
In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon


1
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India
2
Department of Chemistry, Indian Institute of Technology Bombay, Mumbai 400076, India
*
Author to whom correspondence should be addressed.
Atoms 2021, 9(3), 65; https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030065
Submission received: 4 August 2021
/
Revised: 31 August 2021
/
Accepted: 7 September 2021
/
Published: 10 September 2021
(This article belongs to the Special Issue Planar Tetracoordinate Carbon—Fifty Years and Beyond)
Abstract
:Density functional theory (DFT) was used to study the structure, stability, and bonding in some selected neutral pentaatomic systems, viz., CGa2Ge2, CAlGaGe2, and CSiGa2Ge containing planar tetracoordinate carbon. The systems are kinetically stable, as predicted from the ab initio molecular dynamics simulations. The natural bond orbital (NBO) analysis showed that strong electron donation occurs to the central planar carbon atom by the peripheral atoms in all the studied systems. From the nucleus independent chemical shift (NICS) analysis, it is shown that the systems possess both σ- and π- aromaticity. The presence of 18 valence electrons in these systems, in their neutral form, appears to be important for their stability with planar geometries rather than tetrahedral structures. The nature of bonding is understood through the adaptive natural density partitioning analysis (AdNDP), quantum theory of atoms in molecules (QTAIM) analysis, and also via Wiberg bond index (WBI) and electron localization function (ELF).
1. Introduction
Nowadays, planar tetracoordinate carbon (ptC) molecules showed great attention to experimentalists [1,2,3,4,5,6,7] as well as theoreticians [8,9,10,11,12,13,14,15] because it deviates from the usual ideas of tetrahedral tetracoordinate carbon. The tetrahedral carbon concept was given by van’t Hoff and Le Bel in 1874 [16,17]. The idea of the ptC was introduced in 1968 by H. J. Monkhorst as a transition state of methane in a non-dissociative racemization process [18]. After two years, Hoffmann and co-workers showed two strategies for the stabilization of molecules containing ptC and those methods constitute a useful guide to design new ptC molecules [8,19]. The first one is the electronic strategy that involves appropriate substituents with σ-donors and π-acceptors simultaneously to stabilize the ptC species. The second one is the mechanical strategy that involves aromatic systems, cages, and transition metals to design and stabilize ptC systems. Using those two proposals, Schleyer and co-workers, in 1976, designed lithium substituted cyclopropane (C3H3Li2), the first in-silico studied ptC molecule [9]. After that, many theoretical studies from different research groups were reported on ptC systems [12,20,21,22,23,24,25,26,27]. In a theoretical work in 1991, Schleyer and Boldyrev proposed cis and trans geometries of CSi2Al2 molecules containing ptC [28]. In order to understand the stability of the ptC systems, the “18 valence electron rule” was composed and many systems were shown to obey this rule [2]. However, there are some examples in which more or less than 18 valence electrons are present and the ptC systems are still stable [29,30]. One of the most recent theoretical work shows that the CAl4Mg system in its neutral and mono-anionic state show global minima with ptC [30]. The neutral ptC system shows 18 valence electrons and the anionic one possesses 19 valence electrons. Despite their in silico characterization, experimental synthesis and characterization of ptC molecules are limited [1,2,3,4,5,6,7]. In 1999, CAl4−, a pentaatomic monoanionic cluster, was identified in the gas phase [1]. After one year, some new anionic ptC molecules, CAl42−, CAl3Ge−, CAl3Si−, and NaCAl4−, were experimentally detected [7,31,32]. More interestingly, this idea of ptC has been extended to design planar pentacoordinate [33,34,35] and hexacoordinate carbon molecules [36,37,38,39].
Boldyrev and Simons [2] studied on three neutral pentaatomic molecules (CSi2Al2, CSi2Ga2, and CGe2Al2) and showed the planar tetracoordinate central carbon (ptC) in these systems. They used molecular orbital analysis to rationalize the preference of the planar geometry over the tetrahedral one and they concluded that molecules containing one central C and two Si or Ge ligand atoms and two Al or Ga ligand atoms should have stable planar geometries. Inspired by this work, we designed three neutral pentaatomic systems, viz., CGa2Ge2, CAlGaGe2, and CSiGa2Ge, containing 18 valence electrons. Both cis and trans isomers of the systems are considered in this present work. The electronic interaction among the middle carbon atom and the peripheral atoms is responsible for the planar arrangement of these molecules obeying the “electronic strategy method” for their stabilization. All these three systems follow the 18 valence electrons rule to gain stability in planar form. With the help of the DFT method, we studied the geometries, stability, and the nature of bonding in the molecules. Now, the nature of bonding includes molecular orbital analysis, AdNDP analysis, NBO analysis, and electron density analysis.
2. Computational Details
The geometry optimization and the subsequent frequency calculation of all the systems considered in this study were carried out using BP86 functional [40,41] coupled with D3 correction [42]. The BP86 functional is one of the commonly used DFT methods for electronic structure calculations for main group elements. The correlation consistent triple-ζ quality basis set has been taken for this computation. For C, Al, and Si atoms cc-pVTZ basis set and for Ga and Ge atoms cc-pVTZ-PP basis set along with relativistic effective core potential (RECPs), ECP10MDF have been used [43,44,45,46]. All real harmonic vibrational frequencies correspond to the energy minima on the potential energy surfaces, as observed. Using the same computational method the aromaticity analysis of the studied ptC systems was performed, with the help of NICS. All of these computations were performed with the help of the Gaussian 16 program package [47].
The atom-centered density matrix propagation (ADMP) simulation [48,49,50] for the studied systems was performed to obtain information about their kinetic stability at 300 K and 500 K temperatures, and 1 atm pressure over 2000 fs of simulation time. The simulation was performed with the BP86-D3/def2-SVP method by taking the initially optimized geometries.
To know about the charge distribution in the systems and the Wiberg bond indices (WBI) [51] between any two atoms connected through a bond, natural bond orbital (NBO) [52] analysis was performed using NBO 3.1 [52,53] as implemented in Gaussian 16, using the same computational method that was used for the geometry optimization.
3. Results and Discussion
3.1. Geometries
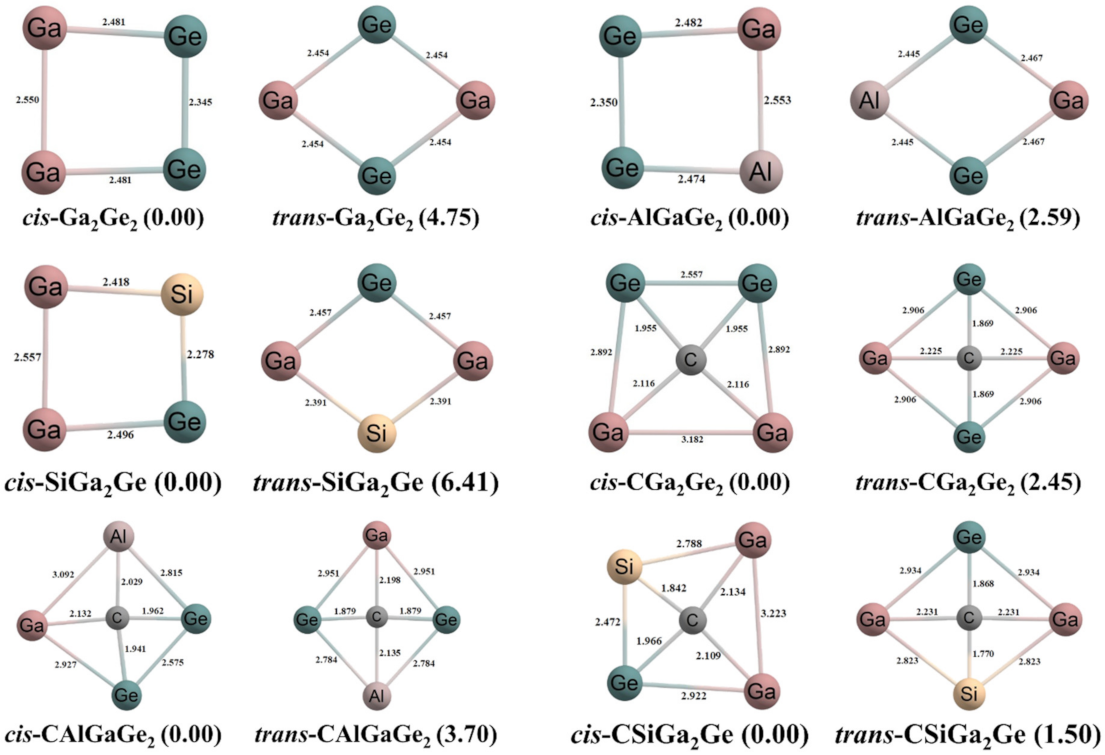
The optimized geometries of the cyclic rings (L) and the ptC systems, both in cis and trans orientations, are given in Figure 1. The L and ptC systems were optimized without any symmetry constraint. In this work, we considered only the singlet state of the ptC systems because the higher spin states correspond to a higher energy than the singlet state. Moreover, the mono-anionic and mono-cationic states of these ptC systems are not planar. So, we considered only neutral states of the studied systems. For all L systems, cis isomers of AlGaGe2 and SiGa2Ge rings have Cs symmetry and the cis Ga2Ge2 ring has C2v symmetry. For AlGaGe2 and SiGa2Ge rings, the trans isomers correspond to the C2v point group of symmetry while, for the Ga2Ge2 ring, the trans isomer has D2h symmetry. The carbon atom at the middle of the rings bonded with four peripheral atoms of each L and the ptC systems were generated. We took the tetrahedral structures of the systems as an initial guess but, after optimization, the structures are converted to more stable planar forms. The energy differences between cis and trans isomers are 2.45, 3.70, and 1.50 kcal/mol for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively. At the second-order Møller−Plesset perturbation (MP2) method [57] with the same basis set, the energy differences are 1.40, 2.27, and 0.72 kcal/mol for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively. So, we considered both isomers throughout the work. The trans isomer of the CGa2Ge2 system has D2h point group of symmetry while the trans isomers of CAlGaGe2 and CSiGa2Ge systems have C2v point group. However, the cis isomers of CAlGaGe2 and CSiGa2Ge systems have Cs symmetry, and the cis CGa2Ge2 system has C2v symmetry. The bond distances for all the bonds in the systems are presented in Figure 1. Boldyrev and Simons [2] explained that tetrahedral structures of their systems are Jahn–Teller unstable and subsequent distortion should lead to a planar structure. For this purpose, they compared the occupancy pattern of the valence MOs of tetrahedral CF4 molecule with the tetrahedral structures of their systems. The CF4 molecule is the 32 valence electronic system and the occupancy pattern of the occupied MOs is 1a121t262a122t261e43t261t16. They assumed that other tetrahedral molecules or nearly tetrahedral structures would follow this occupancy pattern (except for symmetry-imposed degeneracies) and that the 18 valence electronic tetrahedral structures showed a 1a121t262a122t261e2 pattern of occupancy. Due to this partially filled e orbital, the tetrahedral structures of their systems show Jahn–Teller instability and obtain distorted to a planar structure.
Our designed systems are very similar to the systems studied by Boldyrev and Simons. The systems have 18 valence electrons and the carbon atom is present at the central position. So, our systems in tetrahedral structures also show a similar occupancy pattern of the occupied MOs and that is 1a121t262a122t261e2. Due to this partially filled e orbital the tetrahedral structures of our designed systems show Jahn-Teller instability and distorted to a planar structure. So, considering Jahn–Teller distortion the ground state of our systems show planar geometries.
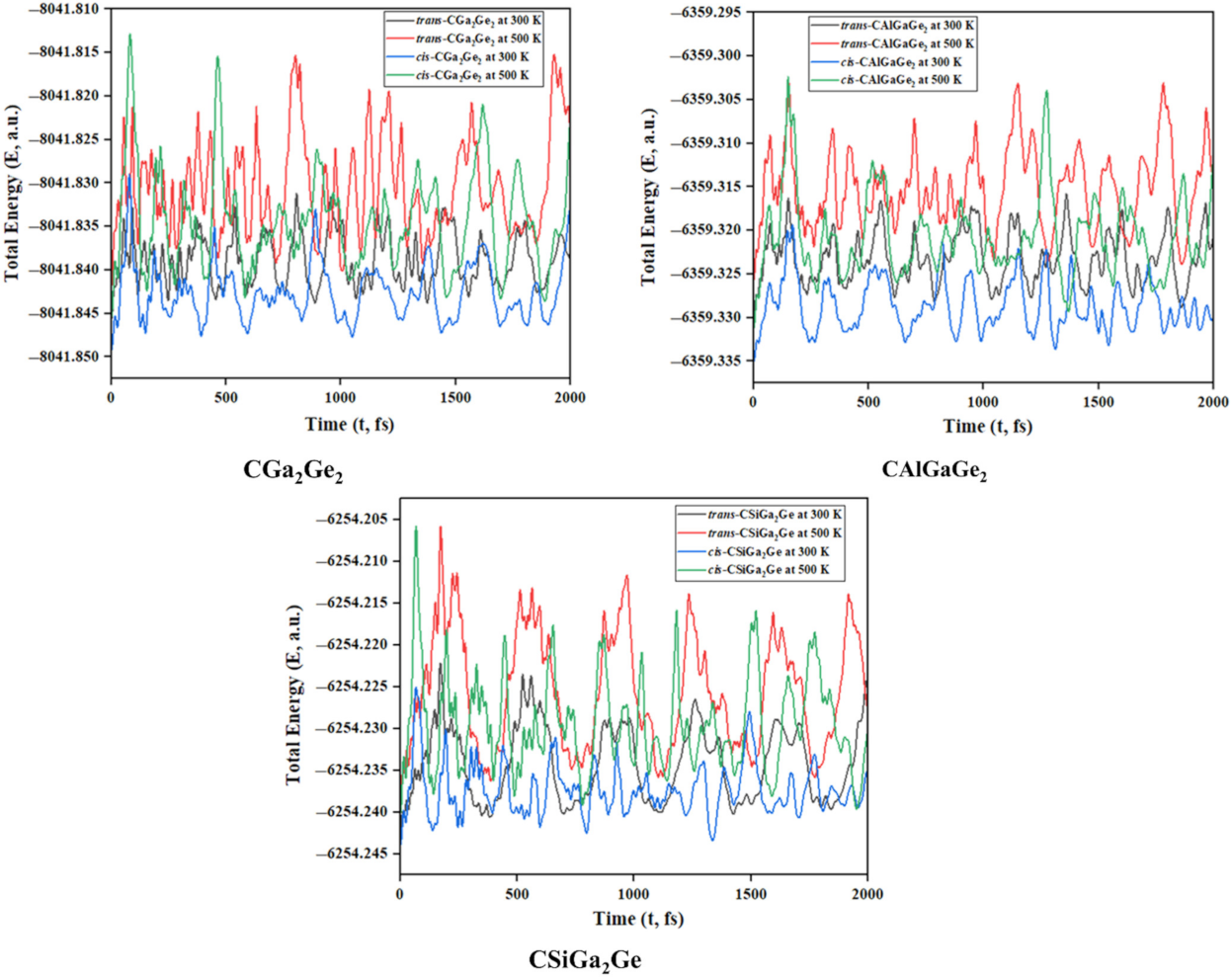
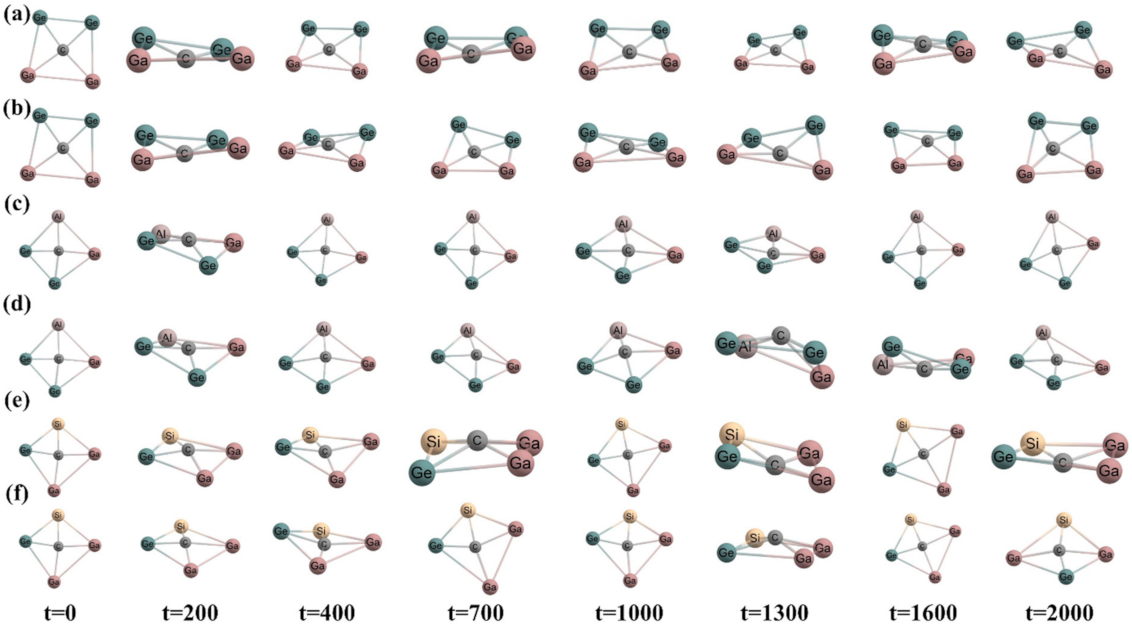
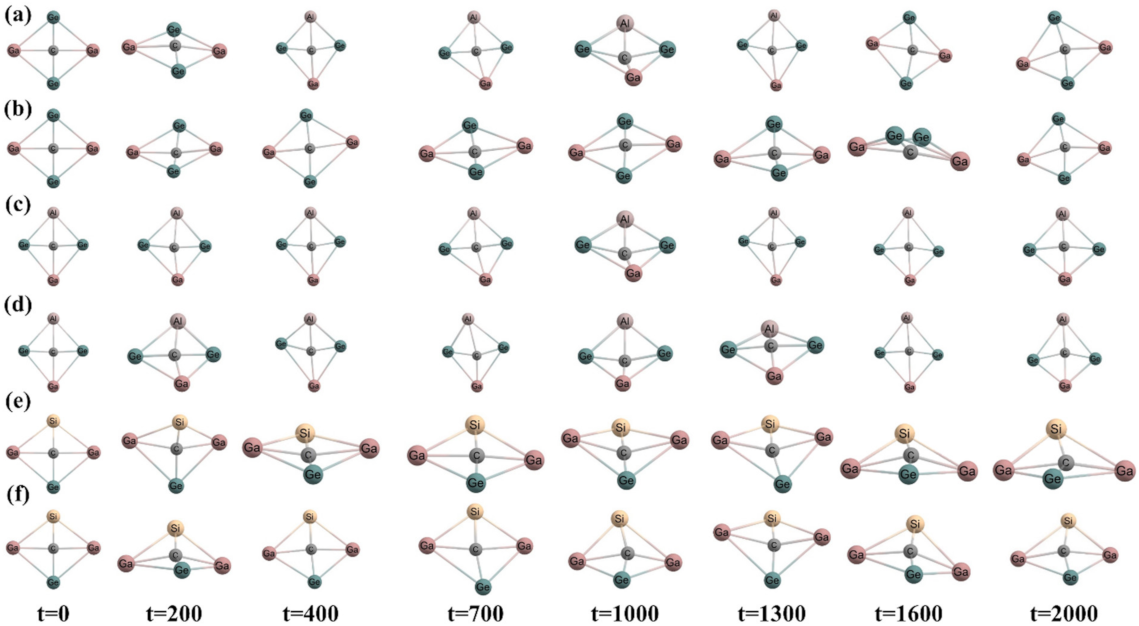
Ab initio molecular dynamics simulations were performed at 300 K and 500 K temperatures and 1 atm pressure over 2000 fs of time to check the kinetic stability for both isomers using ADMP approach. The time evolution of energy of all the systems is presented in Figure 2. The oscillation in the plots arises because of the increase in the nuclear kinetic energy during structural deformation throughout the whole simulation. Some snapshots at different time steps of the simulation for cis and trans isomers are given in Figure 3 and Figure 4, respectively, to show the structural deformation of these systems at these temperatures. The steady fluctuations in energy and consistency in the geometry suggest the kinetic stability of these systems both in cis and trans isomers at these temperatures.
3.2. Molecular Orbitals
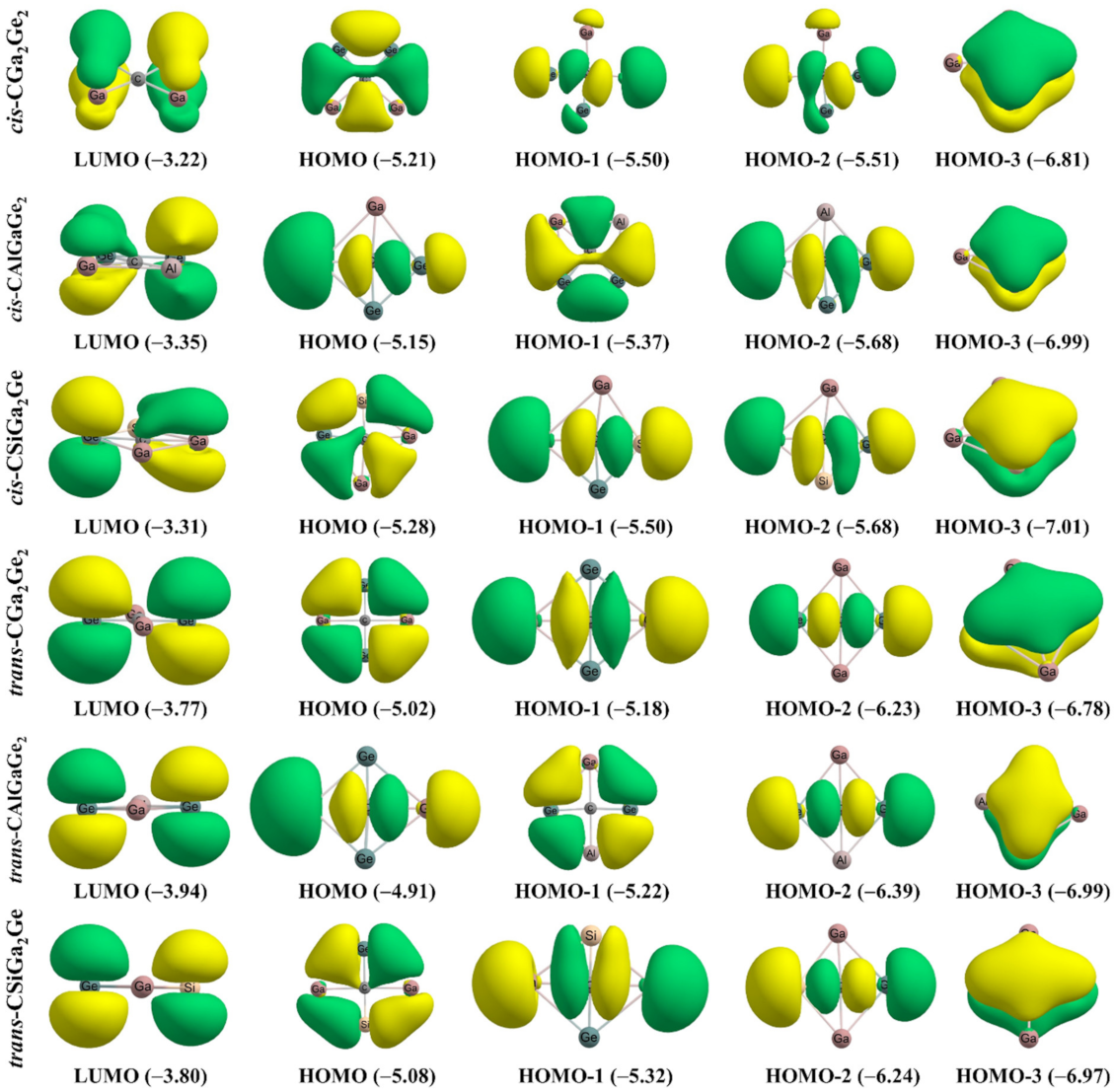
The molecular orbital analysis for the systems was carried out to obtain more information about the bonding. The molecular orbitals along with associated energies for all the systems in cis and trans isomers are presented in Figure 5. The lowest unoccupied molecular orbitals (LUMOs) are the π-type orbitals distributed over the four membered rings. The HOMO-3 molecular orbitals in the systems are π-type molecular orbitals and are delocalized over the whole molecule. In the HOMO-3 π-delocalized MO of the systems, significant contribution comes from the perpendicular 2pz orbital and 2px orbital of central ptC in cis and trans isomers, respectively. This σ- and π- delocalization of electron density support the electronic stabilization in the designed ptC systems. We discussed the electronic delocalization within the systems based on multi-center-2e bonding in the AdNDP section in detail. The highest occupied molecular orbital (HOMO), HOMO-1 and HOMO-2 orbitals, are σ-type molecular orbitals. The energy differences (ΔEH-L) between the HOMO and the LUMO are 1.99, 1.80, 1.97 eV, and 1.26, 0.98, 1.28 eV, for cis and trans isomers for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively. The cis isomers showed higher ΔEH-L values than that of the trans isomers indicating greater stability [58,59,60].
3.3. Natural Bond Orbital (NBO) Analysis
The distribution of natural charge in the studied ptC molecules and the WBI values for a bond was analyzed using the natural bond orbital scheme. This analysis showed that a significant amount of charge transfer occurred from the peripheral atoms to the central carbon atom and the C atom is highly negative in each ptC system. The values of the natural charges on the atoms are shown in Table 1. The natural charges on central C atoms vary from −2.14 |e| to −2.23 |e| in the ptC systems and the peripheral atoms show positive charges. The CAlGaGe2 system showed the highest negative charges and the CGa2Ge2 system showed the lowest negative charges on the central C atom, respectively. The charges on C atoms in the ptC systems are the same or almost the same in both cis and trans isomers but the charges on the peripheral atoms are different for the two isomers. The valence electronic configurations of C atoms in the systems are presented in Table 1. It is well known that, in ptC systems, the central C atoms act as both a σ-acceptor and a π-donor. In our present study, the σ-acceptor is evident from the highly negative charge on C centers in all the systems (see Table 1). From the valence electronic configuration of C centers as shown in Table 1, in the case of cis isomers, the population of 2pz orbital is lower than that of the 2px and 2py orbitals, and, in trans isomers the population of 2px orbital is lower than that of the 2py and 2pz orbitals of all the ptC systems. These lower populations of 2pz and 2px orbitals for cis and trans isomers, respectively, is a consequence of π-donation from the central carbon atom [34]. The cis isomers of the systems lie on the XY plane and the trans isomers of the systems lie on the YZ plane. The valence p orbitals along these perpendicular axes, i.e., the 2pz orbital and 2px orbital of central carbon atom in the cis and trans isomers of the systems, respectively, take part in delocalized π-bonding. The WBI values for C–Al, C–Si, C–Ga, and C–Ge bonds were computed for both isomers of all the ptC systems, and the numerical values are presented in Table 2. The WBI values for the C–Si bonds are 1.09 and 1.18 for cis and trans isomers of CSiGa2Ge system, indicating pure covalent bonds. Similarly for C–Ge bonds, the WBI values are in the range of 0.99 to 1.04 for cis isomers and 1.09 to 1.13 for trans isomers of the systems. These WBIC-Ge values indicate the covalent nature of the bonds. The WBI values for C–Al and C–Ga bonds are comparatively lower than that of the C–Si and C–Ge bonds, indicating the partial covalent character of these bonds. The WBI values for the C–Ga bonds are comparable with that reported in the theoretical study by Zhou et al. [61]. The WBI values for the C–Si and C–Ge bonds in trans isomers are a little bit higher than that in the cis isomers. However, for C–Al and C–Ga bonds, the WBI values in cis isomers are a little bit higher than those in the trans-isomers.
3.4. Adaptive Natural Density Partitioning (AdNDP) Analysis
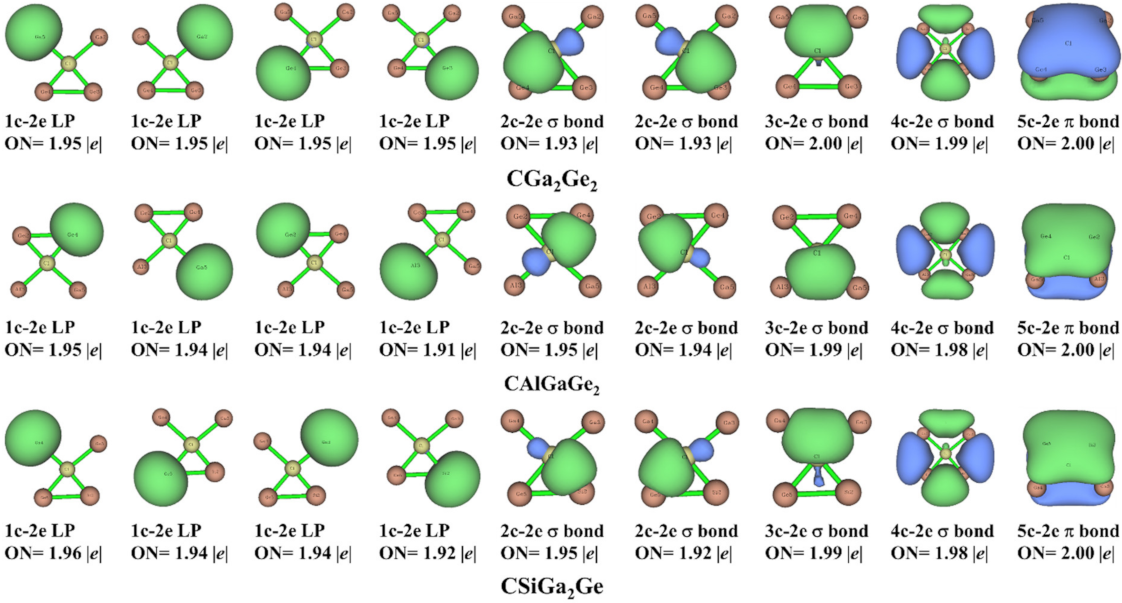
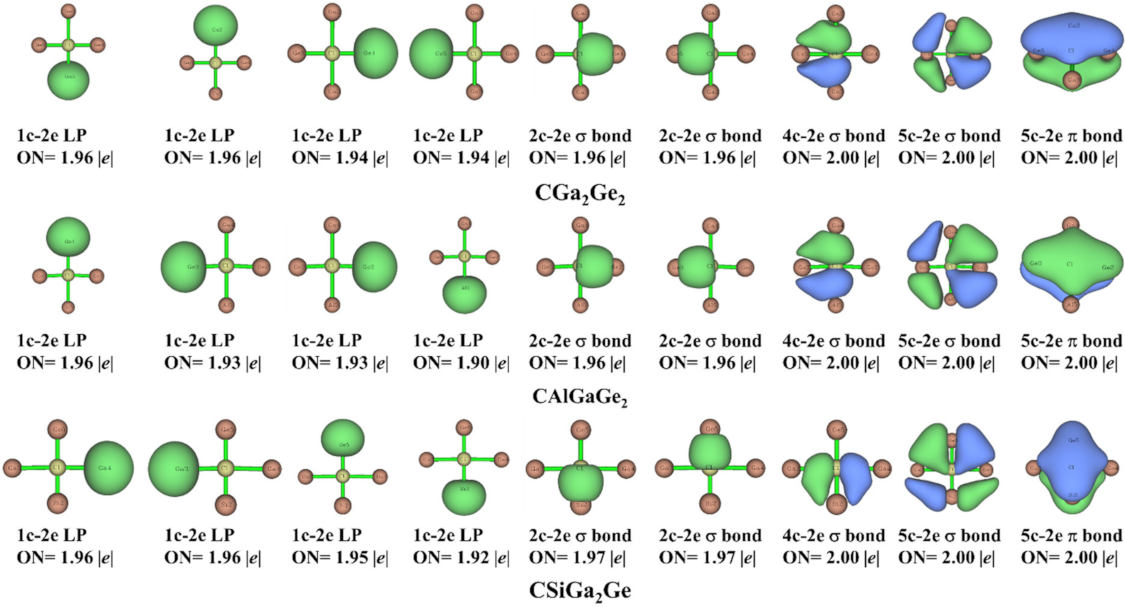
We carried out AdNDP analysis [62,63] in bonding context for both cis and trans isomers of the studied systems. We used this technique to investigate the presence of n-center-two-electron (nc-2e) bonds (n ranges from 1 (lone pair) to the maximum number of atoms (completely delocalized bonding)). Figure 6 and Figure 7 represent the results from this analysis for cis and trans isomers, respectively. The results showed the appearance of four lone pairs (1-center-two-electron bonds) on the four peripheral atoms and the occupation numbers (ON) are in between 1.91–1.96 |e| and 1.90–1.96 |e| in cis and trans isomers, respectively. There are two 2-center-two-electron (2c-2e) σ bonds in both isomers of the systems. Additionally, one 3-center-two-electron (3c-2e) σ bond, one 4-center-two-electron (4c-2e) σ bond, and one 5-center-two-electron (5c-2e) π-bond in cis isomers; and one 4-center-two-electron (4c-2e) σ bond, one 5-center-two-electron (5c-2e) σ bond, and one 5-center-two-electron (5c-2e) π-bond in trans isomers are present for all the systems. The occupation numbers of these nc-2e bonds in the systems under consideration are presented in Figure 6 and Figure 7 for cis and trans isomers, respectively. This analysis shows that in the designed ptC systems the central carbon atom is present in the 2π/6σ framework. Thus, the 2π/6σ frameworks and peripheral 4c-2e σ bonds are crucial for the stability of these ptC species. Both the σ- and π- delocalization of electron densities supported the stability of the designed system in planar form.
3.5. Atoms in Molecule (AIM) Analysis
We generated various electron density descriptors at all the (3, −1) bond critical points (BCPs) of the studied systems, and the numerical values are shown in Table 3. The contour plots of ∇2ρ(r) along the bond paths for the systems are presented in Figure 8. From the presented BCPs in Table 3, the existence of bonds between the middle carbon atom (C) and the four peripheral atoms is confirmed in all the systems. The low ρ(rc) values and positive ∇2ρ(rc) values suggest that there is a closed-shell type of bonding. From the negative values of H(rc) at the corresponding BCPs, it may be argued that the bonds have a partial covalent character. The values of −G(rc)/V(rc) at different BCPs are between 0.5 and 1.0, indicating the absence of a noncovalent interaction.
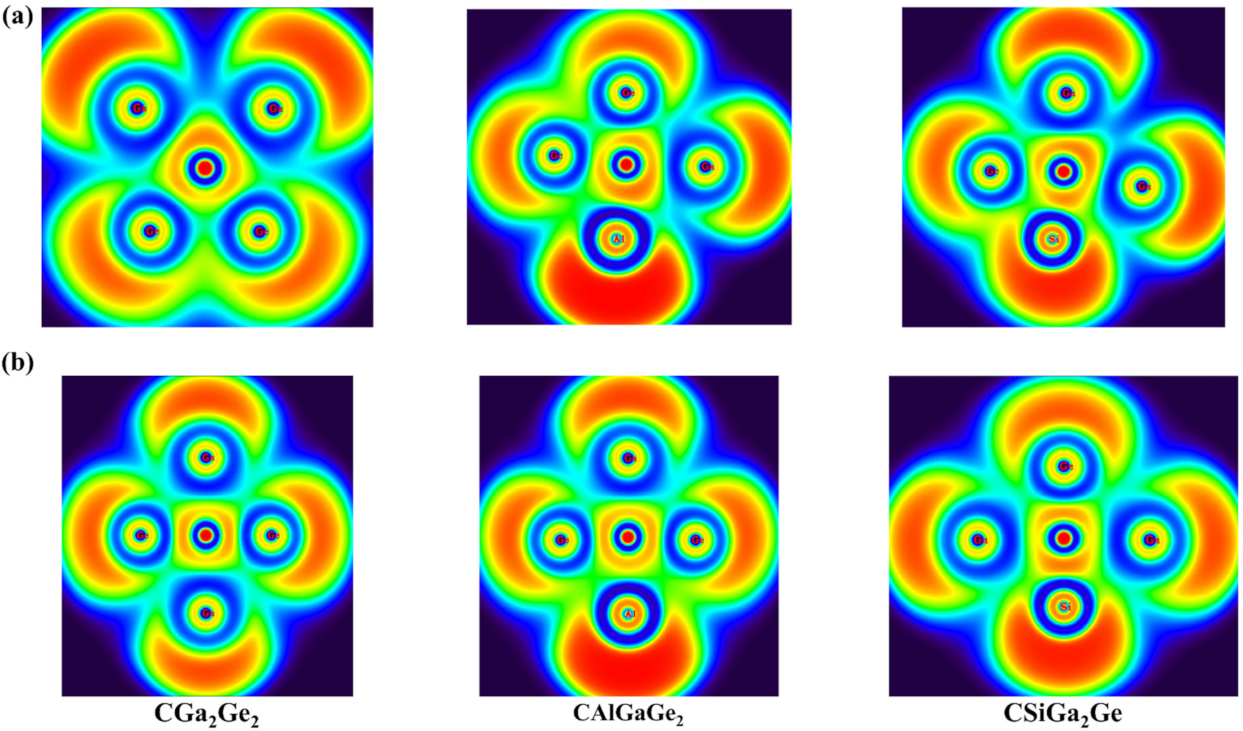
The ELF plots for both the isomers of the systems were generated, and the plots are presented in Figure 9. The ELF plots show the interaction among the middle C atom and the peripheral atoms and the electron densities are delocalized within the whole molecule. These ELF plots are in support of stability of the systems in planar geometries in terms of electron delocalization within the ptC systems.
3.6. Aromaticity Analysis
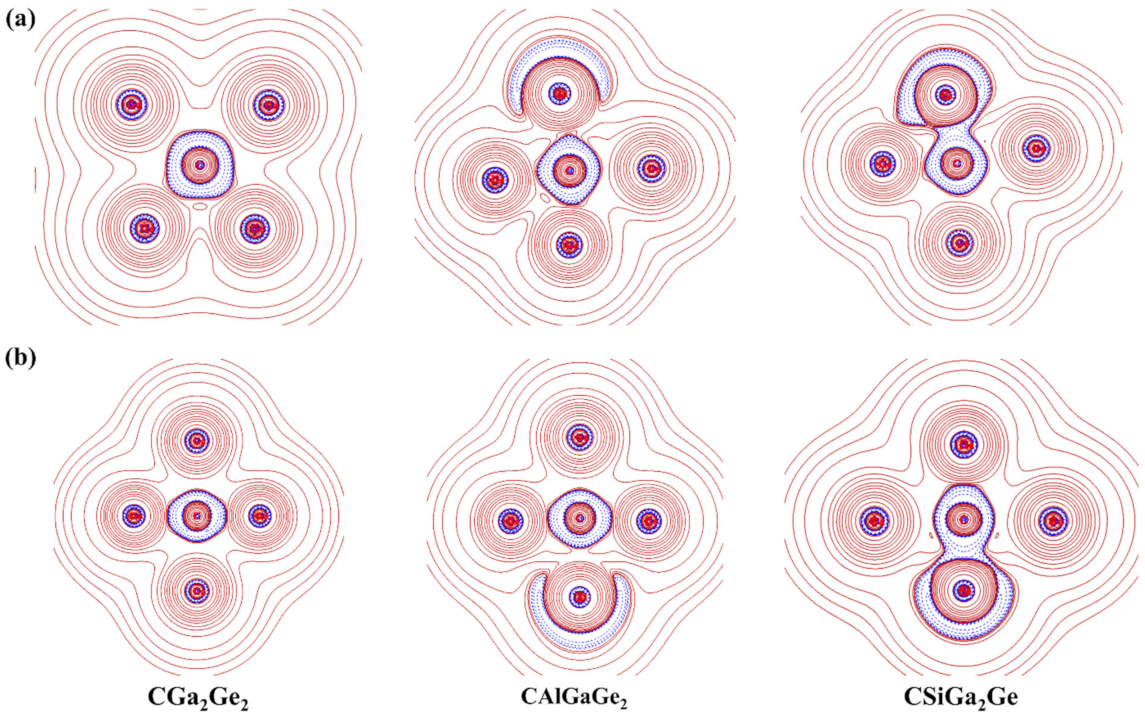
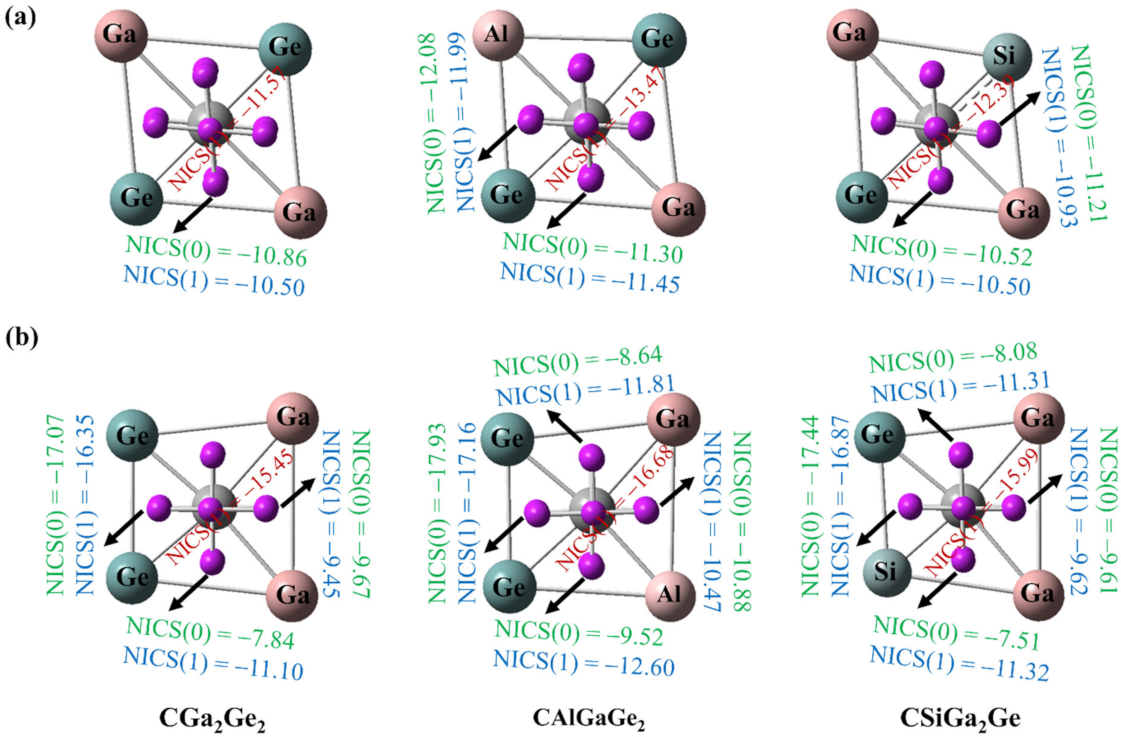
Aromatic molecules are cyclic, planar, conjugated, and have (4n + 2) π electrons. To predict the aromaticity of molecules, NICS is an efficient criterion proposed by Schleyer and co-workers [64,65]. NICS is calculated based on the use of magnetic shielding tensor for a dummy magnetic dipole considered at the center of an under-examination ring. It is called a nucleus-independent chemical shift, because there is no nucleus at the center of the molecular ring to experience the effective magnetic field. Molecules are considered aromatic based on the negative NICS values, and the positive NICS values indicate anti-aromatic systems. The non-aromatic systems have NICS values close to zero.
We computed both NICS(0) and NICS(1) at the middle of each triangle by placing a ghost atom at the plane and 1 Å above the plane. We also computed NICS(1) of the four membered ring by placing the ghost atom at 1 Å above the central C atom for all the systems for cis and trans isomers. The results of this analysis are presented in Figure 10. Both the NICS(0) and NICS(1) values for each triangle and the NICS(1) values at 1 Å above the central C atom of the ring are negative. These negative values of NICS(0) and NICS(1) are the indicators of σ- and π- aromaticity, albeit approximately. In aromatic systems, the electron densities are delocalized throughout the molecule. In all of our designed systems, the electron density is delocalized, as shown from the molecular orbitals, ELF plots, and the multi-center-2e-bonds of all the systems. So, these computed negative NICS values support the stability of the designed ptC systems.
4. Summary and Conclusions
The three neutral ptC systems considered in this work possess 18 valence electrons and show stability in planar form. The stability of both cis and trans isomers of these systems is reported in this work. The NICS calculations showed that the systems possess both σ- and π- aromaticities that support the stability of the designed systems. The ADMP simulation at 300 K and 500 K temperatures over 2000 fs of time revealed the kinetic stability of the molecules. The AIM analysis suggested the presence of bonds between the central C atom and the four peripheral atoms. We believe that this theoretical study will help in designing new ptC molecules.
Experimentally ptC systems were characterized in the gas phase. It is also our hope that the designed ptC systems could be interesting to the experimentalists. Considering all the supported results for the planar geometries of these studied systems, the experimental realization of these ptC systems could be possible in the gas phase or matrix isolation. In isolation, the systems may exist in planar form. When the systems are interacting with other species, the structures of the systems may or may not be planar; however, if the bound species create sufficient steric forces, the systems may exist in planar form.
Author Contributions
Conceptualization, P.D.; methodology, P.D. and P.K.C.; software, P.D.; validation, P.D. and P.K.C.; formal analysis, P.D.; investigation, P.K.C.; resources, P.K.C.; data curation, P.D.; writing—original draft preparation, P.D.; writing—review and editing, P.K.C.; visualization, P.D. and P.K.C.; supervision, P.K.C.; project administration, P.K.C. All authors have read and agreed to the published version of the manuscript.
Funding
DST, New Delhi, India for the J. C. Bose National Fellowship, grant number SR/S2/JCB-09/2009.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in article.
Acknowledgments
PKC thanks DST, New Delhi, India for the J. C. Bose National Fellowship. PD thanks UGC, New Delhi, India for the Research Fellowship.
Conflicts of Interest
The authors declare that they have no conflict of interests regarding the publication of this article, financial, and/or otherwise.
References
- Li, X.; Wang, L.S.; Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and ab Initio Study. J. Am. Chem. Soc. 1999, 121, 6033–6038. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Simons, J. Tetracoordinated Planar Carbon in Pentaatomic Molecules. J. Am. Chem. Soc. 1998, 120, 7967–7972. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chem. Int. Ed. Engl. 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Erker, G. Planar-Tetracoordinate Carbon: Making Stable Anti-van’t Hoff/Le Bel Compounds. Comment. Inorg. Chem. 1992, 13, 111–131. [Google Scholar] [CrossRef]
- Keese, R. Carbon Flatland: Planar Tetracoordinate Carbon and Fenestranes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Yu, S.; Ding, Y.H.; Bowen, K.H. Identifying the Hydrogenated Planar Tetracoordinate Carbon: A Combined Experimental and Theoretical Study of CAl4H and CAl4H−. J. Phys. Chem. Lett. 2017, 8, 2263–2267. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, H.F.; Wang, L.S.; Geske, G.; Boldyrev, A. Pentaatomic Tetracoordinate Planar Carbon, [CAl4]2−: A New Structural Unit and Its Salt Complexes. Angew. Chem. Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; von Ragué Schleyer, P.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Merino, G.; Méndez-Rojas, M.A.; Beltrán, H.I.; Corminboeuf, C.; Heine, T.; Vela, A. Theoretical Analysis of the Smallest Carbon Cluster Containing a Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 2004, 126, 16160–16169. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.; Wang, Z.X.; von Ragué Schleyer, P. Four Decades of the Chemistry of Planar Hypercoordinate Compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Thimmakondu, V.S. Flat Crown Ethers with Planar Tetracoordinate Carbon Atoms. Int. J. Quantum Chem. 2021, 121, e26479. [Google Scholar] [CrossRef]
- Yañez, O.; Vásquez-Espinal, A.; Báez-Grez, R.; Rabanal-León, W.A.; Osorio, E.; Ruiz, L.; Tiznado, W. Carbon Rings Decorated with Group 14 Elements: New Aromatic Clusters Containing Planar Tetracoordinate Carbon. New J. Chem. 2019, 43, 6781–6785. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Karton, A.; Thimmakondu, V.S. From High-Energy C7H2 Isomers with A Planar Tetracoordinate Carbon Atom to an Experimentally Known Carbene. J. Phys. Chem. A 2018, 122, 9054–9064. [Google Scholar] [CrossRef] [Green Version]
- Suresh, C.H.; Frenking, G. Direct 1-3 Metal-Carbon Bonding and Planar Tetracoordinated Carbon in Group 6 Metallacyclobutadienes. Organometallics 2010, 29, 4766–4769. [Google Scholar] [CrossRef]
- van’t Hoff, J.H. A Suggestion Looking to the Extension into Space of the Structural Formulas at Present Used in Chemistry, and a Note upon the Relation between the Optical Activity and the Chemical Constitution of Organic Compounds. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le-Bel, J.A. On the Relations Which Exist between the Atomic Formulas of Organic Compounds and the Rotatory Power of Their Solutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Monkhorst, H.J. Activation Energy for Interconversion of Enantiomers Containing an Asymmetric Carbon Atom without Breaking Bonds. Chem. Commun. 1968, 11, 1111–1112. [Google Scholar] [CrossRef]
- Hoffmann, R. The theoretical design of novel stabilized systems. Pure Appl. Chem. 1971, 28, 181–194. [Google Scholar] [CrossRef]
- Sateesh, B.; Srinivas Reddy, A.; Narahari Sastry, G. Towards Design of the Smallest Planar Tetracoordinate Carbon and Boron Systems. J. Comput. Chem. 2007, 28, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Job, N.; Karton, A.; Thirumoorthy, K.; Cooksy, A.L.; Thimmakondu, V.S. Theoretical Studies of SiC4H2 Isomers Delineate Three Low-Lying Silylidenes are Missing in the Laboratory. J. Phys. Chem. A 2020, 124, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.C.; Feng, L.Y.; Dong, C.; Zhai, H.J. Ternary 12-electron CBe3X3+ (X = H, Li, Na, Cu, Ag) Clusters: Planar Tetracoordinate Carbons and Superalkali Cations. Phys. Chem. Chem. Phys. 2019, 21, 22048–22056. [Google Scholar] [CrossRef]
- Nandula, A.; Trinh, Q.T.; Saeys, M.; Alexandrova, A.N. Origin of Extraordinary Stability of Square-Planar Carbon Atoms in Surface Carbides of Cobalt and Nickel. Angew. Chem. Int. Ed. 2015, 54, 5312–5316. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.H.; Contreras, M.; Ding, Y.H.; Merino, G. Planar Tetracoordinate Carbon versus Planar Tetracoordinate Boron: The Case of CB4 and Its Cation. J. Am. Chem. Soc. 2011, 133, 13228–13231. [Google Scholar] [CrossRef]
- Thirumoorthy, K.; Cooksy, A.; Thimmakondu, V.S. Si2C5H2 Isomers–Search Algorithms Versus Chemical Intuition. Phys. Chem. Chem. Phys. 2020, 22, 5865–5872. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Halla, J.O.C.; Wu, Y.B.; Wang, Z.X.; Islas, R.; Heine, T.; Merino, G. CAl4Be and CAl3Be2−: Global Minima with a Planar Pentacoordinate Carbon Atom. Chem. Commun. 2010, 46, 8776–8778. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.H.; Ding, Y.H.; Cabellos, J.L.; Osorio, E.; Islas, R.; Restrepo, A.; Merino, G. Planar tetracoordinate carbons with a double bond in CAl3E clusters. Phys. Chem. Chem. Phys. 2015, 17, 8769–8775. [Google Scholar] [CrossRef] [PubMed]
- von Ragué Schleyer, P.; Boldyrev, A.I. A new, general strategy for achieving planar tetracoordinate geometries for carbon and other second row periodic elements. J. Chem. Soc. Chem. Commun. 1991, 21, 1536–1538. [Google Scholar] [CrossRef]
- Wu, X.-F.; Cheng, Y.-X.; Guo, J.-C. CLiAl2E and CLi2AlE (E = P, As, Sb, Bi): Planar Tetracoordinate Carbon Clusters with 16 and 14 Valence Electrons. ACS Omega 2019, 4, 21311–21318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Job, N.; Khatun, M.; Thirumoorthy, K.; CH, S.S.R.; Chandrasekaran, V.; Anoop, A.; Thimmakondu, V.S. CAl4Mg0/−: Global Minima with a Planar Tetracoordinate Carbon Atom. Atoms 2021, 9, 24. [Google Scholar] [CrossRef]
- Wang, L.-S.; Boldyrev, A.I.; Li, X.; Simons, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.-S. Beyond Classical Stoichiometry: Experiment and Theory. J. Phys. Chem. A 2001, 105, 10759–10775. [Google Scholar] [CrossRef]
- Pei, Y.; An, W.; Ito, K.; von Ragué Schleyer, P.; Zeng, X.C. Planar Pentacoordinate Carbon in CAl5+: A Global Minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Cabellos, J.L.; Orozco, M.; Merino, G.; Zhao, L.; Chattaraj, P.K. Planar Pentacoordinate Carbon in CGa5+ Derivatives. Phys. Chem. Chem. Phys. 2018, 20, 12350–12355. [Google Scholar] [CrossRef] [PubMed]
- Vassilev-Galindo, V.; Pan, S.; Donald, J.K.; Merino, G. Planar Pentacoordinate Carbons. Nat. Chem. Rev. 2018, 2, 0114. [Google Scholar] [CrossRef]
- Exner, K.; von Ragué Schleyer, P. Planar Hexacoordinate Carbon: A Viable Possibility. Science 2000, 290, 1937–1940. [Google Scholar] [CrossRef]
- Averkiev, B.B.; Zubarev, D.Y.; Wang, L.M.; Huang, W.; Wang, L.S.; Boldyrev, A.I. Carbon Avoids Hypercoordination in CB6−, CB62−, and C2B5− Planar Carbon-Boron Clusters. J. Am. Chem. Soc. 2008, 130, 9248–9250. [Google Scholar] [CrossRef]
- Ito, K.; Chen, Z.; Corminboeuf, C.; Wannere, C.S.; Zhang, X.H.; Li, Q.S.; von Ragué Schleyer, P. Myriad Planar Hexacoordinate Carbon Molecules Inviting Synthesis. J. Am. Chem. Soc. 2007, 129, 1510–1511. [Google Scholar] [CrossRef]
- Zhang, C.F.; Han, S.J.; Wu, Y.B.; Lu, H.G.; Lu, G. Thermodynamic Stability versus Kinetic Stability: Is the Planar Hexacoordinate Carbon Species D3h CN3Mg3+ Viable? J. Phys. Chem. A 2014, 118, 3319–3325. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac-Hartree-Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revis on B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, H.B.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency, energy conservation and choice of initial conditions. J. Chem. Phys. 2001, 115, 10291–10302. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born-Oppenheimer dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef] [Green Version]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO 3.1 QCPE Bulletin. 1990, Volume 10, p. 58. Available online: https://www.scirp.org/(S(czeh2tfqw2orz553k1w0r45))/reference/referencespapers.aspx?referenceid=1595007 (accessed on 1 August 2021).
- Bader, R.F.W. Atoms in Molecules. In A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Cedillo, A.; Parr, R.G.; Arnett, E.M. Appraisal of Chemical Bond Making, Bond Breaking, and Electron Transfer in Solution in the Light of the Principle of Maximum Hardness. J. Org. Chem. 1995, 60, 4707–4714. [Google Scholar] [CrossRef]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef]
- Zhou, K. Theoretical studies on the pentaatomic planar tetracoordinate carbon molecules CGa3Si and CGa3Si−. Comput. Theor. Chem. 2013, 1009, 30–34. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing Intuitively Assessable Chemical Bonding Patterns in Organic Aromatic Molecules via Adaptive Natural Density Partitioning. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; Jiao, H.; Hommes, N.v.E.; Malkin, V.G.; Malkina, O.L. An Evaluation of the Aromaticity of Inorganic Rings: Refined Evidence from Magnetic Properties. J. Am. Chem. Soc. 1997, 119, 12669–12670. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The optimized geometries of the cyclic rings and the ptC systems. Bond lengths are given in Å unit. The values in the parentheses are the relative energies in kcal/mol. The energies of cis isomers are considered to be zero.
Figure 1.
The optimized geometries of the cyclic rings and the ptC systems. Bond lengths are given in Å unit. The values in the parentheses are the relative energies in kcal/mol. The energies of cis isomers are considered to be zero.

Figure 2.
Time evolution of total energy for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively.

Figure 3.
Snapshots at different time steps (time in fs) (a,c,e) at 300 K temperature and (b,d,f) at 500 K temperature of CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively, in cis geometry.
Figure 3.
Snapshots at different time steps (time in fs) (a,c,e) at 300 K temperature and (b,d,f) at 500 K temperature of CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively, in cis geometry.

Figure 4.
Snapshots at different time steps (time in fs) (a,c,e) at 300 K temperature and (b,d,f) at 500 K temperature of CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively, in trans geometry.
Figure 4.
Snapshots at different time steps (time in fs) (a,c,e) at 300 K temperature and (b,d,f) at 500 K temperature of CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems, respectively, in trans geometry.

Figure 5.
Plots of the molecular orbitals of the systems. The values in the parenthesis are the energies of the corresponding orbitals in the eV unit.
Figure 5.
Plots of the molecular orbitals of the systems. The values in the parenthesis are the energies of the corresponding orbitals in the eV unit.

Figure 6.
AdNDP bonding patterns of the systems in cis isomer with the occupation numbers (ONs).

Figure 7.
AdNDP bonding patterns of the systems in trans isomer with the occupation numbers (ONs).

Figure 8.
The plots of the Laplacian of electron density [∇2ρ(r)], blue dashed and red solid lines indicate ∇2ρ(r) < 0 and ∇2ρ(r) > 0 regions, respectively, for (a) cis isomer; (b) trans isomer, for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems.
Figure 8.
The plots of the Laplacian of electron density [∇2ρ(r)], blue dashed and red solid lines indicate ∇2ρ(r) < 0 and ∇2ρ(r) > 0 regions, respectively, for (a) cis isomer; (b) trans isomer, for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems.

Figure 9.
The color-filled plots of the electron localization function (ELF) basin for (a) cis isomer; (b) trans isomer, for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems.
Figure 9.
The color-filled plots of the electron localization function (ELF) basin for (a) cis isomer; (b) trans isomer, for CGa2Ge2, CAlGaGe2, and CSiGa2Ge systems.

Figure 10.
Nucleus independent chemical shifts (NICSs; in ppm) for (a) trans geometries; (b) cis geometries. The values in the red color indicate the NICS (1) values at 1 Å above the central C atom of the ring.
Figure 10.
Nucleus independent chemical shifts (NICSs; in ppm) for (a) trans geometries; (b) cis geometries. The values in the red color indicate the NICS (1) values at 1 Å above the central C atom of the ring.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The natural charges (q, |e|) on the atoms in the rings and the ptC systems and the valence electronic configuration of C atom in the ptC systems.
Table 1.
The natural charges (q, |e|) on the atoms in the rings and the ptC systems and the valence electronic configuration of C atom in the ptC systems.
| Systems | qC | qAl | qSi | qGa | qGe | Valence Electronic Configuration of C |
|---|---|---|---|---|---|---|
| cis-Ga2Ge2 | - | - | - | 0.32 0.32 | −0.32 −0.32 | - |
| trans-Ga2Ge2 | - | - | - | 0.21 0.21 | −0.21 −0.21 | - |
| cis-CGa2Ge2 | −2.14 | - | - | 0.59 0.59 | 0.49 0.49 | 2s1.606 2px1.583 2py1.598 2pz1.315 |
| trans-CGa2Ge2 | −2.14 | - | - | 0.50 0.50 | 0.58 0.58 | 2s1.548 2px1.408 2py1.552 2pz1.596 |
| cis-AlGaGe2 | - | 0.47 | - | 0.36 | −0.42 −0.42 | - |
| trans-AlGaGe2 | - | 0.33 | - | 0.15 | −0.17 −0.30 | - |
| cis-CAlGaGe2 | −2.22 | 0.64 | - | 0.59 | 0.48 0.51 | 2s1.612 2px1.593 2py1.638 2pz1.341 |
| trans-CAlGaGe2 | −2.23 | 0.55 | - | 0.54 | 0.57 0.57 | 2s1.556 2px1.412 2py1.555 2pz1.661 |
| cis-SiGa2Ge | - | - | −0.41 | 0.34 0.34 | −0.27 | - |
| trans-SiGa2Ge | - | - | −0.28 | 0.22 0.25 | −0.19 | - |
| cis-CSiGa2Ge | −2.16 | - | 0.47 | 0.59 0.60 | 0.51 | 2s1.590 2px1.581 2py1.635 2pz1.312 |
| trans-CSiGa2Ge | −2.18 | - | 0.58 | 0.51 0.51 | 0.59 | 2s1.528 2px1.421 2py1.590 2pz1.593 |
Table 2.
Wiberg bond indices (WBI) for some selected bonds in the ptC molecules.
| Complexes | WBI (C–Al) | WBI (C–Si) | WBI (C–Ga) | WBI (C–Ge) |
|---|---|---|---|---|
| cis-CGa2Ge2 | - | - | 0.37 0.37 | 1.04 1.04 |
| trans-CGa2Ge2 | - | - | 0.31 0.31 | 1.13 1.13 |
| cis-CAlGaGe2 | 0.33 | - | 0.34 | 1.02 1.03 |
| trans-CAlGaGe2 | 0.30 | - | 0.31 | 1.09 1.09 |
| cis-CSiGa2Ge | - | 1.09 | 0.34 0.36 | 0.99 |
| trans-CSiGa2Ge | - | 1.18 | 0.29 0.29 | 1.09 |
Table 3.
Electron Density (ρ(rc)), Laplacian of Electron Density (∇2ρ(rc)), Kinetic Energy Density (G(rc)), Potential Energy Density (V(rc)), Total Energy Density (H(rc)) for the ptC systems.
Table 3.
Electron Density (ρ(rc)), Laplacian of Electron Density (∇2ρ(rc)), Kinetic Energy Density (G(rc)), Potential Energy Density (V(rc)), Total Energy Density (H(rc)) for the ptC systems.
| Complexes | BCP | ρ(rc) | ∇2ρ(rc) | G(rc) | V(rc) | H(rc) | ELF | −G(rc)/V(rc) | G(rc)/ρ(rc) |
|---|---|---|---|---|---|---|---|---|---|
| cis-CGa2Ge2 | C-Ga | 0.078 | 0.131 | 0.060 | −0.088 | −0.028 | 0.311 | 0.682 | 0.769 |
| C-Ge | 0.115 | 0.139 | 0.092 | −0.148 | −0.057 | 0.420 | 0.622 | 0.800 | |
| trans-CGa2Ge2 | C-Ga | 0.063 | 0.098 | 0.044 | −0.063 | −0.019 | 0.295 | 0.698 | 0.698 |
| C-Ge | 0.134 | 0.207 | 0.124 | −0.196 | −0.072 | 0.399 | 0.633 | 0.925 | |
| cis-CAlGaGe2 | C-Al | 0.064 | 0.224 | 0.072 | −0.088 | −0.016 | 0.224 | 0.818 | 1.125 |
| C-Ga | 0.075 | 0.125 | 0.057 | −0.083 | −0.026 | 0.125 | 0.687 | 0.760 | |
| C-Ge | 0.112 | 0.137 | 0.089 | −0.144 | −0.055 | 0.417 | 0.618 | 0.795 | |
| trans-CAlGaGe2 | C-Al | 0.053 | 0.152 | 0.052 | −0.065 | −0.014 | 0.149 | 0.800 | 0.981 |
| C-Ga | 0.067 | 0.103 | 0.047 | −0.069 | −0.022 | 0.308 | 0.681 | 0.701 | |
| C-Ge | 0.131 | 0.198 | 0.119 | −0.188 | −0.069 | 0.399 | 0.633 | 0.908 | |
| cis-CSiGa2Ge | C-Si | 0.112 | 0.284 | 0.136 | −0.201 | −0.065 | 0.233 | 0.677 | 1.214 |
| C-Ga | 0.074 | 0.126 | 0.057 | −0.082 | −0.025 | 0.305 | 0.695 | 0.770 | |
| C-Ge | 0.111 | 0.139 | 0.088 | −0.142 | −0.054 | 0.139 | 0.620 | 0.793 | |
| trans-CSiGa2Ge | C-Si | 0.126 | 0.415 | 0.175 | −0.246 | −0.071 | 0.211 | 0.711 | 1.389 |
| C-Ga | 0.061 | 0.099 | 0.043 | −0.062 | −0.018 | 0.287 | 0.694 | 0.705 | |
| C-Ge | 0.135 | 0.209 | 0.126 | −0.199 | −0.073 | 0.400 | 0.633 | 0.933 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Das, P.; Chattaraj, P.K. In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon. Atoms 2021, 9, 65. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030065
AMA Style
Das P, Chattaraj PK. In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon. Atoms. 2021; 9(3):65. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030065
Chicago/Turabian StyleDas, Prasenjit, and Pratim Kumar Chattaraj. 2021. "In Silico Studies on Selected Neutral Molecules, CGa2Ge2, CAlGaGe2, and CSiGa2Ge Containing Planar Tetracoordinate Carbon" Atoms 9, no. 3: 65. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9030065
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.