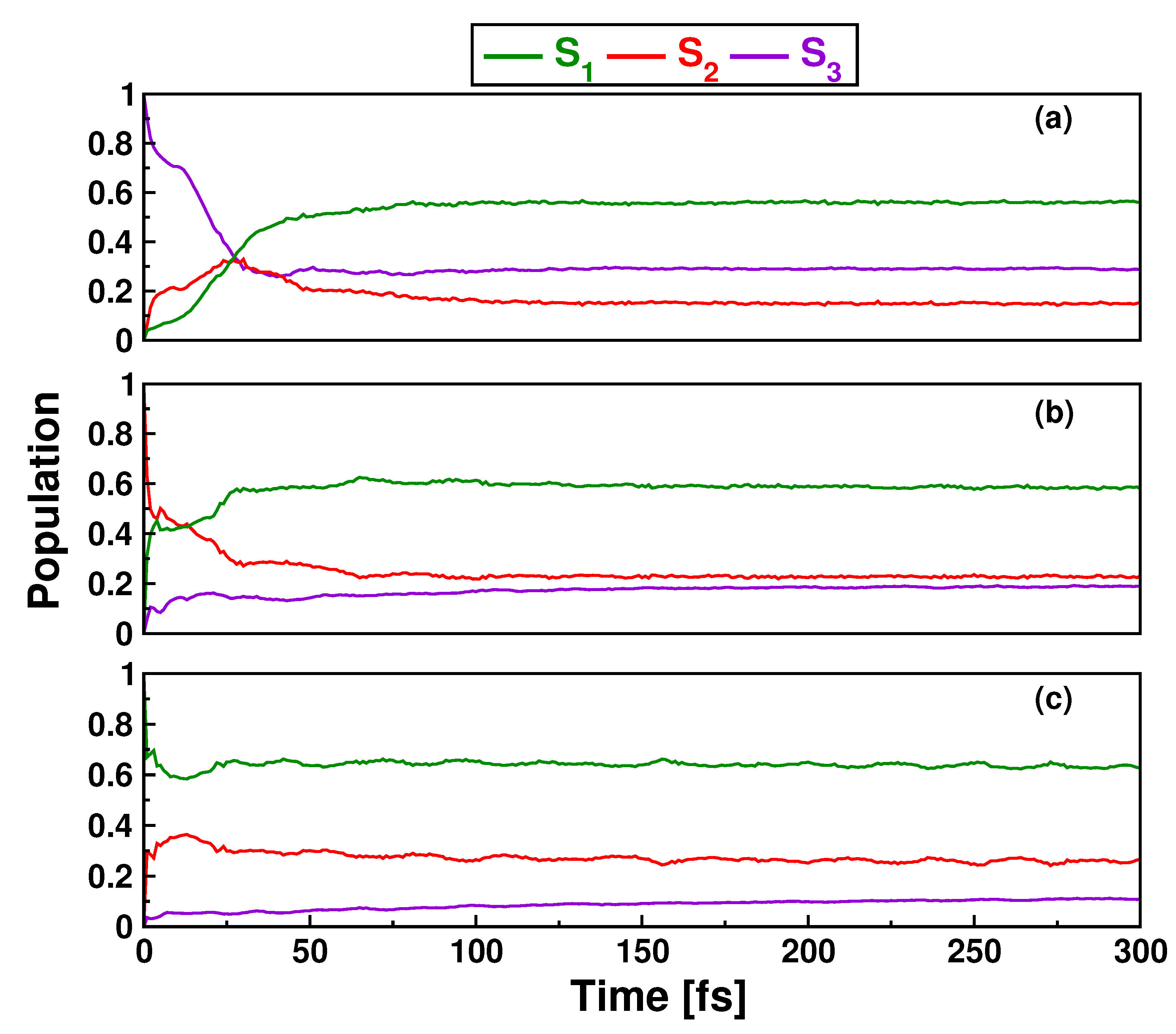
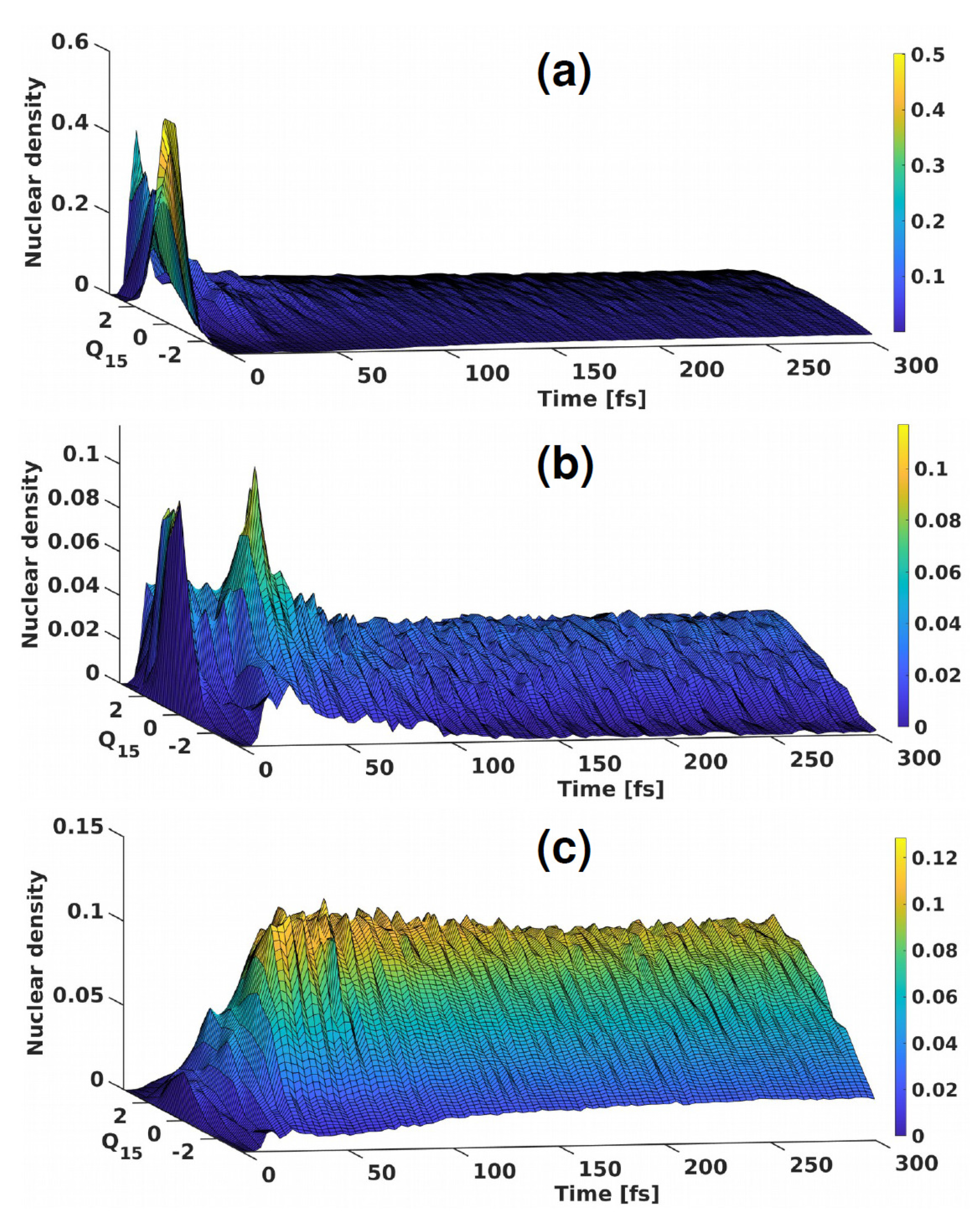
3.2. Singlet Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ptC | Planar tetracoordinated carbon |
| PES | Potential energy surface |
| QVC | Quadratic vibronic coupling |
| MCTDH | Multiconfiguration time-dependent hartree |
| TDDFT | Time-dependent density functional theory |
| EOM-CCSD | Equation of motion coupled cluster with single and double excitations |
| RI-CC2 | Resolution-of-the-identity second-order approximate coupled-cluster |
| singles and doubles | |
| ADC(2) | Algebraic diagrammatic construction method to second-order |
| FC | Franck-Condon |
| MECI | Minimum energy conical intersection |
References
- van’t Hoff, J.H. A suggestion looking to the extension into space of the structural formulas at present used in chemistry, and a note upon the relation between the optical activity and the chemical constitution of organic compounds. Arch. Neerl. Sci. Exact Nat. 1874, 9, 445–454. [Google Scholar]
- Le Bel, J. On the relations which exist between the atomic formulas of organic compounds and the rotatory power of their solutions. Bull. Soc. Chim. 1874, 22, 337–347. [Google Scholar]
- Monkhorst, H.J. Activation energy for interconversion of enantiomers containing an asymmetric carbon atom without breaking bonds. Chem. Commun. 1968, 11, 1111–1112. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar tetracoordinate carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Hoffmann, R. The theoretical design of novel stabilized systems. Pure Appl. Chem. 1971, 28, 181–194. [Google Scholar] [CrossRef]
- McGrath, M.P.; Radom, L. Alkaplanes: A class of neutral hydrocarbons containing a potentially planar tetracoordinate carbon. J. Am. Chem. Soc. 1993, 115, 3320–3321. [Google Scholar] [CrossRef]
- Lyons, J.E.; Rasmussen, D.R.; McGrath, M.P.; Nobes, R.H.; Radom, L. Octaplane: A saturated hydrocarbon with a remarkably low ionization energy leading to a cation with a planar tetracoordinate carbon atom. Angew. Chem. Int. Ed. Engl. 1994, 33, 1667–1668. [Google Scholar] [CrossRef]
- Sorger, K.; Schleyer, P.v.R. Planar and inherently non-tetrahedral tetracoordinate carbon: A status report. J. Mol. Struct. Theochem 1995, 338, 317–346. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chem. Int. Ed. Engl. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Radom, L.; Rasmussen, D.R. The planar carbon story. Pure Appl. Chem. 1998, 70, 1977–1984. [Google Scholar] [CrossRef]
- Siebert, W.; Gunale, A. Compounds containing a planar-tetracoordinate carbon atom as analogues of planar methane. Chem. Soc. Rev. 1999, 28, 367–371. [Google Scholar] [CrossRef]
- Rasmussen, D.R.; Radom, L. Planar-Tetracoordinate Carbon in a Neutral Saturated Hydrocarbon: Theoretical Design and Characterization. Angew. Chem. Int. Ed. 1999, 38, 2875–2878. [Google Scholar] [CrossRef]
- Rasmussen, D.R.; Radom, L. Hemispiroalkaplanes: Hydrocarbon Cage Systems with a Pyramidal-Tetracoordinate Carbon Atom and Remarkable Basicity. Chem. A Eur. J. 2000, 6, 2470–2483. [Google Scholar] [CrossRef]
- Priyakumar, U.D.; Sastry, G.N. A system with three contiguous planar tetracoordinate carbons is viable: A computational study on a C6H62= isomer. Tetrahedron Lett. 2004, 45, 1515–1517. [Google Scholar] [CrossRef]
- Perez, N.; Heine, T.; Barthel, R.; Seifert, G.; Vela, A.; Mendez-Rojas, M.A.; Merino, G. Planar tetracoordinate carbons in cyclic hydrocarbons. Org. Lett. 2005, 7, 1509–1512. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Vela, A.; Heine, T. Recent advances in planar tetracoordinate carbon chemistry. J. Comput. Chem. 2007, 28, 362–372. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated planar carbon in the Al4C-anion. A combined photoelectron spectroscopy and ab initio study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.; Wang, L.; Geske, G.; Boldyrev, A. ZUSCHRIFTEN-Pentaatomic Tetracoordinate Planar Carbon,(CAl4) 2-: A New Structural Unit and Its Salt Complexes Stichworter: Ab-initio-Rechnungen. Aluminium. Charge-Transfer Koordinationschemie. Angew.-Chem.-Ger. Ed. 2000, 112, 3776–3778. [Google Scholar] [CrossRef]
- Wang, L.S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental observation of pentaatomic tetracoordinate planar carbon-containing molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Li, X.; Zhai, H.J.; Wang, L.S. Photoelectron spectroscopy of pentaatomic tetracoordinate planar carbon molecules: CAl3Si- and CAl3Ge-. Chem. Phys. Lett. 2002, 357, 415–419. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Yu, S.; Ding, Y.h.; Bowen, K.H. Identifying the Hydrogenated Planar Tetracoordinate Carbon: A Combined Experimental and Theoretical Study of CAl4H and CAl4H–. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Peralta, N.; Sanchez, M.; Martin-Polo, J.; Islas, R.; Vela, A.; Merino, G. Planar Tetracoordinate Carbons in Cyclic Semisaturated Hydrocarbons. J. Org. Chem. 2008, 73, 7037–7044. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Comparative DFT Study of van der Waals Complexes: Rare-Gas Dimers, Alkaline-Earth Dimers, Zinc Dimer, and Zinc-Rare-Gas Dimers. J. Phys. Chem. A 2006, 110, 5121–5129. [Google Scholar] [CrossRef]
- Sekino, H.; Bartlett, R.J. A linear response, coupled-cluster theory for excitation energy. Int. J. Quantum Chem. 1984, 26, 255–265. [Google Scholar] [CrossRef]
- Stanton, J.F.; Bartlett, R.J. The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties. J. Chem. Phys. 1993, 98, 7029–7039. [Google Scholar] [CrossRef]
- Hättig, C.; Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. [Google Scholar] [CrossRef]
- Trofimov, A.; Krivdina, I.; Weller, J.; Schirmer, J. Algebraic-diagrammatic construction propagator approach to molecular response properties. Chem. Phys. 2006, 329, 1–10. [Google Scholar] [CrossRef]
- TURBOMOLE V7.4 2019, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 11 August 2021).
- Domcke, W.; Yarkony, D.R.; Köppel, H. Conical Intersections; World Scientific: Singapore, 2004. [Google Scholar] [CrossRef]
- Meyer, H.D.; Manthe, U.; Cederbaum, L. The multi-configurational time-dependent Hartree approach. Chem. Phys. Lett. 1990, 165, 73–78. [Google Scholar] [CrossRef]
- Manthe, U.; Meyer, H.; Cederbaum, L.S. Wave-packet dynamics within the multiconfiguration Hartree framework: General aspects and application to NOCl. J. Chem. Phys. 1992, 97, 3199–3213. [Google Scholar] [CrossRef]
- Beck, M.; Jäckle, A.; Worth, G.; Meyer, H.D. The multiconfiguration time-dependent Hartree (MCTDH) method: A highly efficient algorithm for propagating wavepackets. Phys. Rep. 2000, 324, 1–105. [Google Scholar] [CrossRef]
- Worth, G.A.; Beck, M.H.; Jäckle, A.; Meyer, H.D. The MCTDH Package, Version 8.5. 2019. Available online: http://mctdh.uni-hd.de (accessed on 21 April 2021).
- Köppel, H.; Domcke, W.; Cederbaum, L.S. Multimode Molecular Dynamics Beyond the Born-Oppenheimer Approximation. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Amsterdam, The Netherlands, 1984; pp. 59–246. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | S | S | S |
|---|---|---|---|
| TD-B97XD | 4.3417 (B, 0.001) | 4.8192 (B, 0.043) | 5.5606 (B, 0.049) |
| TD-B3LYP | 4.0221 (B, 0.005) | 4.5325 (B, 0.032) | 5.0136 (A, 0.010) |
| TD-CAMB3LYP | 4.3389 (B, 0.001) | 4.7960 (B, 0.045) | 5.5310 (B, 0.045) |
| TD-LC-PBE | 4.5102 (B, 0.000) | 4.9985 (B, 0.050) | 5.7237 (B, 0.037) |
| TD-M06-2X | 4.2035 (B, 0.000) | 4.5731 (B, 0.039) | 5.4983 (A, 0.004) |
| EOM-CCSD | 4.3121 (B, 0.002) | 4.9658 (B, 0.032) | 5.5197 (B, 0.031) |
| ADC(2) | 4.3239 (B, 0.003) | 4.9202 (B, 0.030) | 5.4091 (B ,0.042) |
| RICC2 | 4.2634 (B, 0.004) | 4.8697 (B, 0.027) | 5.3844 (B, 0.046) |
| Stationary Point | Energy (eV) |
|---|---|
| S | 3.6039 |
| S | 4.7940 |
| S | 4.3006 |
| S/S | 4.7970 |
| S/S | 5.6376 |
| S/S | 5.0552 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayakumari, C.M.; Nag, P.; Isukapalli, S.V.K.; Vennapusa, S.R. Exploring the Excited-State Nonadiabatic Effects in the Semisaturated Planar Tetracoordinated Carbon Molecule C7H4. Atoms 2022, 10, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms10010010
Jayakumari CM, Nag P, Isukapalli SVK, Vennapusa SR. Exploring the Excited-State Nonadiabatic Effects in the Semisaturated Planar Tetracoordinated Carbon Molecule C7H4. Atoms. 2022; 10(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms10010010
Chicago/Turabian StyleJayakumari, Chithra Mohan, Probal Nag, Sai Vamsi Krishna Isukapalli, and Sivaranjana Reddy Vennapusa. 2022. "Exploring the Excited-State Nonadiabatic Effects in the Semisaturated Planar Tetracoordinated Carbon Molecule C7H4" Atoms 10, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms10010010