
Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact
by
 ,
,
Mehdi Ayouz
1,*, 
Alexandre Faure
2,
Jonathan Tennyson
3,
Maria Tudorovskaya
4 and
Viatcheslav Kokoouline
5,*
1
LGPM, CentraleSupélec, Université Paris-Saclay, 8-10 rue Joliot-Curie, F-91190 Gif-sur-Yvette, France
2
IPAG, CNRS, Université Grenoble Alpes, F-38000 Grenoble, France
3
Department of Physics and Astronomy, University College London, Gower Street, London WC1E 6BT, UK
4
Quantemol Ltd., 320 City Rd., The Angel, London EC1V 2NZ, UK
5
Department of Physics, University of Central Florida, Orlando, FL 32816, USA
*
Authors to whom correspondence should be addressed.
Atoms 2021, 9(3), 62; https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030062
Submission received: 11 August 2021
/
Revised: 25 August 2021
/
Accepted: 26 August 2021
/
Published: 6 September 2021
(This article belongs to the Special Issue Electron Scattering in Gases –from Cross Sections to Plasma Modeling)
Abstract
:Cross-sections and thermally averaged rate coefficients for vibration (de-)excitation of a water molecule by electron impact are computed; one and two quanta excitations are considered for all three normal modes. The calculations use a theoretical approach that combines the normal mode approximation for vibrational states of water, a vibrational frame transformation employed to evaluate the scattering matrix for vibrational transitions and the UK molecular R-matrix code. The interval of applicability of the rate coefficients is from 10 to 10,000 K. A comprehensive set of calculations is performed to assess uncertainty of the obtained data. The results should help in modelling non-LTE spectra of water in various astrophysical environments.
1. Introduction
The water molecule is fundamental in a variety of research fields, such as biochemistry, meteorology and astrophysics. On Earth, water exists in all three phases (gas, liquid and solid), and life as we know it would not be possible without liquid water. Water is also ubiquitous in astronomical environments, from the Solar System to distant galaxies, where it is observed in both gaseous and solid forms (see ref. [1] for a review). Collisions between free electrons and water molecules thus play an important role in molecular environments as diverse as biological systems, cometary atmospheres and stellar envelopes.
Electron-HO collisions have been extensively studied for many years, both theoretically and experimentally (for a recent review see ref. [2]). Vibrationally elastic and inelastic cross-sections have been measured and computed, and the agreement between experiment and theory is generally good. Rotational and vibrational excitation is dominated by dipole-allowed and transitions, respectively, except possibly in the presence of resonances. We note, however, that cross-sections for individual rotational transitions (vibrationally elastic or inelastic) have not been measured so far. As a result, the best available cross-sections for rotational excitation are those computed by Machado et al. [3] for energies above 7 eV and those of Faure et al. [4] for lower energies, as recommended by refs. [2,5]. Because experiments can hardly distinguish between the two stretching excitations (symmetric and asymmetric) of water, vibrational measurements usually provide cross-sections for bending excitation (010) and for the sum of the two stretching excitations (100) and (001) (in normal mode notations). From their compilation of literature data, Song et al. [2] recommend the experimental vibrational cross-sections obtained by Khakoo et al. [6] for energies above 3 eV and those of Send and Linder [7] for lower energies. The most accurate theoretical data are the cross-sections of ref. [8] obtained by combining the vibrational coupled-channel theory with an interaction potential described as a sum of electrostatic, electron exchange and polarization contributions. The agreement with measurements is generally good for the bending mode but the combined stretching-mode cross-section is about a factor of two smaller than the experimental data for energies below 10 eV. We note that the theoretical data of refs. [4,8] were used by Faure and Josselin [9] to derive rate coefficients in the temperature range of 200–5000 K for use in astrophysical models.
In all previous experimental and theoretical studies, only dipole-allowed vibrational transitions were reported. In the envelopes of giant stars, however, water has been observed in high-energy rotational transitions within several vibrational states, i.e., (010), (100), (001) and (020) [10]. Such environments are not in local thermodynamic equilibrium (LTE), and the observed spectra contain precious information about local physical conditions. For example, one strong maser (microwave amplification by stimulated emission of radiation) transition at 268.149 GHz, arising from in the (020) vibrational state, was detected towards the evolved star VY CMa [11]. In order to extract information from such non-LTE spectra, cross-sections for one-quantum but also two-quantum transitions ( and 2) need to be computed. In addition, rovibrational state-to-state data are required. In all previous works for electron collisions with water, however, vibrational cross-sections were computed for one-quantum transitions only and without considering specific initial and final rotational states. It should be noted, in this context, that Stoecklin and co-workers have recently performed rovibrational state-to-state close-coupling calculations for the quenching of the bending mode (010) of water by (spherical) H [12] and helium atoms [13].
In the present work, new theoretical calculations for the vibrational (de-)excitation of water by electron-impact are performed using the R-matrix theory combined with the vibrational frame transformation. Similar preliminary calculations were presented in ref. [2]. Here, we provide, for the first time, cross-sections for two-quantum transitions and for all three vibrational modes. Rate coefficients are deduced, and simple fits are provided in the temperature range from 10 to 10,000 K for use in models. The theoretical approach is briefly introduced in the next section. The results are presented and discussed in Section 3. Conclusions are summarized in Section 4.
2. Theoretical Approach
The theoretical approach employed in this study is presented in detail in refs. [14,15,16,17]. Here, we sketch below only the main ideas.
2.1. Ab Initio Calculations
Our model employs the fixed-nuclei reactance matrix (K-matrix) obtained numerically using the UK molecular R-Matrix code (UKRMol) [18,19] with the Quantemol-N expert system [20]. The K-matrix for the HO collisions is computed for each geometry configuration of the molecule. It is labelled by the irreducible representations of the molecular point group. The ground-state electronic configuration of HO at its equilibrium geometry of the point group is
Performing the R-matrix calculations, we freeze the 2 core electrons and keep 8 electrons free in the active space of molecular orbitals. A total number of 508 configuration state functions (CSFs) are used for the above-ground state. All the generated states up to 10 eV were retained in the final close-coupling calculation. We employed an R-matrix sphere of radius 10 bohrs and a partial-wave expansion with continuum Gaussian-type orbitals up to .
Several basis sets, including DZP (double zeta-polarization contracted [21]) and cc-pVTZ (correlation-consistent polarized valence triple-zeta [22]) types, were tested to investigate the stability of the target properties, such as the dipole moment and ground state energy. Finally, we chose the cc-pVTZ basis set with the above complete active space (CAS) to perform the scattering calculations. In the following, this calculation will be referred as Model 1.
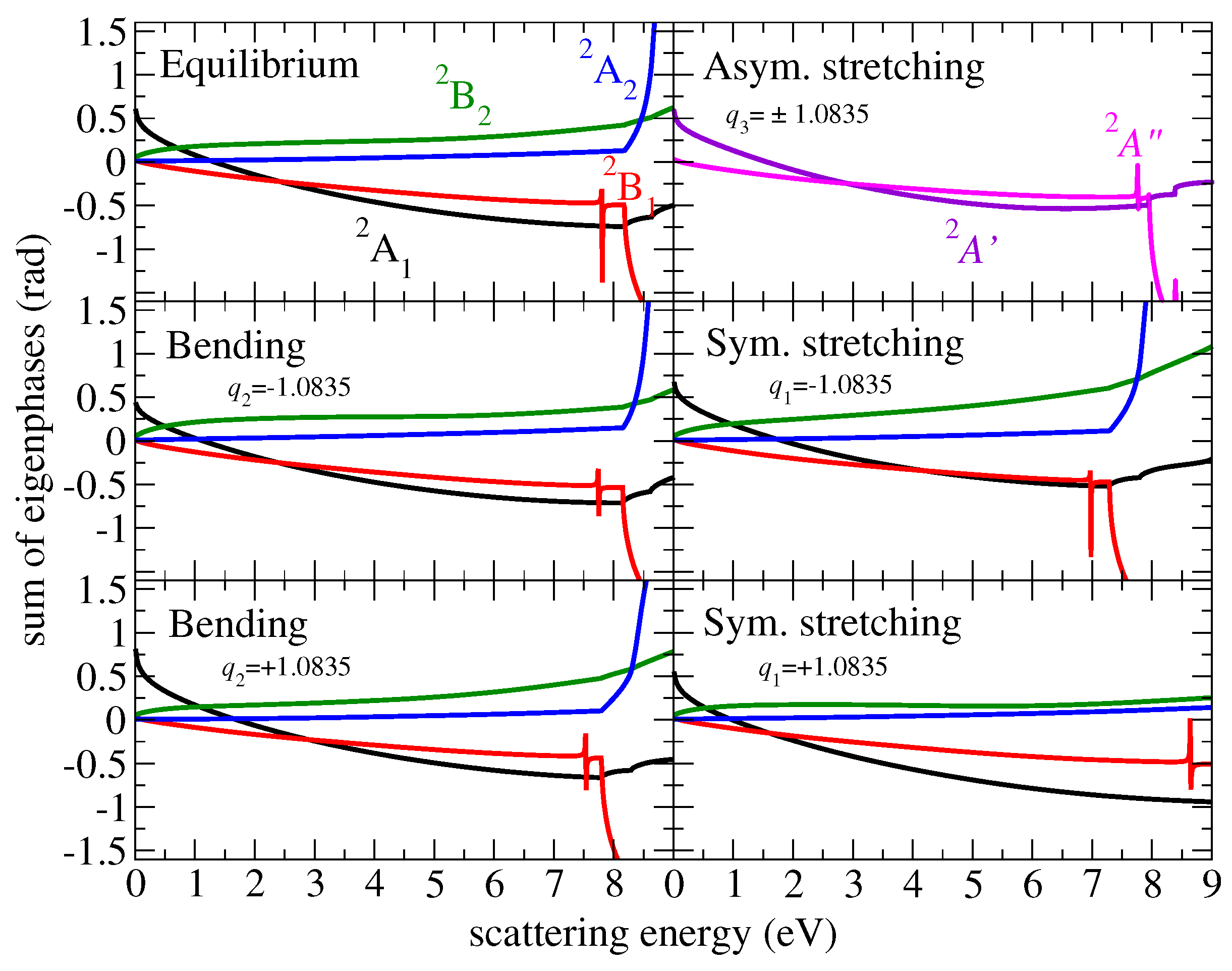
One of the important features of the present theoretical approach is the use of an energy-independent S-matrix. A convenient way to identify a weak or a strong energy dependence of the matrices is the eigenphase sum. Figure 1 displays the eigenphase sum of different irreducible representations at equilibrium and at displacements away from the equilibrium along each normal mode coordinate. Here and below, all normal coordinates are dimensionless. At equilibrium, the lowest resonance is found at 7.8 eV and has the symmetry.
To construct elements of the scattering matrix for transitions from one vibrational level v to another , which are then used to compute the cross−section, one needs the vibrational wave functions of the target molecule. At low collisional energies, the molecule can be characterized by three normal modes of vibration: bending, symmetric stretching and asymmetric stretching with respective frequencies , and and corresponding coordinates . In this study, for the vibrational motion of HO, we use the normal mode coordinates and vibrational wave functions in the normal-mode approximation.
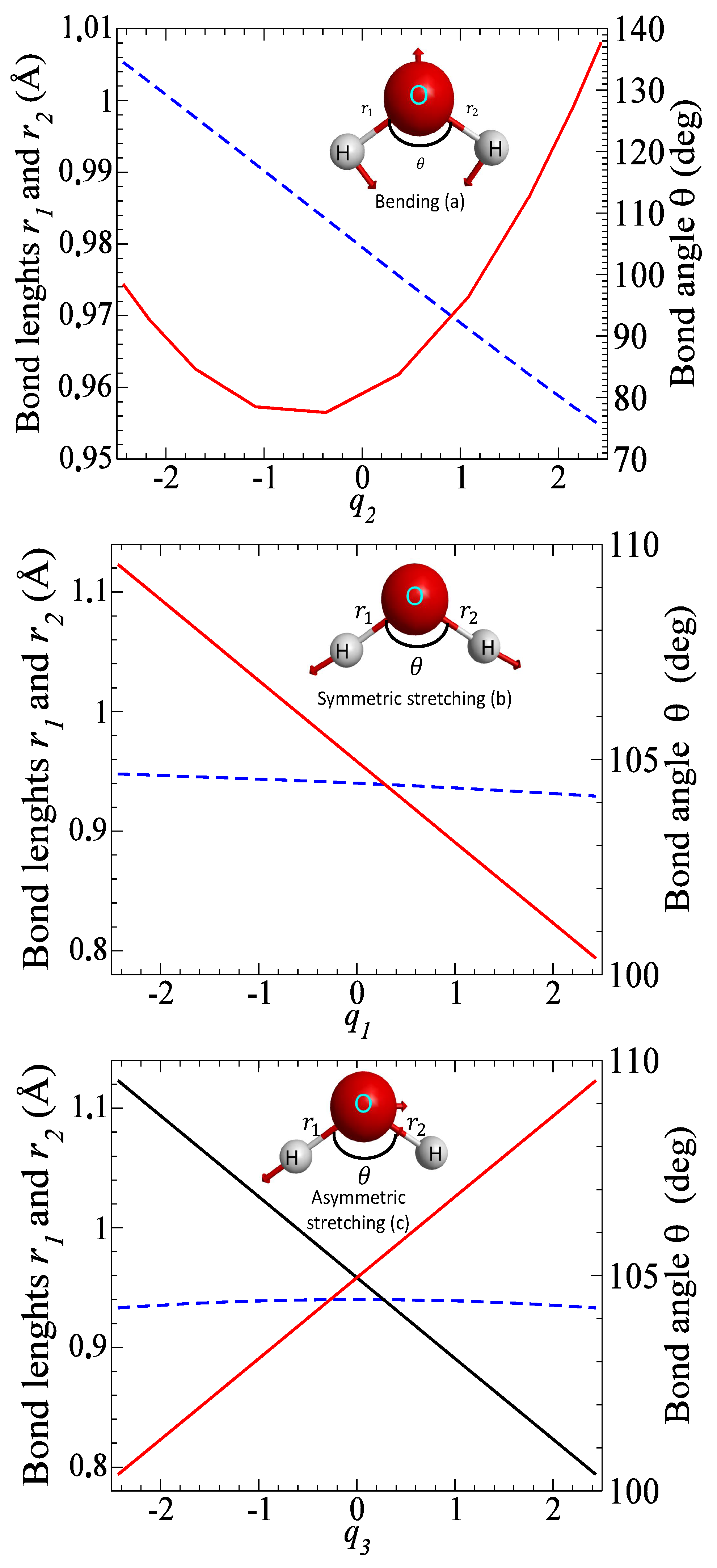
The electronic structure and normal mode frequencies are determined by the complete active space self-consistent field (CASSCF) method using the ab initio quantum chemistry package MOLPRO [23]. The cc-pVTZ basis set is employed for all the atoms. Table 1 gives the optimized geometry and vibrational frequencies, obtained in the present calculation, and compares the results with available experimental data. Figure 2 shows how inter-particle distances , and the bond angle change as functions of normal mode coordinates: bond lengths. Note that displacements along the bending and symmetric stretching modes do not break the C molecular symmetry, while the asymmetric stretching mode reduces the symmetry to the C group.
2.2. Cross-Sections for Vibrational Excitation
The fixed-geometry reactance matrix is transformed to the fixed-geometry scattering matrix . The channels (indexes) of two matrices correspond to different states of target, which could be excited at a given scattering energy, and different partial-wave quantum numbers of the incident electron. For water, the first excited electronic state is at 7.14 eV above the ground vibronic level [2,26]. Here, we consider energies below the excitation of the lowest excited states. Therefore, each channel in the scattering matrix is labeled with the partial-wave indexes only, which are the angular momentum l of the incident electron and its projection on the molecular axis of symmetry.
The scattering matrix is then converted by the vibrational frame transformation
to the matrix in the representation of vibrational channels. Functions are vibrational wave functions of the target molecule.
In this study, we consider excitation of one mode at a time with one or two quanta, while the two other modes are kept in their ground state. Although the integral in the above expression is formally over the three normal-mode coordinates, in practice, we integrate only over one coordinate, which is the one corresponding to the mode that is being excited. For the example, the excitation of the mode i from state to is given with the integral
evaluated over the coordinate , with values of the two other normal-mode coordinates (the matrix depends on all three coordinates) fixed at the equilibrium values, i.e., 0. The integral is evaluated using the Gaussian–Legendre quadrature with 10 points. Functions are eigenfunctions of one-dimensional harmonic oscillator in the dimensionless coordinate , i.e., solutions of the equation
Having the scattering matrix in the vibrational representation, the cross-section for the process is given
where m and are the mass and energy of the incident electron.
3. Results
3.1. Cross Sections
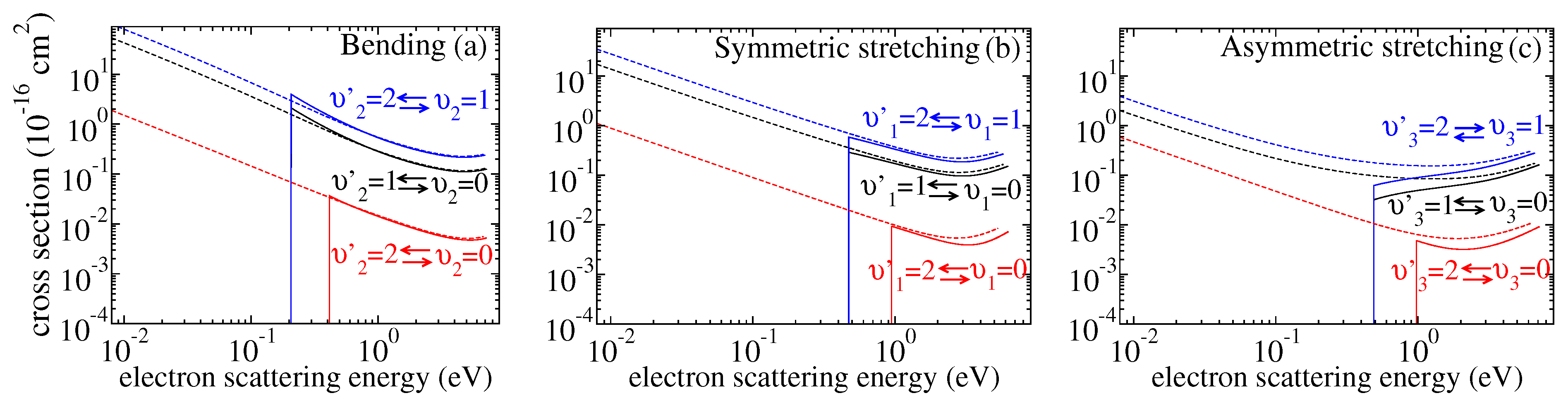
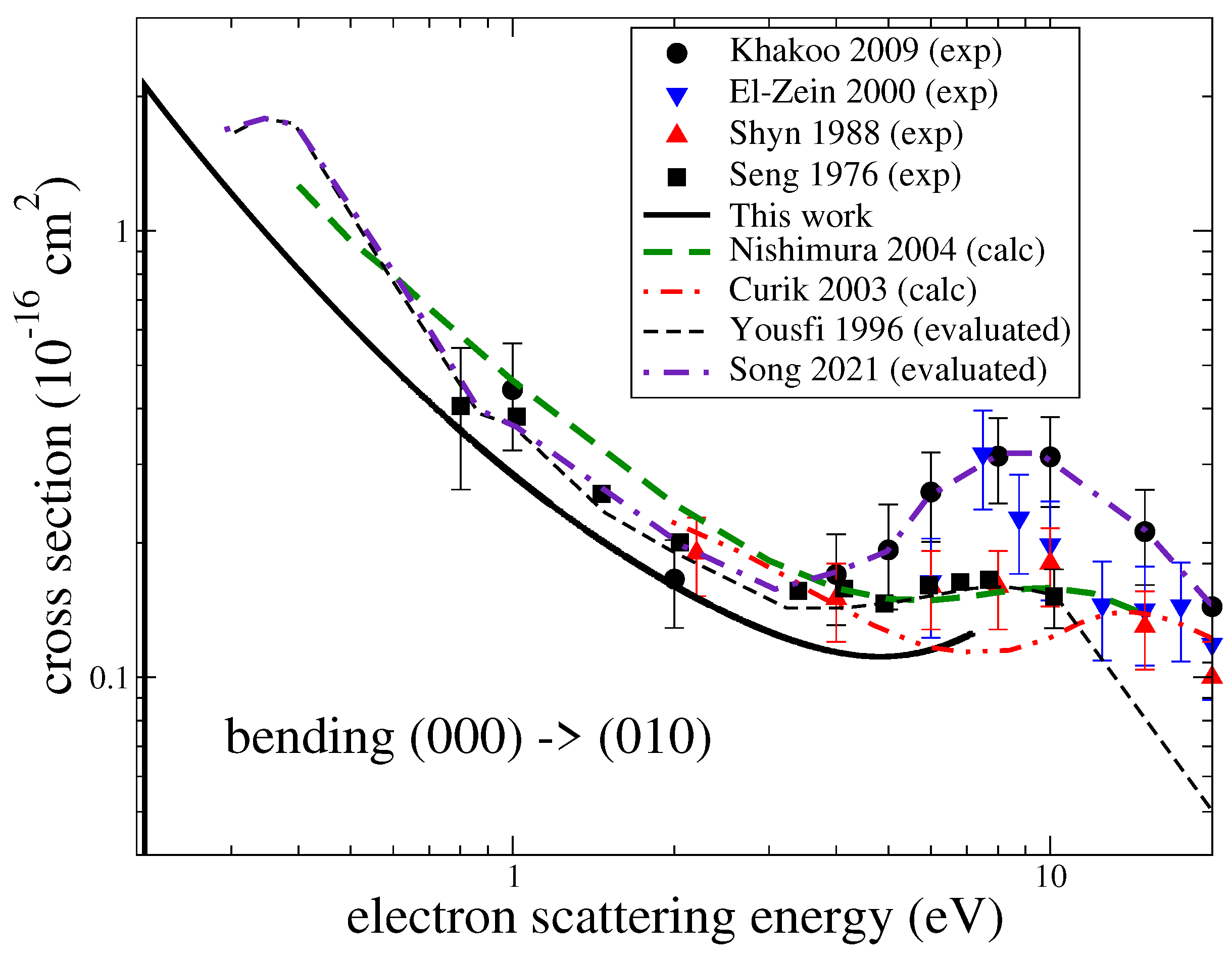
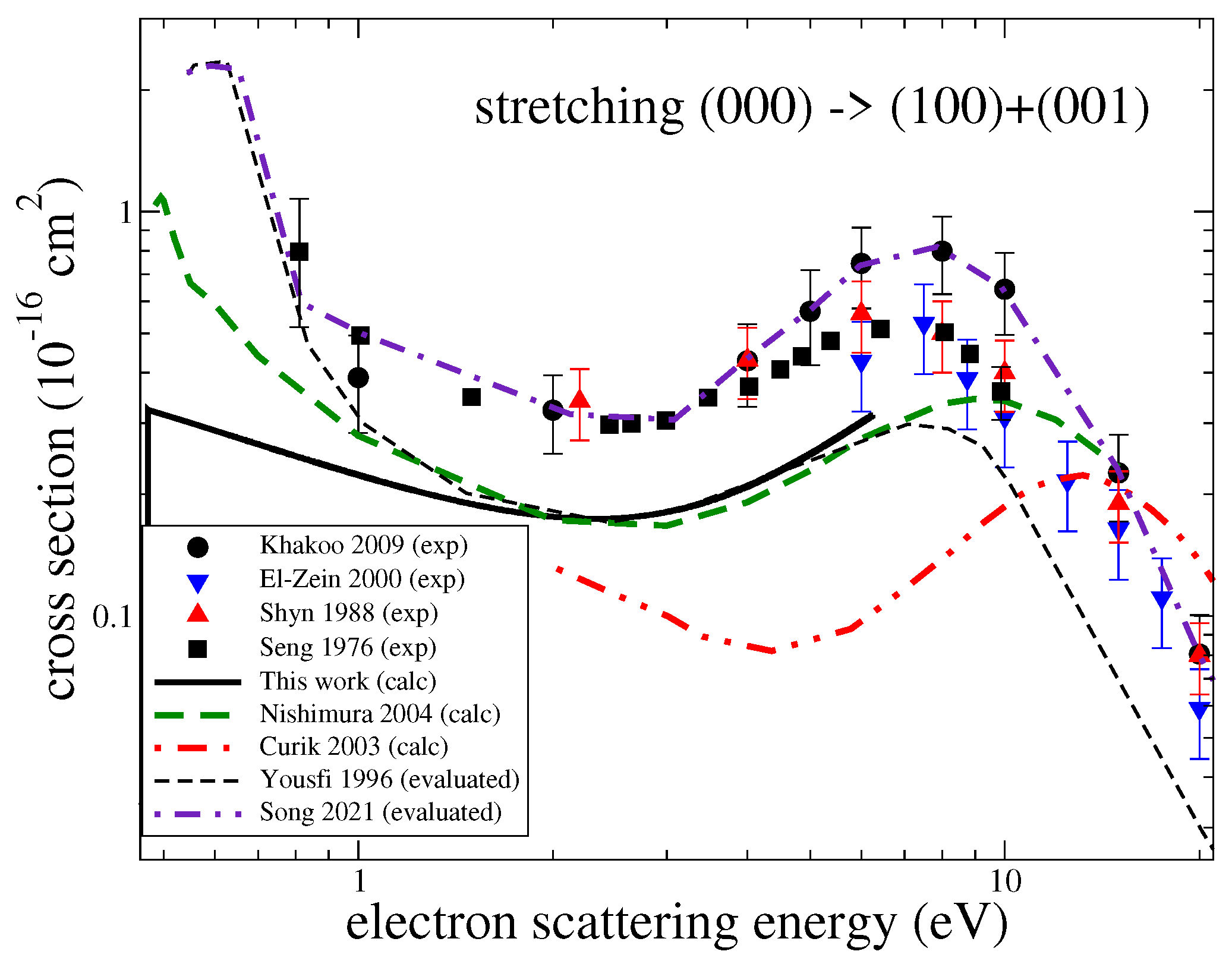
Figure 3 gives the computed cross-sections for transitions between the three lowest vibrational levels for the three modes. Both excitation and de-excitation cross-sections were calculated. There have been several experiments measuring cross-sections for excitation of the ground vibrational level by one quantum. In the experiments, contributions from the two stretching modes were not resolved. Figure 4 and Figure 5 compare the present results with the experimental [6,7,27,28], theoretical [8,29] and previously evaluated and recommended [2,30] data available in the literature. See the review of ref. [2] for details about the data.
For the bending mode and energies below 3 eV, the present cross-section agrees well with the experiment by Seng and Linder [7] but is somewhat below the data obtained from a swarm analysis by Yousfi and Benabdessadok [30]. At energies above 4 eV, the present values are below by about 30% than the swarm data and by a factor of two than the recent experiment by Khakoo et al. [6]. For the stretching mode (Figure 5), all available experimental data generally agree with each other, while the present results and other previous theoretical cross-sections are all systematically below the experimental values.
It is worth mentioning that in the beam experiments, a wide resonant structure near 8 eV is observed. It is especially pronounced in the experiment by Khakoo et al. [6] and less pronounced, manifested rather as a shoulder, in the swarm data by Yousfi and Benabdessadok [30]. In our calculations, we observe four resonances near that energy: two narrow resonances of symmetry at 7.8 eV and of symmetry at 10 eV, and two wider resonances: a resonance at 6.7 eV and a resonance at 11 eV with a width of about 1 eV. Therefore, the resonant structure observed in the experiments (with unresolved rotational structure) can be explained well by the presence of these four resonances. However, it is clear that the theory is unable to reproduce the magnitude of the excitation cross-section correctly, as observed in the experiments. Therefore, it is likely that in the present and previous theoretical calculations, an effect, responsible for a larger vibrational excitation at energies above 3 eV, is not accounted for. One possibility is that in this region of energies, the resonant states, mentioned above, can capture the electron into their vibrational states, which would significantly enhance the excitation cross-section. A similar resonant mechanism was observed in electron-impact vibrational excitation CO [31]. Another consideration is that close-coupling calculations, such as the ones performed here, do not provide a converged treatment of polarization interactions [32]. It is, therefore, possible that the underestimation of polarization effects contributes to the under prediction of the vibrational excitation cross-section.
3.2. Rate Coefficients
The excitation cross-sections were used to compute the thermally averaged rate coefficients (see, for example, Equation (13) of ref. [14]). The coefficients are shown in Figure 6.
Similarly to the previous studies [14,33,34], for a more convenient use in models, the numerical rate coefficients are fitted to the following analytical formula
where
The coefficients () are fitting parameters. The quantity is the (de-)excitation probability. It weakly depends on the scattering energy. In Equation (5), is the threshold energy defined as
3.3. Assessment of Uncertainties
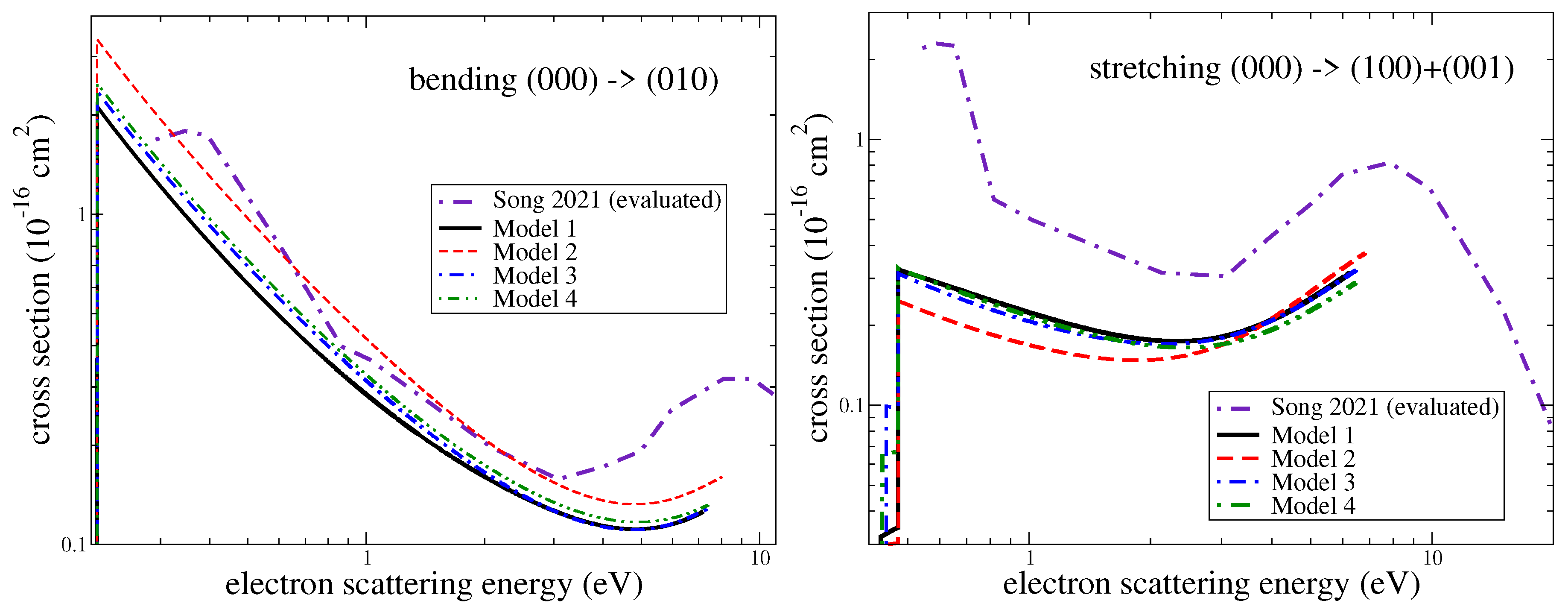
The main source of uncertainty of the present results is due to electron scattering calculations. To assess the uncertainty, we computed the cross-section-varying parameters of the scattering model. The main scattering model (Model 1) is described above. In the second set of calculations (Model 2), the electronic basis was reduced from cc-pVTZ to DZP, and the same CAS (CAS) was employed. In Model 3, we freeze the and molecular orbitals, which leads to a reduced complete active space (CAS) in the configuration interaction calculations with respect to Model 1 by two orbitals. In Model 4, a larger basis cc-pVQZ (correlation-consistent polarized valence quadruple-zeta) and CAS were used. Figure 7 demonstrates a comparison of cross-sections obtained using the models. As evident from the figure, reducing the basis set from cc-pVTZ to DZP (Model 1 vs. Model 2) changes the results by about 30% (stretching) and 50% (bending), while increasing the basis set from cc-pVTZ to cc-pVQZ (Model 1 vs. Model 4) changes the result less, by about 5–15%. Therefore, the convergence of Model 1 with respect to the basis set is about 5% (stretching) and 15% (bending). Changing the CAS (Model 1 vs. Model 3) changes the cross-sections by about 5–10%. Therefore, we estimate the uncertainty of Model 1 to be about 20% for the bending mode and about 10% for the stretching mode.
4. Conclusions
Summarizing the results of the present study, we computed cross-sections for the vibrational excitation of the water molecule by electron impact using a purely ab initio approach. We would like to stress that cross-sections for excitation by two quanta in one collision were obtained and reported for the first time. The uncertainty of the obtained cross-sections is estimated to be 20% for the excitation of the bending mode and 10% for the stretching modes. The resonant structure observed in experimental data near 6–10 eV was characterized using the ab initio calculations. The overall agreement of the present cross-sections with the experiment is within experimental uncertainties (including different experiments) for the bending mode. For the stretching modes, the present theory gives cross-sections somewhat smaller than in the experiment: The difference is slightly larger than the combined uncertainties of the experiment and the theory. Finally, thermally averaged rate coefficients were derived from the obtained cross-sections. The coefficients were fitted to an analytical formula for a convenient use by modellers.
Rotationally resolved vibrational cross-sections are currently being computed and will be reported later.
Finally, we note that the methodology for computing non-resonant vibrational excitation cross-sections employed here has recently been incorporated, with some simplifications, into the QEC (Quantemol Electron Collisions) expert system [35] used to run the new (UKRmol+) UK Molecule R-matrix code [36]. The main simplifications are: (1) The integral of Equation (2) is evaluated using the linear approximation for the scattering matrix near the equilibrium geometry [37,38,39,40]. (2) The electron-scattering calculation for all geometries are performed without taking into account any symmetry of the molecule, i.e., the group of molecular symmetry is used. As here, MOLPRO is used to automatically generate the normal modes.
Author Contributions
Conceptualization, M.A., A.F., J.T., M.T. and V.K.; methodology, M.A., J.T., M.T. and V.K.; software, M.A., A.F., J.T., M.T. and V.K.; validation, M.A., A.F., J.T., M.T. and V.K.; formal analysis, M.A., A.F., J.T., M.T. and V.K.; investigation, M.A., A.F., J.T., M.T. and V.K.; resources, M.A., A.F., J.T. and V.K.; data curation, M.A., J.T., M.T. and V.K.; writing—original draft preparation, M.A., A.F., J.T., M.T. and V.K.; writing—review and editing, M.A., A.F., J.T., M.T. and V.K.; project administration, M.A., A.F., J.T., M.T. and V.K.; funding acquisition, M.A., A.F., J.T. and V.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Thomas Jefferson Fund of the Office for Science and Technology of the Embassy of France in the United States, the National Science Foundation, Grant No. PHY-1806915, and by the program “Accueil des chercheurs étrangers” of CentraleSupélec. A.F. was funded by ANR ‘WATERSTARS’, project ANR-20-CE31-0011.
Data Availability Statement
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van Dishoeck, E.F.; Herbst, E.; Neufeld, D.A. Interstellar Water Chemistry: From Laboratory to Observations. Chem. Rev. 2013, 113, 9043–9085. [Google Scholar] [CrossRef] [Green Version]
- Song, M.Y.; Cho, H.; Karwasz, G.P.; Kokoouline, V.; Nakamura, Y.; Tennyson, J.; Faure, A.; Mason, N.J.; Itikawa, Y. Cross Sections for Electron Collisions with H2O. J. Phys. Chem. Ref. Data 2021, 50, 023103. [Google Scholar] [CrossRef]
- Machado, L.E.; Brescansin, L.M.; Iga, I.; Lee, M.T. Elastic and rotational excitation cross-sections for electron-water collisions in the low- and intermediate-energy ranges. Eur. Phys. J. D 2005, 33, 193–199. [Google Scholar] [CrossRef]
- Faure, A.; Gorfinkiel, J.D.; Tennyson, J. Low-energy electron collisions with water: Elastic and rotationally inelastic scattering. J. Phys. B At. Mol. Phys. 2004, 37, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Itikawa, Y.; Mason, N. Cross Sections for Electron Collisions with Water Molecules. J. Phys. Chem. Ref. Data 2005, 34, 1–22. [Google Scholar] [CrossRef]
- Khakoo, M.; Winstead, C.; McKoy, V. Vibrational excitation of water by electron impact. Phys. Rev. A 2009, 79, 052711. [Google Scholar] [CrossRef] [Green Version]
- Seng, G.; Linder, F. Vibrational excitation of polar molecules by electron impact. II. Direct and resonant excitation in H2O. J. Phys. B At. Mol. Phys. 1976, 9, 2539–2551. [Google Scholar] [CrossRef]
- Nishimura, T.; Gianturco, F. Vibrational excitation of water by low-energy electron scattering: Calculations and experiments. Europhys. Lett. 2004, 65, 179. [Google Scholar] [CrossRef] [Green Version]
- Faure, A.; Josselin, E. Collisional excitation of water in warm astrophysical media. I. Rate coefficients for rovibrationally excited states. Astron. Astrophys. 2008, 492, 257–264. [Google Scholar] [CrossRef]
- Baudry, A.; Humphreys, E.M.L.; Herpin, F.; Torstensson, K.; Vlemmings, W.H.T.; Richards, A.M.S.; Gray, M.D.; De Breuck, C.; Olberg, M. Vibrationally excited water emission at 658 GHz from evolved stars. Astron. Astrophys. 2018, 609, A25. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, E.D.; Dodd, J.L.; Milam, S.N.; Woolf, N.J.; Ziurys, L.M. Comparative Spectra of Oxygen-rich Versus Carbon-rich Circumstellar Shells: VY Canis Majoris and IRC +10216 at 215–285 GHz. Astrophys. J. Lett. 2010, 720, L102–L107. [Google Scholar] [CrossRef] [Green Version]
- Stoecklin, T.; Denis-Alpizar, O.; Clergerie, A.; Halvick, P.; Faure, A.; Scribano, Y. Rigid-Bender Close-Coupling Treatment of the Inelastic Collisions of H2O with para-H2. J. Phys. Chem. A 2019, 123, 5704–5712. [Google Scholar] [CrossRef] [PubMed]
- Stoecklin, T.; Cabrera-González, L.D.; Denis-Alpizar, O.; Páez-Hernández, D. A close coupling study of the bending relaxation of H2O by collision with He. J. Chem. Phys. 2021, 154, 144307. [Google Scholar] [CrossRef]
- Ayouz, M.; Kokoouline, V. Cross Sections and Rate Coefficients for Vibrational Excitation of HeH+ Molecule by Electron Impact. Atoms 2016, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Ayouz, M.; Kokoouline, V. Cross Sections and Rate Coefficients for Rovibrational Excitation of HeH+ Isotopologues by Electron Impact. Atoms 2019, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; dos Santos, S.F.; Yuen, C.H.; Cortona, P.; Kokoouline, V.; Ayouz, M. Theoretical study of electron-induced vibrational excitation of NO2. Plasma Sources Sci. Technol. 2019, 28, 105017. [Google Scholar] [CrossRef]
- Liu, H.; Santos, S.F.d.; Yuen, C.H.; Cortona, P.; Ayouz, M.; Kokoouline, V. Vibrational excitation of N2O by an electron impact and the role of the Renner-Teller effect in the process. Phys. Rev. A 2020, 102, 032808. [Google Scholar] [CrossRef]
- Tennyson, J. Electron–molecule collision calculations using the R-matrix method. Phys. Rep. 2010, 491, 29–76. [Google Scholar] [CrossRef]
- Carr, J.; Galiatsatos, P.; Gorfinkiel, J.; Harvey, A.; Lysaght, M.; Madden, D.; Mašín, Z.; Plummer, M.; Tennyson, J.; Varambhia, H. UKRmol: A low-energy electron- and positron-molecule scattering suite. Eur. Phys. J. D 2012, 66, 58. [Google Scholar] [CrossRef]
- Tennyson, J.; Brown, D.B.; Munro, J.J.; Rozum, I.; Varambhia, H.N.; Vinci, N. Quantemol-N: An expert system for performing electron molecule collision calculations using the R-matrix method. J. Phys. Conf. Ser. 2007, 86, 012001. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Functions for Use in Molecular Calculations. I. Contraction of (9s5p) Atomic Basis Sets for the First-Row Atoms. J. Chem. Phys. 1970, 53, 2823–2833. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Johnson, R.D. NIST Computational Chemistry Comparison and Benchmark Database; NIST Standard Reference Database Number 101; NIST: Gaithersburg, MD, USA, 2010.
- Császár, A.G.; Czako, G.; Furtenbacher, T.; Tennyson, J.; Szalay, V.; Shirin, S.V.; Zobov, N.F.; Polyansky, O.L. On equilibrium structures of the water molecule. J. Chem. Phys. 2005, 122, 214305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, P.; Campbell, L.; Brunger, M. Electron excitation and energy transfer rates for H2O in the upper atmosphere. PMC Phys. B 2009, 2, 1. [Google Scholar] [CrossRef]
- El-Zein, A.; Brunger, M.; Newell, W. Excitation of vibrational quanta in water by electron impact. J. Phys. B At. Mol. Opt. Phys. 2000, 33, 5033. [Google Scholar] [CrossRef]
- Shyn, T.; Cho, S.; Cravens, T. Vibrational-excitation cross sections of water molecules by electron impact. Phys. Rev. A 1988, 38, 678. [Google Scholar] [CrossRef] [PubMed]
- Curik, R.; Carsky, P. Vibrationally inelastic electron scattering on polyatomic molecules by the discrete momentum representation (DMR) method. J. Phys. B At. Mol. Opt. Phys. 2003, 36, 2165. [Google Scholar] [CrossRef]
- Yousfi, M.; Benabdessadok, M.D. Boltzmann equation analysis of electron-molecule collision cross sections in water vapor and ammonia. J. Appl. Phys. 1996, 80, 6619–6630. [Google Scholar] [CrossRef]
- Laporta, V.; Cassidy, C.; Tennyson, J.; Celiberto, R. Electron-impact resonant vibration excitation cross sections and rate coefficients for carbon monoxide. Plasma Sources Sci. Technol. 2012, 21, 045005. [Google Scholar] [CrossRef]
- Jones, M.; Tennyson, J. On the use of pseudostates to calculate molecular polarizabilities. J. Phys. B 2010, 43, 045101. [Google Scholar] [CrossRef]
- Kokoouline, V.; Faure, A.; Tennyson, J.; Greene, C.H. Calculation of rate constants for vibrational and rotational excitation of the H3+ ion by electron impact. Mon. Not. R. Astron. Soc. 2010, 405, 1195–1202. [Google Scholar]
- Jiang, X.; Yuen, C.H.; Cortona, P.; Ayouz, M.; Kokoouline, V. Cross sections for vibronic excitation of CH+ by low-energy electron impact. Phys. Rev. A 2019, 100, 062711. [Google Scholar] [CrossRef]
- Cooper, B.; Tudorovskaya, M.; Mohr, S.; O’Hare, A.; Hanicinec, M.; Dzarasova, A.; Gorfinkiel, J.; Benda, J.; Mašín, Z.; Al-Refaie, A.; et al. Quantemol Electron Collision: An expert system for performing UKRmol+ electron molecule collision calculations. Atoms 2019, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Mašín, Z.; Benda, J.; Gorfinkiel, J.D.; Harvey, A.G.; Tennyson, J. UKRmol+: A suite for modelling of electronic processes in molecules interacting with electrons, positrons and photons using the R-matrix method. Comput. Phys. Comms. 2020, 249, 107092. [Google Scholar] [CrossRef] [Green Version]
- Fonseca dos Santos, S.; Douguet, N.; Kokoouline, V.; Orel, A. Scattering matrix approach to the dissociative recombination of HCO+ and N2H+. J. Chem. Phys. 2014, 140, 164308. [Google Scholar] [CrossRef] [Green Version]
- Douguet, N.; Fonseca dos Santos, S.; Kokoouline, V.; Orel, A. Simplified model to describe the dissociative recombination of linear polyatomic ions of astrophysical interest. EPJ Web Conf. 2015, 84, 07003. [Google Scholar] [CrossRef] [Green Version]
- Kokoouline, V.; Ayouz, M.; Mezei, J.Z.; Hassouni, K.; Schneider, I.F. Theoretical study of dissociative recombination and vibrational excitation of the BF2+ ion by an electron impact. Plasma Sources Sci. Technol. 2018, 27, 115007. [Google Scholar] [CrossRef]
- Ayouz, M.A.; Yuen, C.H.; Balucani, N.; Ceccarelli, C.; Schneider, I.F.; Kokoouline, V. Dissociative electron recombination of NH2CHOH+ and implications for interstellar formamide abundance. Mon. Not. R. Astron. Soc. 2019, 490, 1325–1331. [Google Scholar] [CrossRef]
Figure 1.
The sum of eigenphases as a function of the electron scattering energy for equilibrium geometry and displacements along each normal mode. The eigenphase sums for and of the asymmetric stretching mode are identical. The curves are color coded according the different symmetries of the system (see the left upper panel).
Figure 1.
The sum of eigenphases as a function of the electron scattering energy for equilibrium geometry and displacements along each normal mode. The eigenphase sums for and of the asymmetric stretching mode are identical. The curves are color coded according the different symmetries of the system (see the left upper panel).

Figure 2.
Normal modes of HO. The figure shows the dependence of inter-particle distances , and the bond angle as functions of normal mode coordinates: (a) bending, (b) symmetric stretching and (c) asymmetric stretching. The arrows indicate the direction and magnitude of displacements for each mode. Bond lengths are given with solid lines with values on the left axis, while the bond angle is given with a dashed line with values on the right axis of each panel. Note that the curves of the bond lengths (black) and (red) are indistinguishable for the bending and symmetric stretching modes.
Figure 2.
Normal modes of HO. The figure shows the dependence of inter-particle distances , and the bond angle as functions of normal mode coordinates: (a) bending, (b) symmetric stretching and (c) asymmetric stretching. The arrows indicate the direction and magnitude of displacements for each mode. Bond lengths are given with solid lines with values on the left axis, while the bond angle is given with a dashed line with values on the right axis of each panel. Note that the curves of the bond lengths (black) and (red) are indistinguishable for the bending and symmetric stretching modes.

Figure 3.
The calculated cross sections as functions of the electron scattering energy for the vibrational excitation of for different vibrational states of the three normal modes i (see the text for detailed discussion): (a) cross−sections for the bending mode, (b) for symmetric stretching mode and (c) for asymmetric stretching mode.
Figure 3.
The calculated cross sections as functions of the electron scattering energy for the vibrational excitation of for different vibrational states of the three normal modes i (see the text for detailed discussion): (a) cross−sections for the bending mode, (b) for symmetric stretching mode and (c) for asymmetric stretching mode.

Figure 4.
A comparison of cross sections available in the literature for the excitation of the bending mode . The black solid line is the theoretical result obtained in this study.
Figure 4.
A comparison of cross sections available in the literature for the excitation of the bending mode . The black solid line is the theoretical result obtained in this study.

Figure 5.
A comparison of cross sections available in the literature for the excitation of the stretching modes . The black solid line is the theoretical result obtained in this study.
Figure 5.
A comparison of cross sections available in the literature for the excitation of the stretching modes . The black solid line is the theoretical result obtained in this study.

Figure 6.
Same as Figure 3 for the calculated rate coefficients. Dashed lines represent de-excitation transitions, while the solid lines of the same colour refer to opposite processes.
Figure 6.
Same as Figure 3 for the calculated rate coefficients. Dashed lines represent de-excitation transitions, while the solid lines of the same colour refer to opposite processes.

Figure 7.
Cross sections for the excitation of the bending (left panel) and stretching (right panel) modes obtained using the four different models (see the text). Model 1 is used to produce final results of the study. The three other models, in which the basis set and the CAS were changed compared to Model 1, are used to assess the uncertainty of the results: Model 1—the cc-pVTZ basis and CAS; Model 2—the DZP basis and CAS; Model 3—the cc-pVTZ basis and CAS; Model 4—the cc-pVQZ basis and CAS.
Figure 7.
Cross sections for the excitation of the bending (left panel) and stretching (right panel) modes obtained using the four different models (see the text). Model 1 is used to produce final results of the study. The three other models, in which the basis set and the CAS were changed compared to Model 1, are used to assess the uncertainty of the results: Model 1—the cc-pVTZ basis and CAS; Model 2—the DZP basis and CAS; Model 3—the cc-pVTZ basis and CAS; Model 4—the cc-pVQZ basis and CAS.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The structure and vibrational frequencies (in eV) of HO obtained in this study and compared with experimental data from ref. [24].
Table 1.
The structure and vibrational frequencies (in eV) of HO obtained in this study and compared with experimental data from ref. [24].
| Mode | This Study | Exp. [24,25] |
|---|---|---|
| Bending (010) | 0.207 | 0.198 |
| Symmetric stretching (100) | 0.472 | 0.453 |
| Asymmetric stretching (001) | 0.488 | 0.466 |
| Bond lengths , (Å) | 0.958 | 0.958 |
| Bond angle (Degrees) | 104.44 | 104.50 |
Table 2.
Parameters , and of the polynomial of Equations (5) and (6) between the three lowest vibrational states for the bending mode of HO. The pairs of the final and initial vibrational levels for each normal mode are at the second line in each header of the tables. The third line in each header gives the threshold energies in Equation (7).
Table 2.
Parameters , and of the polynomial of Equations (5) and (6) between the three lowest vibrational states for the bending mode of HO. The pairs of the final and initial vibrational levels for each normal mode are at the second line in each header of the tables. The third line in each header gives the threshold energies in Equation (7).
| (K) | 2403 | 4807 | 0 | 2403 | 0 | 0 |
| 5.76 | 2.23 | 2.63 | 1.08 | 8.33 | 3.99 | |
| −6.32 | −3.05 | 2.24 | −1.19 | 7.07 | 6.83 | |
| 2.76 | 1.95 | −2.97 | 5.30 | −5.27 | −7.34 |
Table 3.
Same as Table 2 for the symmetric stretching mode of HO.
Table 3.
Same as Table 2 for the symmetric stretching mode of HO.
| (K) | 5489 | 10978 | 0 | 5488 | 0 | 0 |
| 2.92 | 1.01 | 1.10 | 5.69 | 6.62 | 2.32 | |
| −5.68 | −1.06 | −8.08 | −1.07 | −1.82 | −1.79 | |
| 4.29 | 7.76 | 1.09 | 8.00 | 2.39 | 2.16 |
Table 4.
Same as Table 2 for the asymmetric stretching mode of HO.
Table 4.
Same as Table 2 for the asymmetric stretching mode of HO.
| (K) | 5673 | 11345 | 0 | 5672 | 0 | 0 |
| 3.56 | 1.66 | 4.27 | 6.64 | 4.49 | 8.57 | |
| −9.80 | −3.59 | −1.51 | −1.82 | −5.01 | −2.94 | |
| 7.05 | 2.45 | 1.66 | 1.31 | 5.15 | 3.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ayouz, M.; Faure, A.; Tennyson, J.; Tudorovskaya, M.; Kokoouline, V. Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact. Atoms 2021, 9, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030062
AMA Style
Ayouz M, Faure A, Tennyson J, Tudorovskaya M, Kokoouline V. Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact. Atoms. 2021; 9(3):62. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030062
Chicago/Turabian StyleAyouz, Mehdi, Alexandre Faure, Jonathan Tennyson, Maria Tudorovskaya, and Viatcheslav Kokoouline. 2021. "Cross Sections and Rate Coefficients for Vibrational Excitation of H2O by Electron Impact" Atoms 9, no. 3: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030062
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.