
MIDESP: Mutual Information-Based Detection of Epistatic SNP Pairs for Qualitative and Quantitative Phenotypes

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.1.1. Bovine Tuberculosis (BT)
2.1.2. Egg Weight (EW)
2.2. Method
2.2.1. Background on Information Theoretic Measures
- is the digamma function;
- for a given sample, , refers to the number of samples for which the genotype x is the same as the genotype of ;
- d is the distance between sample and its kth-nearest neighbor with the same genotype as , defined as the absolute difference between their phenotypes and ;
- is assigned the number of samples where the absolute difference between their phenotypes and the phenotype is less than or equal to d, irrespective of the genotypes.
2.2.2. Identification of Epistatic Interactions between SNP Pairs
2.2.3. Detection of SNPs with Strong Association Signals
2.2.4. Reduction of the Background Associations between SNPs and Phenotype
2.2.5. Validation of the Epistatic Interactions
2.2.6. Implementation
3. Results
3.1. Analysis of Simulated Datasets for Parameter Setting
3.2. Illustration of Background Associations and Its Correction Using APC
3.3. Bovine Tuberculosis Dataset
3.4. Egg Weight Dataset
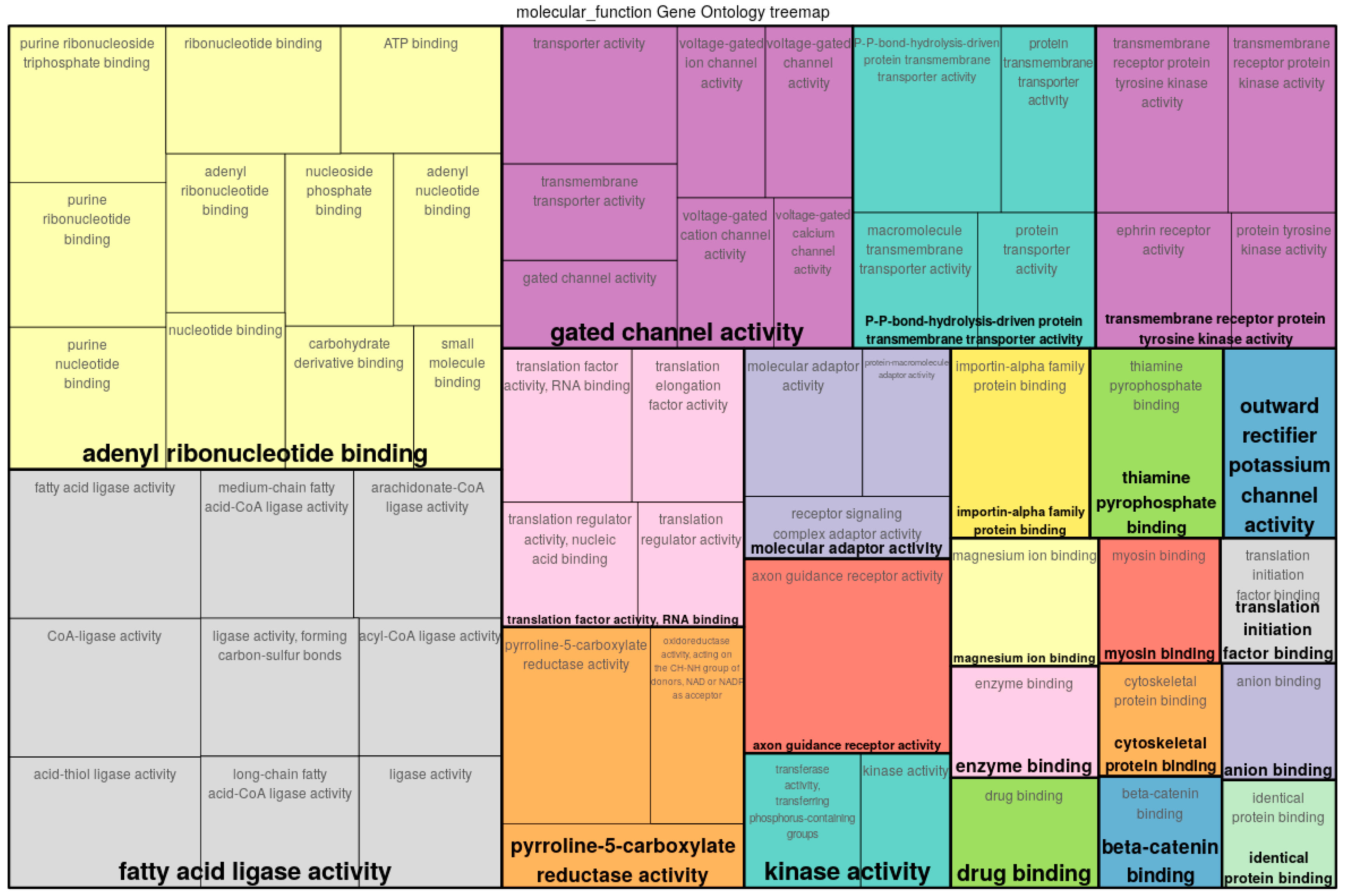
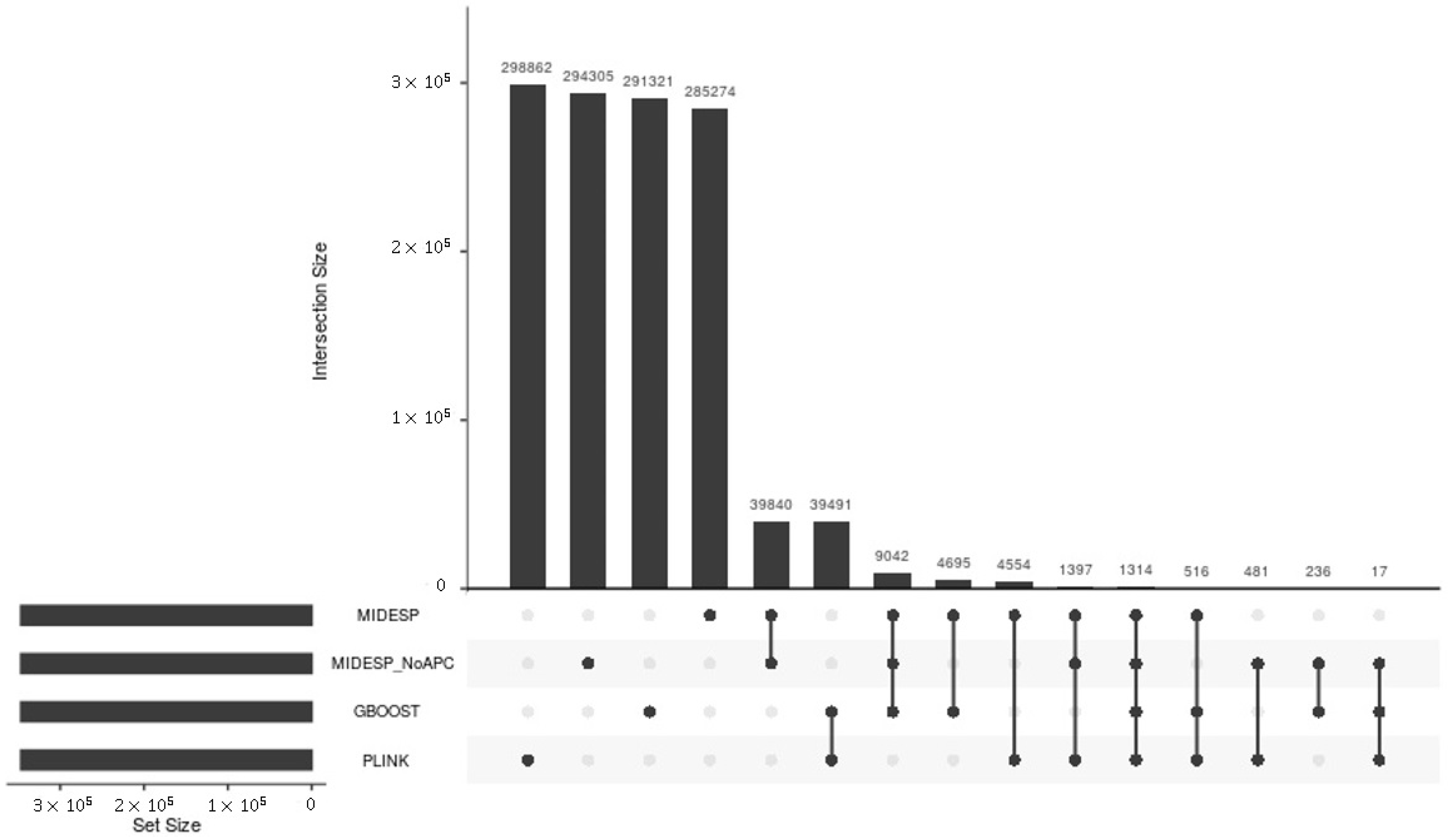
3.5. Comparisons with Existing Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | Single-nucleotide polymorphism |
| GWAS | Genome-wide association studies |
| MI | Mutual information |
| NMI | Normalized mutual information |
| BT | Bovine tuberculosis |
| EW | Egg weight |
| APC | Average product correction |
| FDR | False discovery rate |
| GO | Gene Ontology |
References
- Wei, W.H.; Hemani, G.; Haley, C. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Richards, S.; Carbone, M.A.; Zhu, D.; Anholt, R.R.H.; Ayroles, J.F.; Duncan, L.; Jordan, K.W.; Lawrence, F.; Magwire, M.M.; et al. Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proc. Natl. Acad. Sci. USA 2012, 109, 15553–15559. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.H.; Asselbergs, F.W.; Williams, S.M. Bioinformatics challenges for genome-wide association studies. Bioinformatics 2010, 26, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.H.; Williams, S.M. Epistasis and Its Implications for Personal Genetics. Am. J. Hum. Genet. 2009, 85, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Marchini, J.; Donnelly, P.; Cardon, L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005, 37, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Kanazawa, H.; Fujimoto, S.; Hirata, K. Epistatic effects of multiple receptor genes on pathophysiology of asthma—Its limits and potential for clinical application. Med. Sci. Monit. 2014, 20, 64–71. [Google Scholar]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-Dimensionality Reduction Reveals High-Order Interactions among Estrogen-Metabolism Genes in Sporadic Breast Cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.M.; Ritchie, M.D.; Moore, J.H.; Park, J.Y.; Lee, K.-U.; Shin, H.D.; Lee, H.K.; Park, K.S. Multifactor-dimensionality reduction shows a two-locus interaction associated with Type 2 diabetes mellitus. Diabetologia 2004, 47, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Carlborg, O.; Hocking, P.; Burt, D.; Haley, C. Simultaneous mapping of epistatic QTL in chickens reveals clusters of QTL pairs with similar genetic effects on growth. Genet. Res. 2004, 83 3, 197–209. [Google Scholar] [CrossRef]
- Le Rouzic, A.; Alvarez-Castro, J.M.; Carlborg, O. Dissection of the genetic architecture of body weight in chicken reveals the impact of epistasis on domestication traits. Genetics 2008, 179, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Mackay, T.F.C. Epistasis and quantitative traits: Using model organisms to study gene–gene interactions. Nat. Rev. Genet. 2014, 15, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaust, J.; Hadlich, F.; Weikard, R.; Kuehn, C. Epistatic interactions between at least three loci determine the “rat-tail” phenotype in cattle. Genet. Sel. Evol. 2016, 48, 26. [Google Scholar] [CrossRef] [Green Version]
- Kramer, L.M.; Ghaffar, M.A.A.; Koltes, J.E.; Fritz-Waters, E.R.; Mayes, M.S.; Sewell, A.D.; Weeks, N.T.; Garrick, D.J.; Fernando, R.L.; Ma, L.; et al. Epistatic interactions associated with fatty acid concentrations of beef from angus sired beef cattle. BMC Genom. 2016, 17, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Würschum, T.; Maurer, H.P.; Schulz, B.; Möhring, J.; Reif, J.C. Genome-wide association mapping reveals epistasis and genetic interaction networks in sugar beet. Theor. Appl. Genet. 2011, 123, 109–118. [Google Scholar] [CrossRef]
- Hu, Z.; Li, Y.; Song, X.; Han, Y.; Cai, X.; Xu, S.; Li, W. Genomic value prediction for quantitative traits under the epistatic model. BMC Genet. 2011, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Xu, S.; Cai, X. Whole-Genome Quantitative Trait Locus Mapping Reveals Major Role of Epistasis on Yield of Rice. PLoS ONE 2014, 9, e87330. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, A.; Monir, M.; Meng, X.; Rahaman, M.; Chen, H.; Chen, M. Identification of epistasis loci underlying rice flowering time by controlling population stratification and polygenic effect. DNA Res. 2018, 26, 119–130. [Google Scholar] [CrossRef]
- Mathew, B.; Léon, J.; Sannemann, W.; Sillanpää, M.J. Detection of Epistasis for Flowering Time Using Bayesian Multilocus Estimation in a Barley MAGIC Population. Genetics 2018, 208, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlborg, Ö.; Haley, C.S. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 2004, 5, 618–625. [Google Scholar] [CrossRef]
- Cordell, H.J. Epistasis: What it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 2002, 11, 2463–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.R.; Kardia, S.L.; Ferrell, R.E.; Sing, C.F. A combinatorial partitioning method to identify multilocus genotypic partitions that predict quantitative trait variation. Genome Res. 2001, 11, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culverhouse, R. The Use of the Restricted Partition Method with Case-Control Data. Hum. Hered. 2007, 63, 93–100. [Google Scholar] [CrossRef]
- Li, X. A fast and exhaustive method for heterogeneity and epistasis analysis based on multi-objective optimization. Bioinformatics 2017, 33, 2829–2836. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, S.; Zou, F.; Wang, W. TEAM: Efficient two-locus epistasis tests in human genome-wide association study. Bioinformatics 2010, 26, i217–i227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, J.S. Bayesian inference of epistatic interactions in case-control studies. Nat. Genet. 2007, 39, 1167–1173. [Google Scholar] [CrossRef]
- Tang, W.; Wu, X.; Jiang, R.; Li, Y. Epistatic Module Detection for Case-Control Studies: A Bayesian Model with a Gibbs Sampling Strategy. PLoS Genet. 2009, 5, e1000464. [Google Scholar] [CrossRef] [PubMed]
- Serretti, A.; Smeraldi, E. Neural network analysis in pharmacogenetics of mood disorders. BMC Med. Genet. 2004, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Motsinger-Reif, A.A.; Dudek, S.M.; Hahn, L.W.; Ritchie, M.D. Comparison of approaches for machine-learning optimization of neural networks for detecting gene-gene interactions in genetic epidemiology. Genet. Epidemiol. 2008, 32, 325–340. [Google Scholar] [CrossRef]
- Uppu, S.; Krishna, A.; Gopalan, R. Towards Deep Learning in genome-Wide Association Interaction studies. In Proceedings of the 20th Pacific Asia Conference on Information Systems, PACIS 2016, Chiayi, Taiwan, 27 June–1 July; Volume 20.
- Wang, H.; Yue, T.; Yang, J.; Wu, W.; Xing, E.P. Deep mixed model for marginal epistasis detection and population stratification correction in genome-wide association studies. BMC Bioinform. 2019, 20, 656. [Google Scholar] [CrossRef]
- Xie, Q.; Ratnasinghe, L.D.; Hong, H.; Perkins, R.; Tang, Z.; Hu, N.; Taylor, P.R.; Tong, W. Decision Forest Analysis of 61 Single Nucleotide Polymorphisms in a Case-Control Study of Esophageal Cancer: A novel method. BMC Bioinform. 2005, 6, S4. [Google Scholar] [CrossRef] [Green Version]
- Winham, S.J.; Colby, C.L.; Freimuth, R.R.; Wang, X.; de Andrade, M.; Huebner, M.; Biernacka, J.M. SNP interaction detection with Random Forests in high-dimensional genetic data. BMC Bioinform. 2012, 13, 164. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.A.; Yu, Y.; Cupples, L.A.; Farrer, L.A.; Lunetta, K.L. Performance of random forest when SNPs are in linkage disequilibrium. BMC Bioinform. 2009, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.F.; König, I.R.; Ziegler, A. On safari to Random Jungle: A fast implementation of Random Forests for high-dimensional data. Bioinformatics 2010, 26, 1752–1758. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Koike, A. SNPInterForest: A new method for detecting epistatic interactions. BMC Bioinform. 2011, 12, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Yang, C.; Yang, Q.; Xue, H.; Fan, X.; Tang, N.L.S.; Yu, W. BOOST: A fast approach to detecting gene-gene interactions in genome-wide case-control studies. Am. J. Hum. Genet. 2010, 87, 325–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leem, S.; Jeong, H.; Lee, J.; Wee, K.; Sohn, K. Fast detection of high-order epistatic interactions in genome-wide association studies using information theoretic measure. Comput. Biol. Chem. 2014, 50, 19–28. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Parida, L. Muse: A multi-locus sampling-based epistasis algorithm for quantitative genetic trait prediction. In Pacific Symposium on Biocomputing 2017; World Scientific: Singapore, 2017; pp. 426–437. [Google Scholar]
- Tuo, S. FDHE-IW: A Fast Approach for Detecting High-Order Epistasis in Genome-Wide Case-Control Studies. Genes 2018, 9, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastassiou, D. Computational analysis of the synergy among multiple interacting genes. Mol. Syst. Biol. 2007, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chen, Y.; Kiralis, J.W.; Collins, R.L.; Wejse, C.; Sirugo, G.; Williams, S.M.; Moore, J.H. An information-gain approach to detecting three-way epistatic interactions in genetic association studies. J. Am. Med. Inform. Assoc. 2013, 20, 630–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anunciação, O.; Vinga, S.; Oliveira, A.L. Using Information Interaction to Discover Epistatic Effects in Complex Diseases. PLoS ONE 2013, 8, e76300. [Google Scholar] [CrossRef]
- Wienbrandt, L.; Kassens, J.C.; Hübenthal, M.; Ellinghaus, D. Fast Genome-Wide Third-order SNP Interaction Tests with Information Gain on a Low-cost Heterogeneous Parallel FPGA-GPU Computing Architecture. Procedia Comput. Sci. 2017, 108, 596–605. [Google Scholar] [CrossRef]
- Ponte-Fernández, C.; González-Domínguez, J.; Martín, M.J. Fast search of third-order epistatic interactions on CPU and GPU clusters. Int. J. High Perform. Comput. Appl. 2020, 34, 20–29. [Google Scholar] [CrossRef]
- He, D.; Parida, L. Does encoding matter? A novel view on the quantitative genetic trait prediction problem. BMC Bioinform. 2016, 17, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, J.W.R.; Gao, N.; Cardoso, D.F.; Wimmer, V.; Erbe, M.; Cantet, R.J.C.; Simianer, H. Genomic prediction with epistasis models: On the marker-coding-dependent performance of the extended GBLUP and properties of the categorical epistasis model (CE). BMC Bioinform. 2017, 18, 3. [Google Scholar] [CrossRef] [Green Version]
- Martini, J.W.R.; Rosales, F.; Ha, N.; Heise, J.; Wimmer, V.; Kneib, T. Lost in Translation: On the Problem of Data Coding in Penalized Whole Genome Regression with Interactions. G3 Genes Genomes Genet. 2019, 9, 1117–1129. [Google Scholar] [CrossRef] [Green Version]
- Mangin, B.; Siberchicot, A.; Nicolas, S.; Doligez, A.; This, P.; Cierco-Ayrolles, C. Novel measures of linkage disequilibrium that correct the bias due to population structure and relatedness. Heredity 2012, 108, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Mezmouk, S.; Dubreuil, P.; Bosio, M.; Décousset, L.; Charcosset, A.; Praud, S.; Mangin, B. Effect of population structure corrections on the results of association mapping tests in complex maize diversity panels. Theor. Appl. Genet. 2011, 122, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Ross, B.C. Mutual Information between Discrete and Continuous Data Sets. PLoS ONE 2014, 9, e87357. [Google Scholar] [CrossRef]
- Dunn, S.D.; Wahl, L.M.; Gloor, G.B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics 2007, 24, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Ramzan, F.; Gültas, M.; Bertram, H.; Cavero, D.; Schmitt, A.O. Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations. Genes 2020, 11, 892. [Google Scholar] [CrossRef]
- Ramzan, F.; Klees, S.; Schmitt, A.O.; Cavero, D.; Gültas, M. Identification of Age-Specific and Common Key Regulatory Mechanisms Governing Eggshell Strength in Chicken Using Random Forests. Genes 2020, 11, 464. [Google Scholar] [CrossRef] [PubMed]
- Joiret, M.; Mahachie John, J.M.; Gusareva, E.S.; Van Steen, K. Confounding of linkage disequilibrium patterns in large scale DNA based gene-gene interaction studies. BioData Min. 2019, 12, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, M.L.; Bishop, S.C.; Woolliams, J.A.; Pong-Wong, R.; Allen, A.R.; McBride, S.H.; Ryder, J.J.; Wright, D.M.; Skuce, R.A.; McDowell, S.W.J.; et al. Genome-wide association study identifies novel loci associated with resistance to bovine tuberculosis. Heredity 2014, 112, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Sun, C.; Yan, Y.; Li, G.; Wu, G.; Liu, A.; Yang, N. Genome-Wide Association Analysis of Age-Dependent Egg Weights in Chickens. Front. Genet. 2018, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; John Wiley: New York, NY, USA, 1991. [Google Scholar]
- Dionisio, A.; Menezes, R.; Mendes, D.A. Mutual information: A measure of dependency for nonlinear time series. Phys. A Stat. Mech. Its Appl. 2004, 344, 326–329. [Google Scholar] [CrossRef]
- Kvålseth, T.O. On Normalized Mutual Information: Measure Derivations and Properties. Entropy 2017, 19, 631. [Google Scholar] [CrossRef] [Green Version]
- Gültas, M.; Haubrock, M.; Tüysüz, N.; Waack, S. Coupled mutation finder: A new entropy-based method quantifying phylogenetic noise for the detection of compensatory mutations. BMC Bioinform. 2012, 13, 225. [Google Scholar] [CrossRef] [Green Version]
- Gültas, M.; Düzgün, G.; Herzog, S.; Jäger, S.J.; Meckbach, C.; Wingender, E.; Waack, S. Quantum coupled mutation finder: Predicting functionally or structurally important sites in proteins using quantum Jensen-Shannon divergence and CUDA programming. BMC Bioinform. 2014, 15, 96. [Google Scholar] [CrossRef] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Walsh, B. Multiple comparisons: Bonferroni Corrections and False Discovery Rates. In Lecture Notes for EEB 581; Department of Ecology and Evolutionary Biology, University of Arizona: Tucson, AZ, USA, 2004; pp. 1–17. [Google Scholar]
- Gültas, M. Development of novel Classical and Quantum Information Theory Based Methods for the Detection of Compensatory Mutations in MSAs 2014. Available online: https://hdl.handle.net/11858/00-1735-0000-0022-5EB0-1 (accessed on 14 September 2021).
- Meckbach, C.; Tacke, R.; Hua, X.; Waack, S.; Wingender, E.; Gültas, M. PC-TraFF: Identification of potentially collaborating transcription factors using pointwise mutual information. BMC Bioinform. 2015, 16, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2019, 48, D682–D688. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Mekonnen, Y.A.; Gültas, M.; Effa, K.; Hanotte, O.; Schmitt, A.O. Identification of candidate signature genes and key regulators associated with Trypanotolerance in the Sheko Breed. Front. Genet. 2019, 10, 1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingender, E.; Kel, A. geneXplain–eine integrierte Bioinformatik-Plattform. BIOspektrum 2012, 18, 554–556. [Google Scholar] [CrossRef]
- Cao, X.; Yu, G.; Liu, J.; Jia, L.; Wang, J. Clustermi: Detecting high-order snp interactions based on clustering and mutual information. Int. J. Mol. Sci. 2018, 19, 2267. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Yu, Z.; An, J.; Han, G.; Ma, Y.; Tang, R. A two-stage mutual information based Bayesian Lasso algorithm for multi-locus genome-wide association studies. Entropy 2020, 22, 329. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, C.; Hu, Y. Utilizing mutual information for detecting rare and common variants associated with a categorical trait. PeerJ 2016, 4, e2139. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Wang, Y. Mutual information and linkage disequilibrium based SNP association study by grouping case-control. Genes Genom. 2011, 33, 65–73. [Google Scholar] [CrossRef]
- Speed, D.; Hemani, G.; Johnson, M.R.; Balding, D.J. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012, 91, 1011–1021. [Google Scholar] [CrossRef] [Green Version]
- Kraskov, A.; Stögbauer, H.; Grassberger, P. Estimating mutual information. Phys. Rev. E 2004, 69. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Jeong, H.; Kim, D.; Wee, K.; Park, H.; Kim, S.; Sohn, K. Integrative information theoretic network analysis for genome-wide association study of aspirin exacerbated respiratory disease in Korean population. BMC Med. Genom. 2017, 10, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, D.; Pires, D.; Perdigão, J.; Couto, I.; Portugal, I.; Martins, M.; Amaral, L.; Anes, E.; Viveiros, M. Ion channel blockers as antimicrobial agents, efflux inhibitors, and enhancers of macrophage killing activity against drug resistant Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0149326. [Google Scholar] [CrossRef]
- Viveiros, M.; Martins, M.; Rodrigues, L.; Machado, D.; Couto, I.; Ainsa, J.; Amaral, L. Inhibitors of mycobacterial efflux pumps as potential boosters for anti-tubercular drugs. Expert Rev. Anti-Infect. Ther. 2012, 10, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Viveiros, M.; Couto, I.; Amaral, L. Targeting human macrophages for enhanced killing of intracellular XDR-TB and MDR-TB. Int. J. Tuberc. Lung Dis. 2009, 13, 569–573. [Google Scholar]
- Gupta, S.; Salam, N.; Srivastava, V.; Singla, R.; Behera, D.; Khayyam, K.U.; Korde, R.; Malhotra, P.; Saxena, R.; Natarajan, K. Voltage gated calcium channels negatively regulate protective immunity to Mycobacterium tuberculosis. PLoS ONE 2009, 4, e5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anes, E. Acting on actin during bacterial infection. In Cytoskeleton Structure, Dynamics, Function and Disease; Jimenez-Lopez, J.C., Ed.; IntechOpen: London, UK, 2017; Chapter 13; pp. 257–278. [Google Scholar] [CrossRef]
- Hestvik, A.L.K.; Hmama, Z.; Av-Gay, Y. Mycobacterial manipulation of the host cell. FEMS Microbiol. Rev. 2005, 29, 1041–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guérin, I.; de Chastellier, C. Pathogenic mycobacteria disrupt the macrophage actin filament network. Infect. Immun. 2000, 68, 2655–2662. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt, P.; Marion, S.; Pires, D.; Santos, L.; Lastrucci, C.; Carmo, N.; Blake, J.; Benes, V.; Griffiths, G.; Neyrolles, O.; et al. Actin-binding protein regulation by microRNAs as a novel microbial strategy to modulate phagocytosis by host cells: The case of N-Wasp and miR-142-3p. Front. Cell. Infect. Microbiol. 2013, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yao, Y.; Wu, J.; Deng, Z.; Gu, T.; Tang, X.; Cheng, Y.; Li, G. The mechanism of cytoskeleton protein β-actin and cofilin-1 of macrophages infected by Mycobacterium avium. Am. J. Transl. Res. 2016, 8, 1055. [Google Scholar]
- Levite, M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr. Opin. Pharmacol. 2008, 8, 460–471. [Google Scholar] [CrossRef]
- Pacheco, R.; Riquelme, E.; Kalergis, A.M. Emerging evidence for the role of neurotransmitters in the modulation of T cell responses to cognate ligands. In Central Nervous System Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Central Nervous System Agents); Bentham Science Publishers: Sharjah, United Arab Emirates, 2010; Volume 10, pp. 65–83. [Google Scholar]
- Skinner, M.A.; Parlane, N.; McCarthy, A.; Buddle, B.M. Cytotoxic T-cell responses to Mycobacterium bovis during experimental infection of cattle with bovine tuberculosis. Immunology 2003, 110, 234–241. [Google Scholar] [CrossRef]
- Villarreal-Ramos, B.; McAulay, M.; Chance, V.; Martin, M.; Morgan, J.; Howard, C.J. Investigation of the role of CD8+ T cells in bovine tuberculosis in vivo. Infect. Immun. 2003, 71, 4297–4303. [Google Scholar] [CrossRef] [Green Version]
- Pollock, J.M.; Neill, S.D. Mycobacterium boviss infection and tuberculosis in cattle. Vet. J. 2002, 163, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Finlay, E.K.; Berry, D.P.; Wickham, B.; Gormley, E.P.; Bradley, D.G. A genome wide association scan of bovine tuberculosis susceptibility in Holstein-Friesian dairy cattle. PLoS ONE 2012, 7, e30545. [Google Scholar]
- Pacheco, R.; Gallart, T.; Lluis, C.; Franco, R. Role of glutamate on T-cell mediated immunity. J. Neuroimmunol. 2007, 185, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ganor, Y.; Levite, M. The neurotransmitter glutamate and human T cells: Glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J. Neural Transm. 2014, 121, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- El Masri, R.; Delon, J. RHO GTPases: From new partners to complex immune syndromes. Nat. Rev. Immunol. 2021, 21, 499–513. [Google Scholar] [CrossRef]
- Bokoch, G.M. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005, 15, 163–171. [Google Scholar] [CrossRef]
- Chopra, P.; Koduri, H.; Singh, R.; Koul, A.; Ghildiyal, M.; Sharma, K.; Tyagi, A.K.; Singh, Y. Nucleoside diphosphate kinase of Mycobacterium tuberculosis acts as GTPase-activating protein for Rho-GTPases. FEBS Lett. 2004, 571, 212–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soupene, E.; Kuypers, F.A. Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. 2008, 233, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nys, Y.; Bain, M.; Van Immerseel, F. Improving the Safety and Quality of Eggs and Egg Products: Volume 1: Egg Chemistry, Production and Consumption; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Li, H.; Wang, T.; Xu, C.; Wang, D.; Ren, J.; Li, Y.; Tian, Y.; Wang, Y.; Jiao, Y.; Kang, X.; et al. Transcriptome profile of liver at different physiological stages reveals potential mode for lipid metabolism in laying hens. BMC Genom. 2015, 16, 763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Wei, W.; Xia, M.; Jiang, Z.; He, D.; Li, Z.; Han, H.; Chu, W.; Liu, H.; Chen, J. Molecular characterization, alternative splicing and expression analysis of ACSF 2 and its correlation with egg-laying performance in geese. Anim. Genet. 2016, 47, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zheng, H.; Yang, L.; Li, H.; Tian, Y.; Wang, Y.; Lyu, S.; Brockmann, G.A.; Kang, X.; Liu, X. Dynamic expression profile, regulatory mechanism and correlation with egg-laying performance of ACSF gene family in chicken (Gallus gallus). Sci. Rep. 2018, 8, 8457. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Marques, M.; Cunha, I.; Reis-Henriques, M.A.; Santos, M.M.; Castro, L.F.C. Diversity and history of the long-chain acyl-CoA synthetase (Acsl) gene family in vertebrates. BMC Evol. Biol. 2013, 13, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J.M.; Frahm, J.L.; Li, L.O.; Coleman, R.A. Acyl-coenzyme A synthetases in metabolic control. Curr. Opin. Lipidol. 2010, 21, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brionne, A.; Nys, Y.; Hennequet-Antier, C.; Gautron, J. Hen uterine gene expression profiling during eggshell formation reveals putative proteins involved in the supply of minerals or in the shell mineralization process. BMC Genom. 2014, 15, 220. [Google Scholar] [CrossRef] [Green Version]
- Jonchère, V.; Réhault-Godbert, S.; Hennequet-Antier, C.; Cabau, C.; Sibut, V.; Cogburn, L.A.; Nys, Y.; Gautron, J. Gene expression profiling to identify eggshell proteins involved in physical defense of the chicken egg. BMC Genom. 2010, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Yung, L.S.; Yang, C.; Wan, X.; Yu, W. GBOOST: A GPU-based tool for detecting gene–gene interactions in genome–wide case control studies. Bioinformatics 2011, 27, 1309–1310. [Google Scholar] [CrossRef] [Green Version]
- Hemani, G.; Theocharidis, A.; Wei, W.; Haley, C. EpiGPU: Exhaustive pairwise epistasis scans parallelized on consumer level graphics cards. Bioinformatics 2011, 27, 1462–1465. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Fang, G. MatrixEpistasis: Ultrafast, exhaustive epistasis scan for quantitative traits with covariate adjustment. Bioinformatics 2018, 34, 2341–2348. [Google Scholar] [CrossRef] [Green Version]
- Chatelain, C.; Durand, G.; Thuillier, V.; Augé, F. Performance of epistasis detection methods in semi-simulated GWAS. BMC Bioinform. 2018, 19, 231. [Google Scholar] [CrossRef]
- Niel, C.; Sinoquet, C.; Dina, C.; Rocheleau, G. A survey about methods dedicated to epistasis detection. Front. Genet. 2015, 6, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, P.; Shen, H. MACOED: A multi-objective ant colony optimization algorithm for SNP epistasis detection in genome-wide association studies. Bioinformatics 2014, 31, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, J.; Lim, W.; Jeong, S.; Lee, H.; Cho, Y.; Moon, J.; Kim, N. Genome-wide association and epistatic interactions of flowering time in soybean cultivar. PLoS ONE 2020, 15, e0228114. [Google Scholar] [CrossRef]
- Cui, Z.; Yang, Q.; Zhang, H.; Zhu, Q.; Zhang, Q. Bioinformatics identification of drug resistance-associated gene pairs in Mycobacterium tuberculosis. Int. J. Mol. Sci. 2016, 17, 1417. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Li, Z.; Song, Z.; Chen, J.; Shi, Y. Genome-wide two-locus interaction analysis identifies multiple epistatic SNP pairs that confer risk of prostate cancer: A cross-population study. Int. J. Cancer 2017, 140, 2075–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egli, T.; Vukojevic, V.; Sengstag, T.; Jacquot, M.; Cabezón, R.; Coynel, D.; Freytag, V.; Heck, A.; Vogler, C.; Dominique, J.; et al. Exhaustive search for epistatic effects on the human methylome. Sci. Rep. 2017, 7, 13669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Domínguez, J.; Schmidt, B. GPU-accelerated exhaustive search for third-order epistatic interactions in case–control studies. J. Comput. Sci. 2015, 8, 93–100. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, X.; He, K.; Lu, L.; Cao, Y.; Liu, J.; Hao, J.; Liu, Z.; Chen, L. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics 2012, 28, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, H.; Tian, T. Development of stock correlation networks using mutual information and financial big data. PLoS ONE 2018, 13, e0195941. [Google Scholar] [CrossRef]
- Mohammadi, S.; Desai, V.; Karimipour, H. Multivariate mutual information-based feature selection for cyber intrusion detection. In Proceedings of the 2018 IEEE Electrical Power and Energy Conference (EPEC), Toronto, ON, Canada, 10–11 October 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 1–6. [Google Scholar]
- Vergara, J.R.; Estévez, P.A. A review of feature selection methods based on mutual information. Neural Comput. Appl. 2014, 24, 175–186. [Google Scholar] [CrossRef]
- Wu, J.; Devlin, B.; Ringquist, S.; Trucco, M.; Roeder, K. Screen and clean: A tool for identifying interactions in genome-wide association studies. Genet Epidemiol. 2010, 34, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Salah El-Basyoni, I.; Stephen Baenziger, P.; Crossa, J.; Eskridge, K.M.; Dweikat, I. Prediction of genetic values of quantitative traits with epistatic effects in plant breeding populations. Heredity 2012, 109, 313–319. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Phenotype | #Samples | #SNPs | #SNPs after Filtering | #SNPs after LD Pruning |
|---|---|---|---|---|---|
| Bovine Tuberculosis | Qualitative | 1151 | 617,885 | 616,398 | 358,086 |
| Egg weight | Quantitative | 1063 | 580,961 | 294,705 | 139,101 |
| Dataset | #MIDESP | #MIDESP_NoAPC | #PLINK | #GBOOST | #epiGPU |
|---|---|---|---|---|---|
| BT | 3,799,984 | 3,799,984 | 4,982,695 | 346,632 | - |
| EW | 1,071,463 | 1,071,463 | 1,817,817 | - | 572,914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinrich, F.; Ramzan, F.; Rajavel, A.; Schmitt, A.O.; Gültas, M. MIDESP: Mutual Information-Based Detection of Epistatic SNP Pairs for Qualitative and Quantitative Phenotypes. Biology 2021, 10, 921. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090921
Heinrich F, Ramzan F, Rajavel A, Schmitt AO, Gültas M. MIDESP: Mutual Information-Based Detection of Epistatic SNP Pairs for Qualitative and Quantitative Phenotypes. Biology. 2021; 10(9):921. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090921
Chicago/Turabian StyleHeinrich, Felix, Faisal Ramzan, Abirami Rajavel, Armin Otto Schmitt, and Mehmet Gültas. 2021. "MIDESP: Mutual Information-Based Detection of Epistatic SNP Pairs for Qualitative and Quantitative Phenotypes" Biology 10, no. 9: 921. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090921